
Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke

 , , ,
, , ,
Abstract
1. Introduction
2. Nomenclature
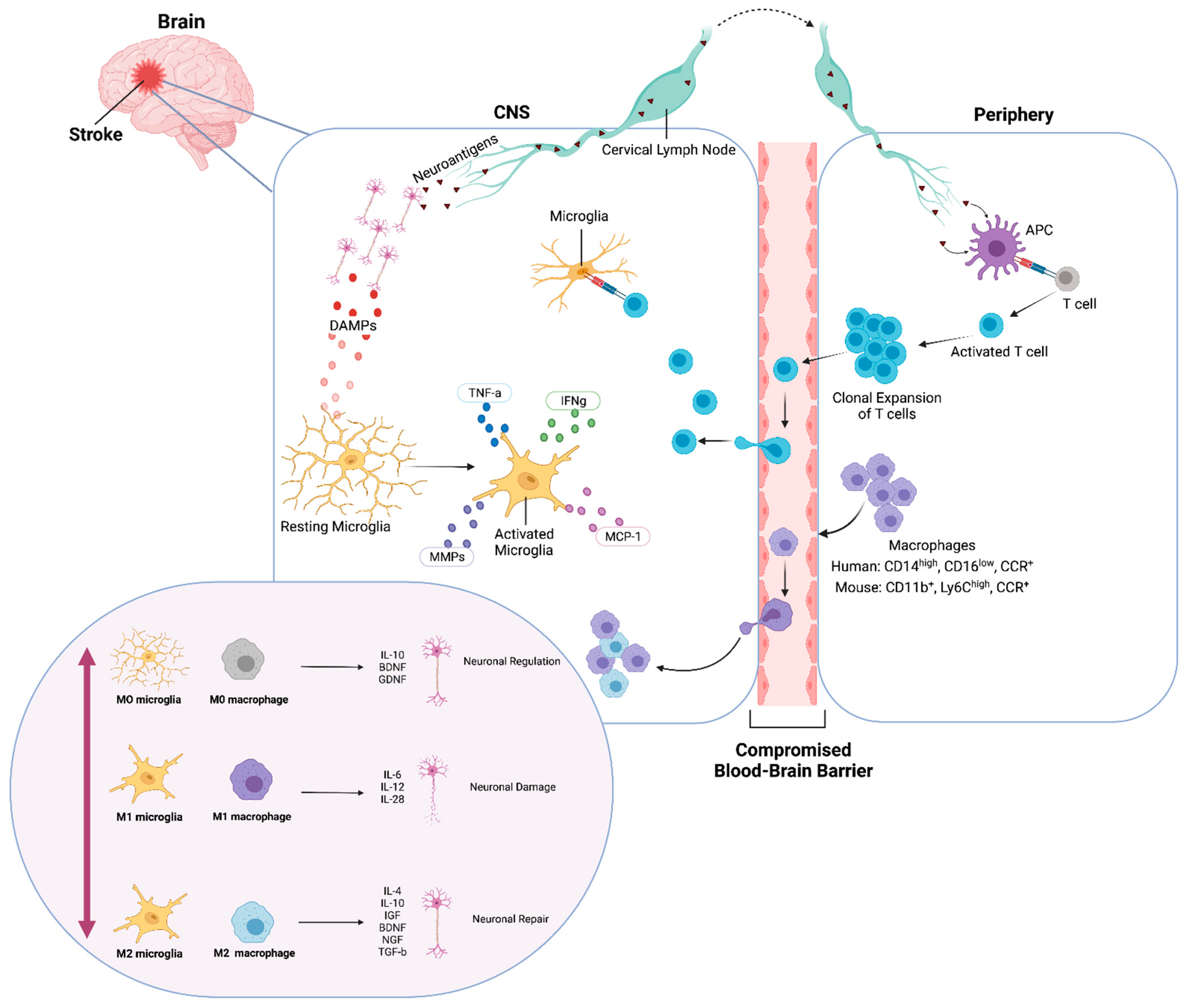
2.1. Microglial Classification
2.2. Macrophage Classification
2.3. Discrepancies in Nomenclature
3. Dichotomous Role of Microglia and Macrophages
3.1. Microglial Activation during Stroke
3.2. Macrophage Activation Profiles
4. Neuroprotection after Stroke
4.1. Angiogenesis
4.2. Synaptic Remodeling
5. Neurogenesis after Stroke
6. Therapeutic Perspectives
6.1. Polarization via Small Molecules
6.2. Stem Cell Therapies
7. Chronic Stroke
Antigen Presentation and Cognitive Decline
8. Microglial Transplantation as a Treatment for Chronic Stroke
8.1. Transplantation of Fetal Microglia
8.2. Transplantation of iPSC Derived Microglia
8.3. Prospects for Transplanting Exogenic Microglia
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef]
- Venkat, P.; Shen, Y.; Chopp, M.; Chen, J. Cell-based and pharmacological neurorestorative therapies for ischemic stroke. Neuropharmacology 2018, 134, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Linfante, I.; Cipolla, M.J. Improving Reperfusion Therapies in the Era of Mechanical Thrombectomy. Transl. Stroke Res. 2016, 7, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Jean, W.C.; Spellman, S.R.; Nussbaum, E.S.; Low, W.C. Reperfusion Injury after Focal Cerebral Ischemia: The Role Inflammation and the therapeutic Horizon. Neurosurgery 1998, 43, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.-Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2016, 157, 247–272. [Google Scholar] [CrossRef]
- Zhao, S.-C.; Ma, L.-S.; Chu, Z.-H.; Xu, H.; Wu, W.-Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Greter, M.; Lelios, I.; Croxford, A.L. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front. Immunol. 2015, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.-M.; Akriditou, M.-A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef]
- Grassivaro, F.; Menon, R.; Acquaviva, M.; Ottoboni, L.; Ruffini, F.; Bergamaschi, A.; Muzio, L.; Farina, C.; Martino, G. Convergence between Microglia and Peripheral Macrophages Phenotype during Development and Neuroinflammation. J. Neurosci. 2019, 40, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Miro-Mur, F.A.; Pedragosa, J.; Zawadzka, M.; Pilanc, P.; Planas, A.M.; Kaminska, B. Defining molecular identity and fates of CNS-border associated macrophages after ischemic stroke in rodents and humans. Neurobiol. Dis. 2020, 137, 104722. [Google Scholar] [CrossRef]
- Ritzel, R.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Abtin, A.; Jain, R.; Mitchell, A.J.; Roediger, B.; Brzoska, A.J.; Tikoo, S.; Cheng, Q.; Ng, L.G.; Cavanagh, L.L.; Von Andrian, U.H.; et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat. Immunol. 2013, 15, 45–53. [Google Scholar] [CrossRef]
- Van Gorp, H.; Delputte, P.L.; Nauwynck, H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010, 47, 1650–1660. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, N.; Yenari, M.A. Mechanisms and Potential Therapeutic Applications of Microglial Activation after Brain Injury. CNS Neurosci. Ther. 2014, 21, 309–319. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.; Ye, Q.; Hassan, S.H.; Zhao, J.; Li, S.; Hu, X.; Leak, R.; Rocha, M.; Wechsler, L.R.; et al. RNA sequencing reveals novel macrophage transcriptome favoring neurovascular plasticity after ischemic stroke. Br. J. Pharmacol. 2019, 40, 720–738. [Google Scholar] [CrossRef]
- Hickman, S.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Chu, S.-H.; Hernandez, M.X.; Fang, M.J.; Modarresi, L.; Selvan, P.; MacGregor, G.R.; Tenner, A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflamm. 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2013, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Kuil, L.E.; Martí, A.L.; Mascaro, A.C.; van den Bosch, J.C.; van den Berg, P.; van der Linde, H.C.; Schoonderwoerd, K.; Ruijter, G.J.G.; van Ham, T.J. Hexb enzyme deficiency leads to lysosomal abnormalities in radial glia and microglia in zebrafish brain development. Glia 2019, 67, 1705–1718. [Google Scholar] [CrossRef]
