
Cellular Senescence in Adrenocortical Biology and Its Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
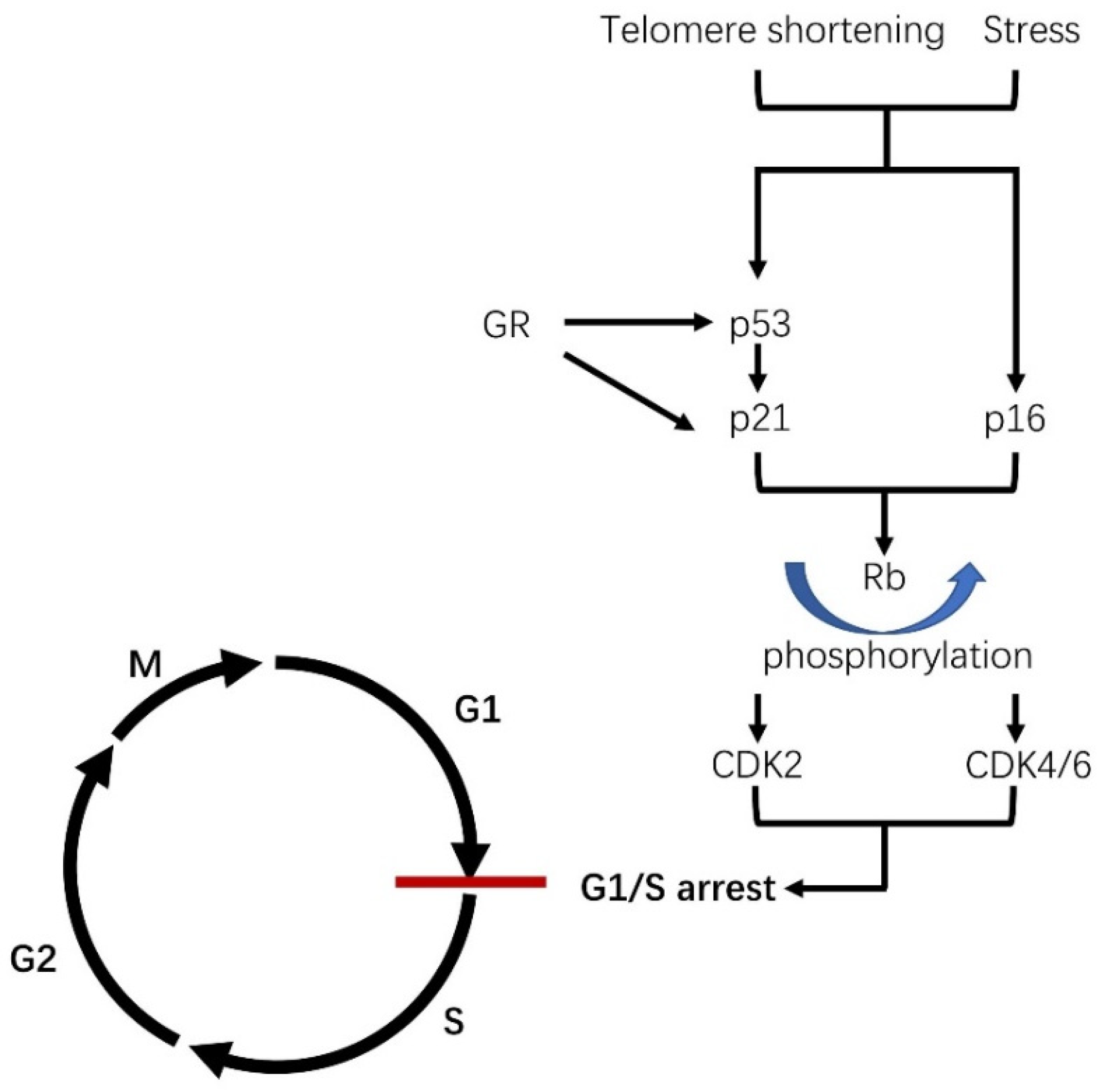
1.1. What Is Cellular Senescence?
1.2. Senescence-Associated Secretory Phenotype (SASP)
1.3. The Role of Cellular Senescence in Age and Its Related Disease
1.4. Physiology in the Adrenal Cortex
2. Age-Related Changes in the Adrenal Cortex
2.1. Age-Related Changes in Aldosterone Production
2.2. Age-Related Changes in Cortisol Production
2.3. Age-Related Changes in Adrenal Androgen Production
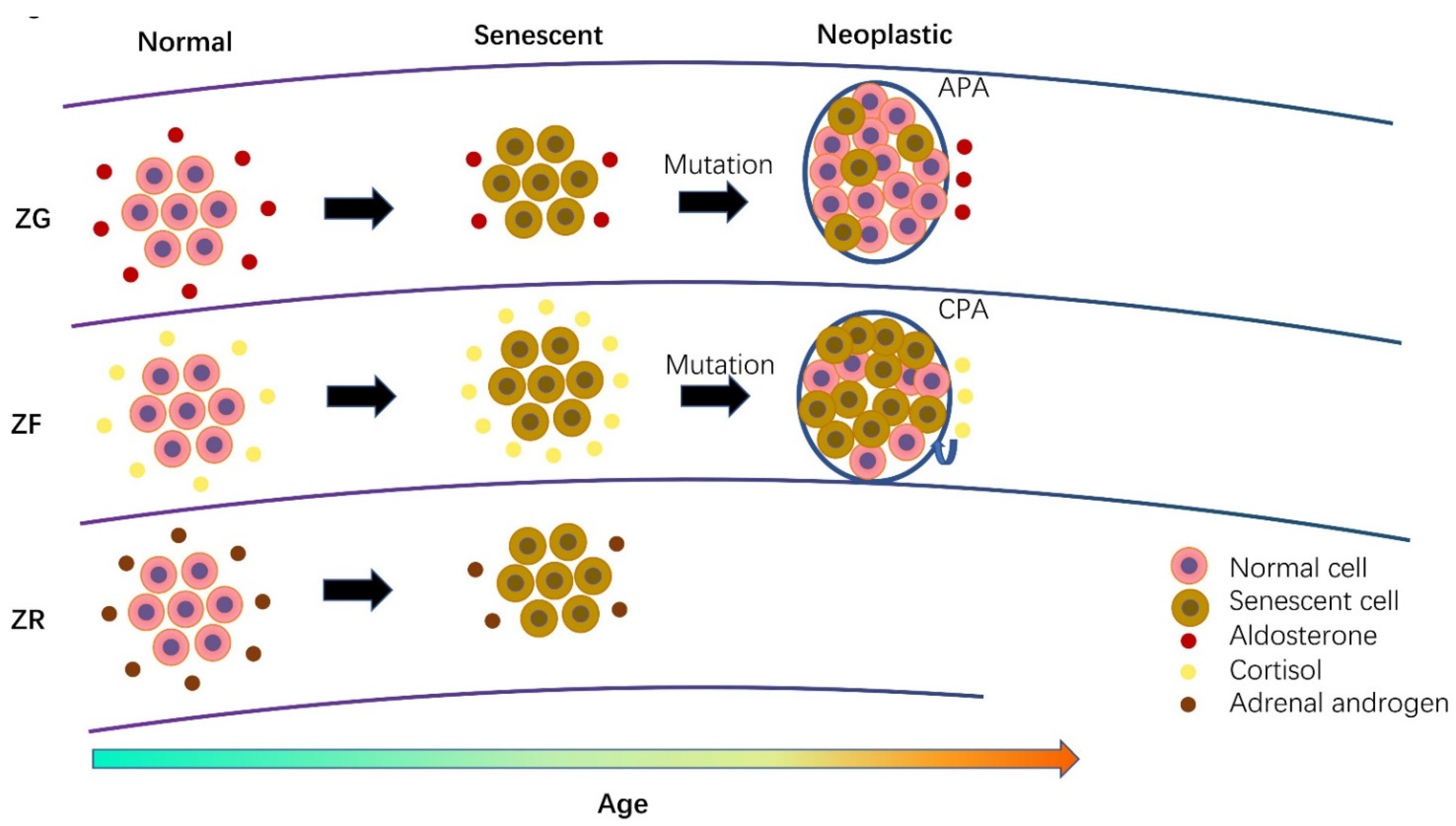
3. Cellular Senescence in Normal Adrenal Cortex and Its Disorders
3.1. Cellular Senescence in Aldosterone-Producing Adenoma
3.2. Cellular Senescence in Cortisol-Producing Adenoma
3.3. Comparison of Senescence between Different Adrenal Hormone-Producing Adenoma
3.4. Cellular Senescence in Adrenocortical Carcinoma
3.5. The Role of Lipofuscin in the Adrenal Gland and Its Related Disorders
3.6. The Role of Cellular Senescence in Adrenal Physiology during Aging
4. Adrenocortical Steroids Induce Distant Cellular Senescence in Other Tissues
4.1. Aldosterone and Glucocorticoid-Induced Cellular Senescence in the Kidneys
4.2. Aldosterone-Induced Cellular Senescence in Vascular Smooth Muscle Cells
4.3. Glucocorticoid-Induced Cellular Senescence in Hepatoma Cells
4.4. Glucocorticoid-Induced Cellular Senescence in the Development of Fetal Lungs and Lung Cancers
4.5. Glucocorticoid-Induced Cellular Senescence in Neural Cells
4.6. Glucocorticoid-Induced Cellular Senescence in Skeletal Damage
4.7. Adrenal Androgen-Induced Cellular Senescence
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Toussaint, O.; Dumont, P.; Dierick, J.F.; Pascal, T.; Frippiat, C.; Chainiaux, F.; Sluse, F.; Eliaers, F.; Remacle, J. Stress-induced premature senescence. Essence of life, evolution, stress, and aging. Ann. N. Y. Acad. Sci. 2000, 908, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. What is and what is not cell senescence. Postepy Biochem. 2018, 64, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Dobbie, J.W. Adrenocortical nodular hyperplasia: The ageing adrenal. J. Pathol. 1969, 99, 1–18. [Google Scholar] [CrossRef]
- Neville, A.M. The nodular adrenal. Invest. Cell Pathol. 1978, 1, 99–111. [Google Scholar]
- Neville, A.M.; O’Hare, M.J. The Human Adrenal Cortex: Pathology and Biology—An Integrated Approach; Springer: Berlin, Germany, 1982. [Google Scholar]
- Neville, A.M.; O’Hare, M.J. Histopathology of the human adrenal cortex. Clin. Endocrinol. Metab. 1985, 14, 791–820. [Google Scholar] [CrossRef]
- Hornsby, P.J. Aging of the human adrenal cortex. Ageing Res. Rev. 2002, 1, 229–242. [Google Scholar] [CrossRef]
- Hornsby, P.J. Aging of the human adrenal cortex. Sci Aging Knowl. Environ. 2004, 2004, re6. [Google Scholar] [CrossRef]
- Pieroni, J.; Yamazaki, Y.; Gao, X.; Tezuka, Y.; Ogata, H.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Satoh, F.; et al. Cellular senescence in human aldosterone-producing adrenocortical cells and related disorders. Biomedicines 2021, 9, 567. [Google Scholar] [CrossRef]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Satoh, F.; Sasano, H. The genotype-based morphology of aldosterone-producing adrenocortical disorders and their association with aging. Endocrinol. Metab. 2021, 36, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Kitawaki, Y.; Nakamura, Y.; Kubota-Nakayama, F.; Yamazaki, Y.; Miki, Y.; Hata, S.; Ise, K.; Kikuchi, K.; Morimoto, R.; Satoh, F.; et al. Tumor microenvironment in functional adrenocortical adenomas: Immune cell infiltration in cortisol-producing adrenocortical adenoma. Hum. Pathol. 2018, 77, 88–97. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Kohno, M.; Hitomi, H.; Kitada, K.; Fujisawa, Y.; Yatabe, J.; Yatabe, M.; Felder, R.A.; Ohsaki, H.; Rafiq, K.; et al. Aldosterone/mineralocorticoid receptor stimulation induces cellular senescence in the kidney. Endocrinology 2011, 152, 680–688. [Google Scholar] [CrossRef]