- Abo-Ouf, H.; Hooper, A.W.; White, E.J.; van Rensburg, H.J.; Trigatti, B.L.; Igdoura, S.A. Deletion of tumor necrosis factor-α ameliorates neurodegeneration in Sandhoff disease mice. Hum. Mol. Genet. 2013, 22, 3960–3975. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2015, 36, 39–49. [Google Scholar] [CrossRef]
- Tanaka, J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neuroimmunol. Neuroinflamm. 2020, 2020, 73–91. [Google Scholar] [CrossRef][Green Version]
- Amici, S.A.; Dong, J.; Guerau-De-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [PubMed]
- Cosenza-Nashat, M.A.; Kim, M.-O.; Zhao, M.-L.; Suh, H.-S.; Lee, S.C. CD45 Isoform Expression in Microglia and Inflammatory Cells in HIV-1 Encephalitis. Brain Pathol. 2006, 16, 256–265. [Google Scholar] [CrossRef]
- Hellwig, S.; Brioschi, S.; Dieni, S.; Frings, L.; Masuch, A.; Blank, T.; Biber, K. Altered microglia morphology and higher resilience to stress-induced depression-like behavior in CX3CR1-deficient mice. Brain Behav. Immun. 2016, 55, 126–137. [Google Scholar] [CrossRef]
- Dos Anjos Cassado, A. F4/80 as a Major Macrophage Marker: The Case of the Peritoneum and Spleen. In Results and Problems in Cell Differentiation; Springer: Amsterdam, The Netherlands, 2017; Volume 62, pp. 161–179. [Google Scholar] [CrossRef]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Imai, Y.; Sasaki, Y.; Kohsaka, S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 2004, 88, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169+ and TCR+ Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- O’Neill, A.S.G.; Berg, T.K.V.D.; Mullen, G.E.D. Sialoadhesin—A macrophage-restricted marker of immunoregulation and inflammation. Immunology 2013, 138, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Claflin, J.; Wang, X.; Lengi, A.; Kikuchi, T. Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int. J. Parasitol. 2005, 35, 83–90. [Google Scholar] [CrossRef]
- Bertani, F.R.; Mozetic, P.; Fioramonti, M.; Iuliani, M.; Ribelli, G.; Pantano, F.; Santini, D.; Tonini, G.; Trombetta, M.; Businaro, L.; et al. Classification of M1/M2-polarized human macrophages by label-free hyperspectral reflectance confocal microscopy and multivariate analysis. Sci. Rep. 2017, 7, 8965. [Google Scholar] [CrossRef]
- Zhang, Y.; Sime, W.; Juhas, M.; Sjölander, A. Crosstalk between colon cancer cells and macrophages via inflammatory mediators and CD47 promotes tumour cell migration. Eur. J. Cancer 2013, 49, 3320–3334. [Google Scholar] [CrossRef]
- Terra, X.; Quintero, Y.; Auguet, T.; Porras, J.A.; Hernández, M.; Sabench, F.; Aguilar, C.; Luna, A.M.; Del Castillo, D.; Richart, C. FABP 4 is associated with inflammatory markers and metabolic syndrome in morbidly obese women. Eur. J. Endocrinol. 2011, 164, 539–547. [Google Scholar] [CrossRef]
- Steen, K.A.; Xu, H.; Bernlohr, D.A. FABP4/aP2 Regulates Macrophage Redox Signaling and Inflammasome Activation via Control of UCP2. Mol. Cell. Biol. 2017, 37, e00282-16. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Nappi, G.; Bernardi, G.; Bagetta, G.; Corasaniti, M.T. Post-ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J. 2008, 276, 13–26. [Google Scholar] [CrossRef]
- Stevens, S.L.; Bao, J.; Hollis, J.; Lessov, N.S.; Clark, W.M.; Stenzel-Poore, M.P. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002, 932, 110–119. [Google Scholar] [CrossRef]
- Schilling, M.; Strecker, J.-K.; Ringelstein, E.B.; Schäbitz, W.-R.; Kiefer, R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009, 1289, 79–84. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and Macrophage Plasticity in Tissue Repair and Regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Tian, P.-X.; Han, F.; Zheng, J.; Xia, X.-X.; Xue, W.-J.; Ding, X.-M.; Ding, C.-G. Comparison of the characteristics of macrophages derived from murine spleen, peritoneal cavity, and bone marrow. J. Zhejiang Univ. Sci. B 2017, 18, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hallenbeck, J.M.; Ruetzler, C.; Bol, D.; Thomas, K.; Berman, N.E.J.; Vogel, S.N. Overexpression of Monocyte Chemoattractant Protein 1 in the Brain Exacerbates Ischemic Brain Injury and is Associated with Recruitment of Inflammatory Cells. J. Cereb. Blood Flow Metab. 2003, 23, 748–755. [Google Scholar] [CrossRef]
- Schuette-Nuetgen, K.; Strecker, J.-K.; Minnerup, J.; Ringelstein, E.B.; Schilling, M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp. Neurol. 2012, 233, 849–858. [Google Scholar] [CrossRef]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte Chemoattractant Protein-1 Deficiency is Protective in a Murine Stroke Model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.; Andjelkovic, A.V. Absence of the Chemokine Receptor CCR2 Protects Against Cerebral Ischemia/Reperfusion Injury in Mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Perego, C.; Fumagalli, S.; De Simoni, M.-G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Crane, M.J.; Daley, J.M.; Van Houtte, O.; Brancato, S.K.; Jr, W.L.H.; Albina, J.E. The Monocyte to Macrophage Transition in the Murine Sterile Wound. PLoS ONE 2014, 9, e86660. [Google Scholar] [CrossRef]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, J.; Wang, R.; Jiang, M.; Ye, Q.; Smith, A.D.; Chen, J.; Shi, Y. Macrophages reprogram after ischemic stroke and promote efferocytosis and inflammation resolution in the mouse brain. CNS Neurosci. Ther. 2019, 25, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Cell Biol. 2009, 185, i6. [Google Scholar] [CrossRef]
- Zhao, X.; Eyo, U.B.; Murugan, M.; Wu, L.-J.; Murguan, M. Microglial interactions with the neurovascular system in physiology and pathology. Dev. Neurobiol. 2018, 78, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Mao, X.; Jin, K.; Greenberg, D.A. Vascular endothelial growth factor-B expression in postischemic rat brain. Vasc. Cell 2013, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; van Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2014, 129, 279–295. [Google Scholar] [CrossRef]
- Nie, J.; Yang, X. Modulation of Synaptic Plasticity by Exercise Training as a Basis for Ischemic Stroke Rehabilitation. Cell. Mol. Neurobiol. 2016, 37, 5–16. [Google Scholar] [CrossRef]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.W.H.; Koizumi, K.S.S.; Moorhouse, A.; Yoshimura, A.W.I.Y.; Nabekura, A.M. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.; Duan, X.; Tian, X.; Shen, H.; Sun, Q.; Chen, G. NADPH Oxidase: A Potential Target for Treatment of Stroke. Oxid. Med. Cell. Longev. 2016, 2016, 5026984. [Google Scholar] [CrossRef]
- Zhang, J.; Malik, A.; Choi, H.B.; Ko, R.W.; Dissing-Olesen, L.; MacVicar, B.A. Microglial CR3 Activation Triggers Long-Term Synaptic Depression in the Hippocampus via NADPH Oxidase. Neuron 2014, 82, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.; Wu, L.-J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.R.; Schoenfeld, T.J.; Karlsson, R.-M.; Bannerman, D.M.; Cameron, H.A. Ongoing neurogenesis in the adult dentate gyrus mediates behavioral responses to ambiguous threat cues. PLoS Biol. 2017, 15, e2001154. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Meinl, E.; Hohlfeld, R. Neuro-Immune Crosstalk in CNS Diseases. Results Probl. Cell Differ. 2010, 51, 197–216. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef]
- Lin, R.; Iacovitti, L. Classic and novel stem cell niches in brain homeostasis and repair. Brain Res. 2015, 1628, 327–342. [Google Scholar] [CrossRef]
- Otero, L.; Zurita, M.; Bonilla, C.; Rico, M.A.; Aguayo, C.; Rodriguez, A.; Vaquero, J. Endogenous neurogenesis after intracerebral hemorrhage. Histol. Histopathol. 2012, 27, 303–315. [Google Scholar] [CrossRef]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and Inflammation after Ischemic Stroke: What is Known and Where We Go from Here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Kreuzberg, M.; Kanov, E.; Timofeev, O.; Schwaninger, M.; Monyer, H.; Khodosevich, K. Increased subventricular zone-derived cortical neurogenesis after ischemic lesion. Exp. Neurol. 2010, 226, 90–99. [Google Scholar] [CrossRef]