- Crochemore, C.; Michaelidis, T.M.; Fischer, D.; Loeffler, J.P.; Almeida, O.F. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. FASEB J. 2002, 16, 761–770. [Google Scholar] [CrossRef]
- Bird, A.D.; Choo, Y.L.; Hooper, S.B.; McDougall, A.R.; Cole, T.J. Mesenchymal glucocorticoid receptor regulates the development of multiple cell layers of the mouse lung. Am. J. Respir. Cell Mol. Biol. 2014, 50, 419–428. [Google Scholar] [CrossRef]
- Cha, H.H.; Cram, E.J.; Wang, E.C.; Huang, A.J.; Kasler, H.G.; Firestone, G.L. Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J. Biol. Chem. 1998, 273, 1998–2007. [Google Scholar] [CrossRef]
- Li, H.; Qian, W.; Weng, X.; Wu, Z.; Li, H.; Zhuang, Q.; Feng, B.; Bian, Y. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS ONE 2012, 7, e37030. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P.; Toussaint, O. Telomeres and telomerase: A modern fountain of youth? Rejuvenation Res. 2004, 7, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wu, S.B.; Wu, Y.T.; Wei, Y.H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460. [Google Scholar] [CrossRef]
- Mena, S.; Ortega, A.; Estrela, J.M. Oxidative stress in environmental-induced carcinogenesis. Mutat. Res. 2009, 674, 36–44. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Ann. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Sengupta, S.; Wasylyk, B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen, and estrogen receptors. Ann. N. Y. Acad. Sci. 2004, 1024, 54–71. [Google Scholar] [CrossRef]
- Cram, E.J.; Ramos, R.A.; Wang, E.C.; Cha, H.H.; Nishio, Y.; Firestone, G.L. Role of the CCAAT/enhancer binding protein-alpha transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J. Biol. Chem. 1998, 273, 2008–2014. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Ann. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Sikora, E. Activation-induced and damage-induced cell death in aging human T cells. Mech. Ageing Dev. 2015, 151, 85–92. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Gruber, H.E.; Ingram, J.A.; Norton, H.J.; Hanley, E.N., Jr. Senescence in cells of the aging and degenerating intervertebral disc: Immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine 2007, 32, 321–327. [Google Scholar] [CrossRef]
- Geng, Y.Q.; Guan, J.T.; Xu, X.H.; Fu, Y.C. Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem. Biophys. Res. Commun. 2010, 396, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Deleuze, S.; Zuo, Y.; Potthoff, S.A.; Ma, L.J.; Fogo, A.B. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. J. Am. Soc. Nephrol. 2009, 20, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, H.; Gary-Bobo, G.; Alifano, M.; Marcos, E.; Saker, M.; Vienney, N.; Amsellem, V.; Maitre, B.; Chaouat, A.; Chouaid, C.; et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ. Res. 2011, 109, 543–553. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Farr, J.N. Cellular senescence in age-related disorders. Transl. Res. 2020, 226, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Melk, A.; Schmidt, B.M.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef]
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular senescence in the kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Caroccia, B.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; Rossi, G.P. The biology of normal zona glomerulosa and aldosterone-producing adenoma: Pathological implications. Endocr. Rev. 2018, 39, 1029–1056. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yamazaki, Y.; Tezuka, Y.; Satoh, F.; Sasano, H. Expression of CYP11B2 in aldosterone-producing adrenocortical adenoma: Regulatory mechanisms and clinical significance. Tohoku J. Exp. Med. 2016, 240, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Sasano, H.; Sasano, N.; Okamoto, M. Immunohistochemical demonstration of cholesterol side-chain cleavage cytochrome P-450 in bovine and human adrenals. Pathol. Res. Pract. 1989, 184, 337–342. [Google Scholar] [CrossRef]