- Lindvall, O.; Kokaia, Z. Neurogenesis following Stroke Affecting the Adult Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a019034. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, J.; Lin, X.; Wang, L.; Shao, B.; Jin, K.; Wang, Y.; Yang, G.-Y. Neural Stem Cell Protects Aged Rat Brain from Ischemia–Reperfusion Injury through Neurogenesis and Angiogenesis. J. Cereb. Blood Flow Metab. 2014, 34, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Nada, S.E.; Tulsulkar, J.; Shah, Z.A. Heme Oxygenase 1-Mediated Neurogenesis Is Enhanced by Ginkgo biloba (EGb 761®) After Permanent Ischemic Stroke in Mice. Mol. Neurobiol. 2013, 49, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, P.E.; Porritt, M.J.; Martinello, P.; Parish, C.L.; Liberatore, G.T.; Donnan, G.A.; Howells, D.W. Macrophages and Microglia Produce Local Trophic Gradients That Stimulate Axonal Sprouting Toward but Not beyond the Wound Edge. Mol. Cell. Neurosci. 2002, 21, 436–453. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Ferretti, M.T. Function and Dysfunction of Microglia during Brain Development: Consequences for Synapses and Neural Circuits. Front. Synaptic Neurosci. 2017, 9, 9. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L. Microglia and Beyond: Innate Immune Cells as Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.; Sanberg, P.R.; Bickford, P.; Kaneko, Y.; Borlongan, C.V. Long-Term Upregulation of Inflammation and Suppression of Cell Proliferation in the Brain of Adult Rats Exposed to Traumatic Brain Injury Using the Controlled Cortical Impact Model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Rotschafer, J.H.; Hu, S.; Little, M.; Erickson, M.; Low, W.C.; Cheeran, M.C. Modulation of neural stem/progenitor cell proliferation during experimental Herpes Simplex encephalitis is mediated by differential FGF-2 expression in the adult brain. Neurobiol. Dis. 2013, 58, 144–155. [Google Scholar] [CrossRef]
- Fan, W.; Dai, Y.; Xu, H.; Zhu, X.; Cai, P.; Wang, L.; Sun, C.; Hu, C.; Zheng, P.; Zhao, B. Caspase-3 Modulates Regenerative Response After Stroke. Stem Cells 2013, 32, 473–486. [Google Scholar] [CrossRef]
- Kempermann, G.; Gage, F.H.; Aigner, L.; Song, H.; Curtis, M.A.; Thuret, S.; Kuhn, H.-G.; Jessberger, S.; Frankland, P.W.; Cameron, H.A.; et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 2018, 23, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Ron-Harel, N.; Gal, H.; Deczkowska, A.; Shifrut, E.; Ndifon, W.; Mirlas-Neisberg, N.; Cardon, M.; Vaknin, I.; Cahalon, L.; et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc. Natl. Acad. Sci. USA 2013, 110, 2264–2269. [Google Scholar] [CrossRef]
- Hu, S.; Rotschafer, J.H.; Lokensgard, J.R.; Cheeran, M.C.-J. Activated CD8+ T Lymphocytes Inhibit Neural Stem/Progenitor Cell Proliferation: Role of Interferon-Gamma. PLoS ONE 2014, 9, e105219. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, Y.; Won, S.J.; Neumann, M.; Hu, D.; Zhou, L.; Weinstein, P.R.; Liu, J. Chronic Treatment with Minocycline Preserves Adult New Neurons and Reduces Functional Impairment After Focal Cerebral Ischemia. Stroke 2007, 38, 146–152. [Google Scholar] [CrossRef]
- Song, D.; Zhang, X.; Chen, J.; Liu, X.; Xue, J.; Zhang, L.; Lan, X. Wnt canonical pathway activator TWS119 drives microglial anti-inflammatory activation and facilitates neurological recovery following experimental stroke. J. Neuroinflamm. 2019, 16, 1–17. [Google Scholar] [CrossRef]
- Laksitorini, M.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/β-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 19718. [Google Scholar] [CrossRef]
- Fumagalli, S.; Perego, C.; Pischiutta, F.; Zanier, E.; de Simoni, M.G. The Ischemic Environment Drives Microglia and Macrophage Function. Front. Neurol. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Gimeno-Bayón, J.; López-López, A.; Rodríguez, M.; Mahy, N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef]
- Li, C.; Bian, Y.; Feng, Y.; Tang, F.; Wang, L.; Hoi, M.P.M.; Ma, D.; Zhao, C.; Lee, S.M.Y. Neuroprotective Effects of BHDPC, a Novel Neuroprotectant, on Experimental Stroke by Modulating Microglia Polarization. ACS Chem. Neurosci. 2019, 10, 2434–2449. [Google Scholar] [CrossRef]
- Cai, W.; Liu, S.; Hu, M.; Sun, X.; Qiu, W.; Zheng, S.; Hu, X.; Lu, Z. Post-stroke DHA Treatment Protects Against Acute Ischemic Brain Injury by Skewing Macrophage Polarity Toward the M2 Phenotype. Transl. Stroke Res. 2018, 9, 669–680. [Google Scholar] [CrossRef]
- Lei, X.; Li, H.; Li, M.; Dong, Q.; Zhao, H.; Zhang, Z.; Sun, B.; Mao, L. The novel Nrf2 activator CDDO-EA attenuates cerebral ischemic injury by promoting microglia/macrophage polarization toward M2 phenotype in mice. CNS Neurosci. Ther. 2020, 27, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, H.; Xiang, F.; Xu, L.; Dong, Z. β-Caryophyllene protects against ischemic stroke by promoting polarization of microglia toward M2 phenotype via the TLR4 pathway. Life Sci. 2019, 237, 116915. [Google Scholar] [CrossRef]
- Korhonen, P.; Kanninen, K.M.; Lehtonen, S.; Lemarchant, S.; Puttonen, K.; Oksanen, M.; Dhungana, H.; Loppi, S.; Pollari, E.; Wojciechowski, S.; et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav. Immun. 2015, 49, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cao, D.; Guo, C.; Liu, M.; Tao, Y.; Zhou, J.; Wang, F.; Zhao, Y.; Wei, J.; Zhang, Y.; et al. Berberine Facilitates Angiogenesis Against Ischemic Stroke Through Modulating Microglial Polarization via AMPK Signaling. Cell. Mol. Neurobiol. 2019, 39, 751–768. [Google Scholar] [CrossRef]
- Benedek, G.; Vandenbark, A.A.; Alkayed, N.J.; Offner, H. Partial MHC class II constructs as novel immunomodulatory therapy for stroke. Neurochem. Int. 2016, 107, 138–147. [Google Scholar] [CrossRef]
- Brown, J.; Kingsbury, C.; Lee, J.; Vandenbark, A.A.; Meza-Romero, R.; Offner, H.; Borlongan, C.V. Spleen participation in partial MHC class II construct neuroprotection in stroke. CNS Neurosci. Ther. 2020, 26, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Castelli, V.; Bonsack, B.; Coats, A.B.; Navarro-Torres, L.; Garcia-Sanchez, J.; Kingsbury, C.; Nguyen, H.; Vandenbark, A.A.; Meza-Romero, R.; et al. A Novel Partial MHC Class II Construct, DRmQ, Inhibits Central and Peripheral Inflammatory Responses to Promote Neuroprotection in Experimental Stroke. Transl. Stroke Res. 2020, 11, 831–836. [Google Scholar] [CrossRef]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Berry, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar] [CrossRef]
- Yu, G.; Liang, Y.; Zheng, S.; Zhang, H. Inhibition of Myeloperoxidase by N-Acetyl Lysyltyrosylcysteine Amide Reduces Oxidative Stress-Mediated Inflammation, Neuronal Damage, and Neural Stem Cell Injury in a Murine Model of Stroke. J. Pharmacol. Exp. Ther. 2018, 364, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; McBride, D.W.; Zhang, J.H. Axl activation attenuates neuroinflammation by inhibiting the TLR/TRAF/NF-κB pathway after MCAO in rats. Neurobiol. Dis. 2017, 110, 59–67. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, W.-N.; Matei, N.; Li, X.; Pang, J.-W.; Mo, J.; Chen, S.-P.; Tang, J.-P.; Yan, M.; Zhang, J.H. Ezetimibe Attenuates Oxidative Stress and Neuroinflammation via the AMPK/Nrf2/TXNIP Pathway after MCAO in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 4717258. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, H.; Du, Q.; Shen, J. Targeting Myeloperoxidase (MPO) Mediated Oxidative Stress and Inflammation for Reducing Brain Ischemia Injury: Potential Application of Natural Compounds. Front. Physiol. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Sakaguchi, M.; Kuroiwa, T.; Yamasaki, M.; Kanemura, Y.; Shizuko, I.; Shimazaki, T.; Onodera, M.; Okano, H.; Mizusawa, H. Human neural stem/progenitor cells, expanded in long-term neurosphere culture, promote functional recovery after focal ischemia in Mongolian gerbils. J. Neurosci. Res. 2004, 78, 215–223. [Google Scholar] [CrossRef]
- Fukunaga, A.; Uchida, K.; Hara, K.; Kuroshima, Y.; Kawase, T. Differentiation and angiogenesis of central nervous system stem cells implanted with mesenchyme into ischemic rat brain. Cell Transplant. 1999, 8, 435–441. [Google Scholar] [CrossRef]
- Daadi, M.M.; Li, Z.; Arac, A.; Grueter, B.; Sofilos, M.; Malenka, R.C.; Wu, J.C.; Steinberg, G.K. Molecular and Magnetic Resonance Imaging of Human Embryonic Stem Cell–Derived Neural Stem Cell Grafts in Ischemic Rat Brain. Mol. Ther. 2009, 17, 1282–1291. [Google Scholar] [CrossRef]
- Muir, K.W.; Bulters, D.; Willmot, M.; Sprigg, N.; Dixit, A.; Ward, N.; Tyrrell, P.; Majid, A.; Dunn, L.; Bath, P.; et al. Intracerebral implantation of human neural stem cells and motor recovery after stroke: Multicentre prospective single-arm study (PISCES-2). J. Neurol. Neurosurg. Psychiatry 2020, 91, 396–401. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Wang, L.; Lu, M.; Zhang, X.; Chopp, M. Therapeutic benefit of intracerebral transplantation of bone marrow stromal cells after cerebral ischemia in rats. J. Neurol. Sci. 2001, 189, 49–57. [Google Scholar] [CrossRef]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Bates, D.; King, B.; Case, C. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: A phase 1/2a study. Stroke 2016, 47, 1817–1824. [Google Scholar] [CrossRef]