- Sasano, H.; White, P.C.; New, M.I.; Sasano, N. Immunohistochemistry of cytochrome P-450 21-hydroxylase: Microscopic examination of the enzyme in the bovine adrenal cortex and kidney. Endocrinology 1988, 122, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.F.; Rege, J.; Auchus, R.J.; Rainey, W.E. 11-Oxygenated androgens in health and disease. Nat. Rev. Endocrinol. 2020, 16, 284–296. [Google Scholar] [CrossRef]
- Laragh, J.H.; Baer, L.; Brunner, H.R.; Buhler, F.R.; Sealey, J.E.; Vaughan, E.D., Jr. Renin, angiotensin and aldosterone system in pathogenesis and management of hypertensive vascular disease. Am. J. Med. 1972, 52, 633–652. [Google Scholar] [CrossRef]
- Laragh, J.H.; Sealey, J.E. The plasma renin test reveals the contribution of body sodium-volume content (V) and renin-angiotensin (R) vasoconstriction to long-term blood pressure. Am. J. Hypertens. 2011, 24, 1164–1180. [Google Scholar] [CrossRef]
- Patel, S.; Sealey, J.E. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- Miller, W.L. The hypothalamic-pituitary-adrenal axis: A brief history. Horm. Res. Paediatr. 2018, 89, 212–223. [Google Scholar] [CrossRef]
- Auchus, R.J.; Rainey, W.E. Adrenarche—Physiology, biochemistry and human disease. Clin. Endocrinol. 2004, 60, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Yuen, B.H.; Mincey, E.K. Human chorionic gonadotropin, prolactin, estriol, and dehydroepiandrosterone sulfate concentrations in cord blood of premature and term newborn infants: Relationship to the sex of the neonate. Am. J. Obstet. Gynecol. 1987, 156, 396–400. [Google Scholar] [CrossRef]
- Quinn, T.; Greaves, R.; Badoer, E.; Walker, D. DHEA in prenatal and postnatal life: Implications for brain and behavior. Vitam. Horm. 2018, 108, 145–174. [Google Scholar]
- De Peretti, E.; Forest, M.G. Unconjugated dehydroepiandrosterone plasma levels in normal subjects from birth to adolescence in human: The use of a sensitive radioimmunoassay. J. Clin. Endocrinol. Metab. 1976, 43, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, J.R.; Forest, M.G.; de Peretti, E.; Sempe, M.; Collu, R.; Bertrand, J. Plasma adrenal and gonadal sex steroids in human pubertal development. J. Clin. Endocrinol. Metab. 1976, 42, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Gruenwald, P. Embryonic and postnatal development of the adrenal cortex, particularly the zona glomerulosa and accessory nodules. Anat. Rec. 1946, 95, 391–421. [Google Scholar] [CrossRef]
- Hatano, O.; Takakusu, A.; Nomura, M.; Morohashi, K. Identical origin of adrenal cortex and gonad revealed by expression profiles of Ad4BP/SF-1. Genes Cells 1996, 1, 663–671. [Google Scholar] [CrossRef]
- Luo, X.; Ikeda, Y.; Parker, K.L. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef]
- Keegan, C.E.; Hammer, G.D. Recent insights into organogenesis of the adrenal cortex. Trends Endocrinol. Metab. 2002, 13, 200–208. [Google Scholar] [CrossRef]
- Johannisson, E. The foetal adrenal cortex in the human. Its ultrastructure at different stages of development and in different functional states. Acta Endocrinol. 1968, 58, 4297360. [Google Scholar]
- Xing, Y.; Lerario, A.M.; Rainey, W.; Hammer, G.D. Development of adrenal cortex zonation. Endocrinol. Metab. Clin. North Am. 2015, 44, 243–274. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, T.; Sahut-Barnola, I.; Septier, A.; Montanier, N.; Plotton, I.; Roucher-Boulez, F.; Ducros, V.; Lefrancois-Martinez, A.M.; Pointud, J.C.; Zubair, M.; et al. PKA signaling drives reticularis differentiation and sexually dimorphic adrenal cortex renewal. JCI Insight 2018, 3, e98394. [Google Scholar] [CrossRef]
- Freedman, B.D.; Kempna, P.B.; Carlone, D.L.; Shah, M.; Guagliardo, N.A.; Barrett, P.Q.; Gomez-Sanchez, C.E.; Majzoub, J.A.; Breault, D.T. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev. Cell 2013, 26, 666–673. [Google Scholar] [CrossRef]
- Hammer, G.D.; Basham, K.J. Stem cell function and plasticity in the normal physiology of the adrenal cortex. Mol. Cell Endocrinol. 2020, 519, 111043. [Google Scholar] [CrossRef] [PubMed]