- Luan, X.; Qiu, H.; Hong, X.; Wu, C.; Zhao, K.; Chen, H.; Zhu, Z.; Li, X.; Shen, H.; He, J. High serum nerve growth factor concentrations are associated with good functional outcome at 3 months following acute ischemic stroke. Clin. Chim. Acta 2018, 488, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.-J.; Li, Z.; Qiu, J.-Y.; Zheng, X.-X.; Bian, T.-T.; Gao, F.-L.; Yu, Y.-Y.; Yang, D.-Z.; Tang, D.-Q. Screening for Potential Bioactive Components in Ginkgo biloba Extract by the Rat Renal Tubular Epithelial Cell Extraction and LC-MS/MS. Comb. Chem. High Throughput Screen. 2015, 18, 514–523. [Google Scholar] [CrossRef]
- Tang, W.; Lv, X.; Huang, J.; Wang, B.; Lin, L.; Shen, Y.; Yao, Y. Neuroprotective Effect of Stroke Pretreated Mesenchymal Stem Cells Against Cerebral Ischemia/Reperfusion Injury in Rats. World Neurosurg. 2021. [Google Scholar] [CrossRef]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.-C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Metheny, L.; Caimi, P.; De Lima, M. Cord Blood Transplantation: Can We Make it Better? Front. Oncol. 2013, 3, 238. [Google Scholar] [CrossRef]
- Gupta, A.O.; Wagner, J.E. Umbilical Cord Blood Transplants: Current Status and Evolving Therapies. Front. Pediatr. 2020, 8, 570282. [Google Scholar] [CrossRef] [PubMed]
- Shiao, M.L.; Yuan, C.; Crane, A.T.; Voth, J.; Juliano, M.; Stone, L.L.H.; Nan, Z.; Zhang, Y.; Kuzmin-Nichols, N.; Sanberg, P.R.; et al. Immunomodulation with Human Umbilical Cord Blood Stem Cells Ameliorates Ischemic Brain Injury—A Brain Transcriptome Profiling Analysis. Cell Transplant. 2019, 28, 864–873. [Google Scholar] [CrossRef]
- Stone, L.L.H.; Xiao, F.; Rotschafer, J.; Nan, Z.; Juliano, M.; Sanberg, C.D.; Sanberg, P.R.; Kuzmin-Nichols, N.; Grande, A.; Cheeran, M.; et al. Amelioration of Ischemic Brain Injury in Rats with Human Umbilical Cord Blood Stem Cells: Mechanisms of Action. Cell Transplant. 2016, 25, 1473–1488. [Google Scholar] [CrossRef]
- Levine, D.A.; Galecki, A.T.; Langa, K.; Unverzagt, F.W.; Kabeto, M.U.; Giordani, B.; Wadley, V.G. Trajectory of Cognitive Decline After Incident Stroke. JAMA 2015, 314, 41–51. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Pösel, C.; Uri, A.; Schulz, I.; Boltze, J.; Weise, G.; Wagner, D.-C. Flow cytometric characterization of brain dendritic cell subsets after murine stroke. Exp. Transl. Stroke Med. 2014, 6, 11. [Google Scholar] [CrossRef][Green Version]
- Felger, J.C.; Abe, T.; Kaunzner, U.W.; Gottfried-Blackmore, A.; Gal-Toth, J.; McEwen, B.S.; Iadecola, C.; Bulloch, K. Brain dendritic cells in ischemic stroke: Time course, activation state, and origin. Brain Behav. Immun. 2010, 24, 724–737. [Google Scholar] [CrossRef]
- Schwab, J.; Nguyen, T.; Meyermann, R.; Schluesener, H. Human focal cerebral infarctions induce differential lesional interleukin-16 (IL-16) expression confined to infiltrating granulocytes, CD8+ T-lymphocytes and activated microglia/macrophages. J. Neuroimmunol. 2001, 114, 232–241. [Google Scholar] [CrossRef]
- Miro-Mur, F.A.; Urra, X.; Gallizioli, M.; Chamorro, A.; Planas, A.M. Antigen Presentation after Stroke. Neurotherapeutics 2016, 13, 719–728. [Google Scholar] [CrossRef]
- Gottfried-Blackmore, A.; Kaunzner, U.W.; Idoyaga, J.; Felger, J.C.; McEwen, B.S.; Bulloch, K. Acute in vivo exposure to interferon- enables resident brain dendritic cells to become effective antigen presenting cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20918–20923. [Google Scholar] [CrossRef]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.A.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Dando, S.J.; Naranjo Golborne, C.; Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. A case of mistaken identity: CD11c-eYFP+ cells in the normal mouse brain parenchyma and neural retina display the phenotype of microglia, not dendritic cells. Glia 2016, 64, 1331–1349. [Google Scholar] [CrossRef] [PubMed]