- Omata, K.; Anand, S.K.; Hovelson, D.H.; Liu, C.J.; Yamazaki, Y.; Nakamura, Y.; Ito, S.; Satoh, F.; Sasano, H.; Rainey, W.E.; et al. Aldosterone-producing cell clusters frequently harbor somatic mutations and accumulate with age in normal adrenals. J. Endocr. Soc. 2017, 1, 787–799. [Google Scholar] [CrossRef]
- Nishimoto, K.; Tomlins, S.A.; Kuick, R.; Cani, A.K.; Giordano, T.J.; Hovelson, D.H.; Liu, C.J.; Sanjanwala, A.R.; Edwards, M.A.; Gomez-Sanchez, C.E.; et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. USA 2015, 112, E4591–E4599. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, P.; de Myttenaere-Bursztein, S.; Maxwell, M.H.; de Lima, J. Effect on aging on plasma renin and aldosterone in normal man. Kidney Int. 1975, 8, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Noth, R.H.; Lassman, M.N.; Tan, S.Y.; Fernandez-Cruz, A., Jr.; Mulrow, P.J. Age and the renin-aldosterone system. Arch. Intern. Med. 1977, 137, 1414–1417. [Google Scholar] [CrossRef]
- Tsunoda, K.; Abe, K.; Goto, T.; Yasujima, M.; Sato, M.; Omata, K.; Seino, M.; Yoshinaga, K. Effect of age on the renin-angiotensin-aldosterone system in normal subjects: Simultaneous measurement of active and inactive renin, renin substrate, and aldosterone in plasma. J. Clin. Endocrinol. Metab. 1986, 62, 384–389. [Google Scholar] [CrossRef]
- Rakotondrazafy, J.; Brudieux, R. Age-related change in plasma aldosterone response to exogenous angiotensin II in the rat. Horm Res. 1993, 39, 156–160. [Google Scholar] [CrossRef]
- Giacche, M.; Vuagnat, A.; Hunt, S.C.; Hopkins, P.N.; Fisher, N.D.; Azizi, M.; Corvol, P.; Williams, G.H.; Jeunemaitre, X. Aldosterone stimulation by angiotensin II: Influence of gender, plasma renin, and familial resemblance. Hypertension 2000, 35, 710–716. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crane, M.G.; Harris, J.J. Effect of aging on renin activity and aldosterone excretion. J. Lab. Clin. Med. 1976, 87, 947–959. [Google Scholar] [PubMed]
- Hegstad, R.; Brown, R.D.; Jiang, N.S.; Kao, P.; Weinshilboum, R.M.; Strong, C.; Wisgerhof, M. Aging and aldosterone. Am. J. Med. 1983, 74, 442–448. [Google Scholar] [CrossRef]
- Kerstens, M.N.; Kobold, A.C.; Volmer, M.; Koerts, J.; Sluiter, W.J.; Dullaart, R.P. Reference values for aldosterone-renin ratios in normotensive individuals and effect of changes in dietary sodium consumption. Clin. Chem. 2011, 57, 1607–1611. [Google Scholar] [CrossRef]
- Nanba, K.; Vaidya, A.; Rainey, W.E. Aging and adrenal aldosterone production. Hypertension 2018, 71, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Vaidya, A.; Williams, G.H.; Zheng, I.; Else, T.; Rainey, W.E. Age-related autonomous aldosteronism. Circulation 2017, 136, 347–355. [Google Scholar] [CrossRef]
- Brown, J.M.; Robinson-Cohen, C.; Luque-Fernandez, M.A.; Allison, M.A.; Baudrand, R.; Ix, J.H.; Kestenbaum, B.; de Boer, I.H.; Vaidya, A. The spectrum of subclinical primary aldosteronism and incident hypertension: A cohort study. Ann. Intern. Med. 2017, 167, 630–641. [Google Scholar] [CrossRef]
- Chung, S.; Son, G.H.; Kim, K. Circadian rhythm of adrenal glucocorticoid: Its regulation and clinical implications. Biochim. Biophys. Acta 2011, 1812, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowski, M. Adrenal cortex—The next biological clock? Neuroendocrinol. Lett. 2005, 26, 193–195. [Google Scholar] [PubMed]
- Fries, E.; Dettenborn, L.; Kirschbaum, C. The cortisol awakening response (CAR): Facts and future directions. Int. J. Psychophysiol. 2009, 72, 67–73. [Google Scholar] [CrossRef]
- Van Cauter, E.; Leproult, R.; Kupfer, D.J. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J. Clin. Endocrinol. Metab. 1996, 81, 2468–2473. [Google Scholar]
- Kern, W.; Dodt, C.; Born, J.; Fehm, H.L. Changes in cortisol and growth hormone secretion during nocturnal sleep in the course of aging. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51, M3–M9. [Google Scholar] [CrossRef]
- Hatzinger, M.; Z’Brun, A.; Hemmeter, U.; Seifritz, E.; Baumann, F.; Holsboer-Trachsler, E.; Heuser, I.J. Hypothalamic-pituitary-adrenal system function in patients with Alzheimer’s disease. Neurobiol. Aging 1995, 16, 205–209. [Google Scholar] [CrossRef]
- Gaffey, A.E.; Bergeman, C.S.; Clark, L.A.; Wirth, M.M. Aging and the HPA axis: Stress and resilience in older adults. Neurosci. Biobehav. Rev. 2016, 68, 928–945. [Google Scholar] [CrossRef]
- Gupta, D.; Morley, J.E. Hypothalamic-pituitary-adrenal (HPA) axis and aging. Compr. Physiol. 2014, 4, 1495–1510. [Google Scholar]
- Pavlov, E.P.; Harman, S.M.; Chrousos, G.P.; Loriaux, D.L.; Blackman, M.R. Responses of plasma adrenocorticotropin, cortisol, and dehydroepiandrosterone to ovine corticotropin-releasing hormone in healthy aging men. J. Clin. Endocrinol. Metab. 1986, 62, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Cravello, L.; Muzzoni, B.; Casarotti, D.; Paltro, M.; Solerte, S.B.; Fioravanti, M.; Cuzzoni, G.; Pontiggia, B.; Magri, F. Age-related changes of the hypothalamic-pituitary-adrenal axis: Pathophysiological correlates. Eur. J. Endocrinol. 2001, 144, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Seeman, T.E.; Robbins, R.J. Aging and hypothalamic-pituitary-adrenal response to challenge in humans. Endocr. Rev. 1994, 15, 233–260. [Google Scholar]
- Wilkinson, C.W.; Peskind, E.R.; Raskind, M.A. Decreased hypothalamic-pituitary-adrenal axis sensitivity to cortisol feedback inhibition in human aging. Neuroendocrinology 1997, 65, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F.; Grasser, A.; Friess, E.; Wiedemann, K. Steroid effects on central neurons and implications for psychiatric and neurological disorders. Ann. N. Y. Acad. Sci. 1994, 746, 345–359. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Weiss, J.M.; Schwartz, L.S. Selective retention of corticosterone by limbic structures in rat brain. Nature 1968, 220, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Meaney, M.J.; Aitken, D.H.; Bhatnagar, S.; Sapolsky, R.M. Postnatal handling attenuates certain neuroendocrine, anatomical, and cognitive dysfunctions associated with aging in female rats. Neurobiol. Aging 1991, 12, 31–38. [Google Scholar] [CrossRef]
- Martocchia, A.; Stefanelli, M.; Falaschi, G.M.; Toussan, L.; Ferri, C.; Falaschi, P. Recent advances in the role of cortisol and metabolic syndrome in age-related degenerative diseases. Aging Clin. Exp. Res. 2016, 28, 17–23. [Google Scholar] [CrossRef]
- Dmitrieva, N.O.; Almeida, D.M.; Dmitrieva, J.; Loken, E.; Pieper, C.F. A day-centered approach to modeling cortisol: Diurnal cortisol profiles and their associations among U.S. adults. Psychoneuroendocrinology 2013, 38, 2354–2365. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Satoh, F.; Sasano, H. Gender differences in human adrenal cortex and its disorders. Mol. Cell Endocrinol. 2021, 526, 111177. [Google Scholar] [CrossRef]
- Labrie, F.; Belanger, A.; Cusan, L.; Gomez, J.L.; Candas, B. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J. Clin. Endocrinol. Metab. 1997, 82, 2396–2402. [Google Scholar] [CrossRef]
- Ohashi, M.; Kato, K.; Nawata, H.; Ibayashi, H. Adrenocortical responsiveness to graded ACTH infusions in normal young and elderly human subjects. Gerontology 1986, 32, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.R., Jr.; Mixon, R.L.; Brissie, R.M.; Grizzle, W.E. Aging alters zonation in the adrenal cortex of men. J. Clin. Endocrinol. Metab. 1997, 82, 3898–3901. [Google Scholar] [CrossRef]
- Wolkersdorfer, G.W.; Ehrhart-Bornstein, M.O.N.K.; Brauer, S.I.L.K.E.; Marx, C.H.R.I.S.T.I.A.N.; Scherbaum, W.A.; Bornstein, S.R. Differential regulation of apoptosis in the normal human adrenal gland. J. Clin. Endocrinol. Metab. 1996, 81, 4129–4136. [Google Scholar]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Autophagy, organelles and ageing. J. Pathol. 2007, 211, 134–143. [Google Scholar] [CrossRef]
- Rege, J.; Rainey, W.E. The steroid metabolome of adrenarche. J. Endocrinol. 2012, 214, 133–143. [Google Scholar] [CrossRef][Green Version]
- Baulieu, E.E. Dehydroepiandrosterone (DHEA): A fountain of youth? J. Clin. Endocrinol. Metab. 1996, 81, 3147–3151. [Google Scholar] [CrossRef]