- Gregerson, D.S.; Sam, T.N.; McPherson, S.W. The antigen-presenting activity of fresh, adult parenchymal microglia and perivascular cells from retina. J. Immunol. 2004, 172, 6587–6597. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Løbner, M.; Cédile, O.; Owens, T. Comparison of microglia and infiltrating CD11c+ cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 57. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- Parachikova, A.; Agadjanyan, M.; Cribbs, D.; Blurton-Jones, M.; Perreau, V.; Rogers, J.; Beach, T.; Cotman, C. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol. Aging 2007, 28, 1821–1833. [Google Scholar] [CrossRef]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- Planas, A.M.; Gómez-Choco, M.; Urra, X.; Gorina, R.; Caballero, M.; Chamorro, Á. Brain-Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke. J. Immunol. 2012, 188, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Jauch, E.C.; Lindsell, C.; Broderick, J.; Fagan, S.C.; Tilley, B.C.; Levine, S.R. Association of serial biochemical markers with acute ischemic stroke: The National Institute of Neurological Disorders and Stroke recombinant tissue plasminogen activator Stroke Study. Stroke 2006, 37, 2508–2513. [Google Scholar] [CrossRef]
- Doyle, K.; Quach, L.N.; Solé, M.; Axtell, R.C.; Nguyen, T.-V.V.; Soler-Llavina, G.J.; Jurado, S.; Han, J.; Steinman, L.; Longo, F.M.; et al. B-Lymphocyte-Mediated Delayed Cognitive Impairment following Stroke. J. Neurosci. 2015, 35, 2133–2145. [Google Scholar] [CrossRef]
- Bornstein, N.; Aronovich, B.; Korczyn, A.; Shavit, S.; Michaelson, D.; Chapman, J. Antibodies to brain antigens following stroke. Neurology 2001, 56, 529–530. [Google Scholar] [CrossRef] [PubMed]
- Prüss, H.; Iggena, D.; Baldinger, T.; Prinz, V.; Meisel, A.; Endres, M.; Dirnagl, U.; Schwab, J. Evidence of intrathecal immunoglobulin synthesis in stroke: A cohort study. Arch. Neurol. 2012, 69, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, K.; Dziennis, S.; Palmateer, J.; Ren, X.; Vandenbark, A.A.; Offner, H.; Herson, P.S.; Hurn, P.D. Recombinant T Cell Receptor Ligands Improve Outcome After Experimental Cerebral Ischemia. Transl. Stroke Res. 2011, 2, 404–410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, W.; Casper, A.; Libal, N.L.; Murphy, S.J.; Bodhankar, S.; Offner, H.; Alkayed, N.J. Preclinical evaluation of recombinant T cell receptor ligand RTL1000 as a therapeutic agent in ischemic stroke. Transl. Stroke Res. 2014, 6, 60–68. [Google Scholar] [CrossRef]
- Wang, J.; Ye, Q.; Xu, J.; Benedek, G.; Zhang, H.; Yang, Y.; Liu, H.; Meza-Romero, R.; Vandenbark, A.A.; Offner, H.; et al. DRα1-MOG-35-55 Reduces Permanent Ischemic Brain Injury. Transl. Stroke Res. 2016, 8, 284–293. [Google Scholar] [CrossRef]
- Kou, D.; Li, T.; Liu, H.; Liu, C.; Yin, Y.; Wu, X.; Yu, T. Transplantation of rat-derived microglial cells promotes functional recovery in a rat model of spinal cord injury. Braz. J. Med. Biol. Res. 2018, 51, e7076. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Yanagisawa, D.; Morikawa, S.; Morita, M.; Inubushi, T.; Tsuchiya, D.; Chishiro, S.; Saeki, M.; Taniguchi, T.; et al. Microglial transplantation increases amyloid-β clearance in Alzheimer model rats. FEBS Lett. 2007, 581, 475–478. [Google Scholar] [CrossRef]
- Wei, L.; Wei, Z.Z.; Jiang, M.Q.; Mohamad, O.; Yu, S.P. Stem cell transplantation therapy for multifaceted therapeutic benefits after stroke. Prog. Neurobiol. 2017, 157, 49–78. [Google Scholar] [CrossRef]
- Thiel, A.; Cechetto, D.F.; Heiss, W.-D.; Hachinski, V.; Whitehead, S.N. Amyloid Burden, Neuroinflammation, and Links to Cognitive Decline after Ischemic Stroke. Stroke 2014, 45, 2825–2829. [Google Scholar] [CrossRef]
- Basu, A.; Lazovic, J.; Krady, J.K.; Mauger, D.T.; Rothstein, R.P.; Smith, M.B.; Levison, S. Interleukin-1 and the Interleukin-1 Type 1 Receptor are Essential for the Progressive Neurodegeneration that Ensues Subsequent to a Mild Hypoxic/Ischemic Injury. Br. J. Pharmacol. 2005, 25, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H.; Zhang, H.; Ye, Q.; Wang, J.; Yang, B.; Mao, L.; Zhu, W.; Leak, R.; Xiao, B.; et al. ST2/IL-33-Dependent Microglial Response Limits Acute Ischemic Brain Injury. J. Neurosci. 2017, 37, 4692–4704. [Google Scholar] [CrossRef]