- Beer, N.A.; Jakubowicz, D.J.; Beer, R.M.; Nestler, J.E. Disparate effects of insulin reduction with diltiazem on serum dehydroepiandrosterone sulfate levels in obese hypertensive men and women. J. Clin. Endocrinol. Metab. 1994, 79, 1077–1081. [Google Scholar]
- Nonaka, K.; Aida, J.; Takubo, K.; Yamazaki, Y.; Takakuma, S.; Kakizaki, M.; Matsuda, Y.; Ishikawa, N.; Ishiwata, T.; Chong, J.M.; et al. Correlation between differentiation of adrenocortical zones and telomere lengths measured by Q-FISH. J. Clin. Endocrinol. Metab. 2019, 104, 5642–5650. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Nakamura, Y.; Omata, K.; Ise, K.; Tezuka, Y.; Ono, Y.; Morimoto, R.; Nozawa, Y.; Gomez-Sanchez, C.E.; Tomlins, S.A.; et al. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J. Clin. Endocrinol. Metab. 2017, 102, 1182–1192. [Google Scholar] [CrossRef]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Pieroni, J.; Ishii, K.; Atsumi, N.; Ono, Y.; Omata, K.; Morimoto, R.; Nakamura, Y.; et al. Intratumoral heterogeneity of the tumor cells based on in situ cortisol excess in cortisol-producing adenomas; An association among morphometry, genotype and cellular senescence approximately. J. Steroid Biochem. Mol. Biol. 2020, 204, 105764. [Google Scholar] [CrossRef]
- Nishimoto, K.; Nakagawa, K.; Li, D.; Kosaka, T.; Oya, M.; Mikami, S.; Shibata, H.; Itoh, H.; Mitani, F.; Yamazaki, T.; et al. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 2010, 95, 2296–2305. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Gomez-Sanchez, C.E.; Rainey, W.E.; Giordano, T.J.; Lam, A.K.; Marker, A.; Mete, O.; Yamazaki, Y.; Zerbini, M.C.N.; Beuschlein, F.; et al. International histopathology consensus for unilateral primary aldosteronism. J. Clin. Endocrinol. Metab. 2021, 106, 42–54. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Aulinas, A.; Ramirez, M.J.; Barahona, M.J.; Valassi, E.; Resmini, E.; Mato, E.; Santos, A.; Crespo, I.; Bell, O.; Surralles, J.; et al. Telomere length analysis in Cushing’s syndrome. Eur. J. Endocrinol. 2014, 171, 21–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suwa, T.; Yang, L.; Hornsby, P.J. Telomerase activity in primary cultures of normal adrenocortical cells. J. Endocrinol. 2001, 170, 677–684. [Google Scholar] [CrossRef]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Onodera, Y.; Ogata, H.; Omata, K.; Morimoto, R.; Nakamura, Y.; Satoh, F.; Sasano, H. The crosstalk between aldosterone and calcium metabolism in primary aldosteronism: A possible calcium metabolism-associated aberrant “neoplastic” steroidogenesis in adrenals. J. Steroid Biochem. Mol. Biol. 2019, 193, 105434. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed]
- Roman, S. Adrenocortical carcinoma. Curr. Opin. Oncol. 2006, 18, 36–42. [Google Scholar] [CrossRef]
- Nakamura, Y.; Maekawa, T.; Felizola, S.J.; Satoh, F.; Qi, X.; Velarde-Miranda, C.; Plonczynski, M.W.; Ise, K.; Kikuchi, K.; Rainey, W.E.; et al. Adrenal CYP11B1/2 expression in primary aldosteronism: Immunohistochemical analysis using novel monoclonal antibodies. Mol. Cell Endocrinol. 2014, 392, 73–79. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Omata, K.; Tezuka, Y.; Ono, Y.; Morimoto, R.; Adachi, Y.; Ise, K.; Nakamura, Y.; Gomez-Sanchez, C.E.; Shibahara, Y.; et al. Tumor cell subtypes based on the intracellular hormonal activity in KCNJ5-mutated aldosterone-producing adenoma. Hypertension 2018, 72, 632–640. [Google Scholar] [CrossRef]
- Ono, Y.; Yamazaki, Y.; Omata, K.; Else, T.; Tomlins, S.A.; Rhayem, Y.; Williams, T.A.; Reincke, M.; Carling, T.; Monticone, S.; et al. Histological characterization of aldosterone-producing adrenocortical adenomas with different somatic mutations. J. Clin. Endocrinol. Metab. 2020, 105, e282–e289. [Google Scholar] [CrossRef]
- Morio, H.; Terano, T.; Yamamoto, K.; Tomizuka, T.; Oeda, T.; Saito, Y.; Tamura, Y.; Sasano, H. Serum levels of dehydroepiandrosterone sulfate in patients with asymptomatic cortisol producing adrenal adenoma: Comparison with adrenal Cushing’s syndrome and non-functional adrenal tumor. Endocr. J. 1996, 43, 387–396. [Google Scholar] [CrossRef][Green Version]
- Babinska, A.; Sworczak, K.; Wisniewski, P.; Nalecz, A.; Jaskiewicz, K. The role of immunohistochemistry in histopathological diagnostics of clinically “silent” incidentally detected adrenal masses. Exp. Clin. Endocrinol. Diabetes 2008, 116, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.S.; Morais, T.; Costa, M.M.; Monteiro, M.P.; Pignatelli, D. The emerging role of the molecular marker p27 in the differential diagnosis of adrenocortical tumors. Endocr. Connect. 2013, 2, 137–145. [Google Scholar] [CrossRef]
- Mannelli, M.; Gelmini, S.; Arnaldi, G.; Becherini, L.; Bemporad, D.; Crescioli, C.; Pazzagli, M.; Mantero, F.; Serio, M.; Orlando, C. Telomerase activity is significantly enhanced in malignant adrenocortical tumors in comparison to benign adrenocortical adenomas. J. Clin. Endocrinol. Metab. 2000, 85, 468–470. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Zambetti, G.P.; Malkin, D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol. Cell Endocrinol. 2012, 351, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, J.; Patsalis, N.; Fendrich, V.; Langer, P.; Saeger, W.; Chaloupka, B.; Ramaswamy, A.; Fassnacht, M.; Bartsch, D.K.; Slater, E.P. Clinical impact of TP53 alterations in adrenocortical carcinomas. Langenbecks Arch. Surg. 2012, 397, 209–216. [Google Scholar] [CrossRef] [PubMed]
- McNicol, A.M.; Nolan, C.E.; Struthers, A.J.; Farquharson, M.A.; Hermans, J.; Haak, H.R. Expression of p53 in adrenocortical tumours: Clinicopathological correlations. J. Pathol. 1997, 181, 146–152. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. P53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: Influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar] [CrossRef]
- Peifer, M.; Polakis, P. WNT signaling in oncogenesis and embryogenesis—A look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Salomon, A.; Keramidas, M.; Maisin, C.; Thomas, M. Loss of beta-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget 2015, 6, 11421–11433. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ding, G.; Zhou, Z.; Feng, C. β-catenin-driven adrenocortical carcinoma is characterized with immune exclusion. OncoTargets Ther. 2018, 11, 2029–2036. [Google Scholar] [CrossRef]
- Landwehr, L.S.; Altieri, B.; Schreiner, J.; Sbiera, I.; Weigand, I.; Kroiss, M.; Fassnacht, M.; Sbiera, S. Interplay between glucocorticoids and tumor-infiltrating lymphocytes on the prognosis of adrenocortical carcinoma. J. Immunother. Cancer 2020, 8, e000469. [Google Scholar] [CrossRef]
- Puglisi, S.; Perotti, P.; Pia, A.; Reimondo, G.; Terzolo, M. Adrenocortical carcinoma with hypercortisolism. Endocrinol. Metab. Clin. North Am. 2018, 47, 395–407. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of formation and increase with age. APMIS 1998, 106, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Munnell, J.F.; Getty, R. Rate of accumulation of cardiac lipofuscin in the aging canine. J. Gerontol. 1968, 23, 154–158. [Google Scholar] [CrossRef]
- Nakano, M.; Gotoh, S. Accumulation of cardiac lipofuscin depends on metabolic rate of mammals. J. Gerontol. 1992, 47, B126–B129. [Google Scholar] [CrossRef]
- Sheehy, M.R.; Greenwood, J.G.; Fielder, D.R. Lipofuscin as a record of “rate of living” in an aquatic poikilotherm. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, B327–B336. [Google Scholar] [CrossRef]
- Angelousi, A.; Szarek, E.; Shram, V.; Kebebew, E.; Quezado, M.; Stratakis, C.A. Lipofuscin accumulation in cortisol-producing adenomas with and without PRKACA mutations. Horm. Metab. Res. 2017, 49, 786–792. [Google Scholar] [CrossRef]
- Reichel, W. Lipofuscin pigment accumulation and distribution in five rat organs as a function of age. J. Gerontol. 1968, 23, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, K.; Horvath, E.; Feldman, P.S. Pigmented adenoma of adrenal cortex associated with Cushing’s syndrome: Light and electron microscopic study. Urology 1976, 7, 641–645. [Google Scholar] [CrossRef]