- Narantuya, D.; Nagai, A.; Sheikh, A.M.; Masuda, J.; Kobayashi, S.; Yamaguchi, S.; Kim, S.U. Human Microglia Transplanted in Rat Focal Ischemia Brain Induce Neuroprotection and Behavioral Improvement. PLoS ONE 2010, 5, e11746. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Yano, S.; Mitaki, S.; Haque, M.A.; Yamaguchi, S.; Nagai, A. A Mesenchymal stem cell line (B10) increases angiogenesis in a rat MCAO model. Exp. Neurol. 2018, 311, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Narantuya, D.; Nagai, A.; Sheikh, A.M.; Wakabayashi, K.; Shiota, Y.; Watanabe, T.; Masuda, J.; Kobayashi, S.; Kim, S.U.; Yamaguchi, S. Microglia transplantation attenuates white matter injury in rat chronic ischemia model via matrix metalloproteinase-2 inhibition. Brain Res. 2010, 1316, 145–152. [Google Scholar] [CrossRef]
- Quarta, A.; Le Blon, D.; D’Aes, T.; Pieters, Z.; Taj, S.H.; Miro-Mur, F.A.; Luyckx, E.; Van Breedam, E.; Daans, J.; Goossens, H.; et al. Murine iPSC-derived microglia and macrophage cell culture models recapitulate distinct phenotypical and functional properties of classical and alternative neuro-immune polarisation. Brain Behav. Immun. 2019, 82, 406–421. [Google Scholar] [CrossRef]
- Svoboda, D.S.; Barrasa, M.I.; Shu, J.; Rietjens, R.; Zhang, S.; Mitalipova, M.; Berube, P.; Fu, D.; Shultz, L.D.; Bell, G.W.; et al. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 25293–25303. [Google Scholar] [CrossRef]
- Xu, R.; Li, X.; Boreland, A.; Posyton, A.; Kwan, K.; Hart, R.P.; Jiang, P. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tatarishvili, J.; Oki, K.; Monni, E.; Koch, P.; Memanishvili, T.; Buga, A.-M.; Verma, V.; Popa-Wagner, A.; Brüstle, O.; Lindvall, O.; et al. Human induced pluripotent stem cells improve recovery in stroke-injured aged rats. Restor. Neurol. Neurosci. 2014, 32, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Saito, H.; Shinozaki, Y.; Shigetomi, E.; Miwa, H.; Yoneda, S.; Tanimura, M.; Omachi, S.; Asaki, T.; Takahashi, K.; et al. Transnasal transplantation of human induced pluripotent stem cell-derived microglia to the brain of immunocompetent mice. Glia 2021, 69, 2332–2348. [Google Scholar] [CrossRef] [PubMed]
- Imai, F.; Suzuki, H.; Oda, J.; Ninomiya, T.; Ono, K.; Sano, H.; Sawada, M. Neuroprotective Effect of Exogenous Microglia in Global Brain Ischemia. Br. J. Pharmacol. 2006, 27, 488–500. [Google Scholar] [CrossRef]
- Kanazawa, M.; Miura, M.; Toriyabe, M.; Koyama, M.; Hatakeyama, M.; Ishikawa, M.; Nakajima, T.; Onodera, O.; Takahashi, T.; Nishizawa, M.; et al. Microglia preconditioned by oxygen-glucose deprivation promote functional recovery in ischemic rats. Sci. Rep. 2017, 7, 42582. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Z.; Qu, G.; Wang, H.; Lin, L.; Li, X.; Xie, X.; Lei, Y.; He, X.; Chen, Y.; et al. Hypoxic Preconditioning Enhances the Efficacy of Mesenchymal Stem Cells-Derived Conditioned Medium in Switching Microglia toward Anti-inflammatory Polarization in Ischemia/Reperfusion. Cell. Mol. Neurobiol. 2020, 41, 505–524. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Type | Marker | References |
|---|---|---|
| Microglia | C1qa | [23,24] |
| CD11b+CD45low | [21,22] | |
| CD11b+CD45int | [21,22] | |
| Fcrls | [24,25] | |
| Hexb | [26,27] | |
| P2ry12 | [17,24] | |
| P2ry13 | [17,24] | |
| Tmem119 | [17,28] | |
| Microglia and Macrophage | CD115 | [20,21] |
| CD11b | [20,21] | |
| CD45 | [29,30,31] | |
| CD68 | [30,31] | |
| Cx3cr1 | [24,32] | |
| F4/80 | [21,33] | |
| Iba1 | [34,35] | |
| Macrophage | CD163 | [16,18,19] |
| CD169 | [36,37] | |
| CD11b+CD45high | [21,38] | |
| CD206high | [39,40] | |
| Fabp4 | [41,42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Var, S.R.; Shetty, A.V.; Grande, A.W.; Low, W.C.; Cheeran, M.C. Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke. Cells 2021, 10, 3555. https://doi.org/10.3390/cells10123555
Var SR, Shetty AV, Grande AW, Low WC, Cheeran MC. Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke. Cells. 2021; 10(12):3555. https://doi.org/10.3390/cells10123555
Chicago/Turabian StyleVar, Susanna R., Anala V. Shetty, Andrew W. Grande, Walter C. Low, and Maxim C. Cheeran. 2021. "Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke" Cells 10, no. 12: 3555. https://doi.org/10.3390/cells10123555
APA StyleVar, S. R., Shetty, A. V., Grande, A. W., Low, W. C., & Cheeran, M. C. (2021). Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke. Cells, 10(12), 3555. https://doi.org/10.3390/cells10123555