- Cheng, B.; Tserng, K.Y.; Kowal, J.; Buekers, K.S.; Abraham, S.; Gerhart, J.P. Characterization and identification of an adrenal age-related nonpolar fluorescent substance. Endocrinology 1996, 137, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Kamalanathan, S.; Mahesh, D.M.; Muruganandham, K.; Basu, D. Black adrenal adenoma: Distinction from PPNAD. BMJ Case Rep. 2012, 2012, bcr0320126076. [Google Scholar] [CrossRef]
- Horvath, A.; Stratakis, C. Primary pigmented nodular adrenocortical disease and Cushing’s syndrome. Arq. Bras. Endocrinol. Metab. 2007, 51, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Balazs, M. Functioning “black adenoma” of the adrenal gland with emphasis on ultrastructural studies. Zentralbl. Pathol. 1991, 137, 151–156. [Google Scholar] [PubMed]
- Kawai, K.; Shigematsu, K.; Matsuo, K.; Tsuchiyama, H.; Saito, Y. Multiple pigmented adrenal cortical nodules associated with Cushing’s syndrome. Histochemical, ultrastructural and quantitative studies. Acta Pathol. Jpn. 1984, 34, 827–837. [Google Scholar]
- Valente, M.; Pennelli, N.; Segato, P.; Bevilacqua, L.; Thiene, G. Androgen producing adrenocortical carcinoma. Virchows Arch. A Pathol. Anat. Histol. 1978, 378, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.A.; Pagnin, E.; Davis, P.A.; Armanini, D.; Mormino, P.; Rossi, G.P.; Pessina, A.C. Oxidative stress-related proteins in a Conn’s adenoma tissue. Relevance for aldosterone’s prooxidative and proinflammatory activity. J. Endocrinol. Invest. 2010, 33, 48–53. [Google Scholar] [CrossRef]
- Petramala, L.; Pignatelli, P.; Carnevale, R.; Zinnamosca, L.; Marinelli, C.; Settevendemmie, A.; Concistre, A.; Tonnarini, G.; de Toma, G.; Violi, F.; et al. Oxidative stress in patients affected by primary aldosteronism. J. Hypertens. 2014, 32, 2022–2029. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, H.; Zhang, J.; Zhang, H.; Zeng, Y.; Fang, S.; Li, P.; Zhang, Y.; Lin, X.; Wang, L.; et al. Increased oxidative stress, inflammation and fibrosis in perirenal adipose tissue of patients with cortisol-producing adenoma. Adipocyte 2019, 8, 347–356. [Google Scholar] [CrossRef]
- Ogata, H.; Yamazaki, Y.; Tezuka, Y.; Gao, X.; Omata, K.; Ono, Y.; Kawasaki, Y.; Tanaka, T.; Nagano, H.; Wada, N.; et al. Renal injuries in primary aldosteronism: Quantitative histopathological analysis of 19 patients with primary adosteronism. Hypertension 2021, 78, 411–421. [Google Scholar] [CrossRef]
- Andrade, L.; Rodrigues, C.E.; Gomes, S.A.; Noronha, I.L. Acute kidney injury as a condition of renal senescence. Cell Transplant. 2018, 27, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 2009, 76, 604–613. [Google Scholar] [CrossRef]
- Yu, F.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Identification of the functional domain of p21(WAF1/CIP1) that protects cells from cisplatin cytotoxicity. Am. J. Physiol. Renal. Physiol. 2005, 289, F514–F520. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.M. Acute kidney injury induces dramatic p21 upregulation via a novel, glucocorticoid-activated, pathway. Am. J. Physiol. Renal Physiol. 2019, 316, F674–F681. [Google Scholar] [CrossRef]
- Galla, J.H. IgA nephropathy. Kidney Int. 1995, 47, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Sasano, H.; Watanabe, N.; Sasano, N. Corticosteroid binding in human artery. Tohoku J. Exp. Med. 1986, 150, 117–125. [Google Scholar] [CrossRef]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Suzuki, T.; Satoh, F.; Sasano, H. Pathology of aldosterone biosynthesis and its action. Tohoku J. Exp. Med. 2021, 254, 1–15. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Li, J.M.; Sakata, A.; Fujita, T.; Tsukuda, K.; Iwai, M.; Horiuchi, M. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc Res. 2007, 76, 506–516. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhong, B.; Wu, Q.; Tong, J.; Zhu, T.; Zhang, M. Effect of aldosterone on senescence and proliferation inhibition of endothelial progenitor cells induced by Sirtuin 1 (SIRT1) in pulmonary arterial hypertension. Med. Sci. Monit. 2020, 26, e920678. [Google Scholar] [CrossRef]
- Park, J.H.; Oh, E.J.; Choi, Y.H.; Kang, C.D.; Kang, H.S.; Kim, D.K.; Kang, K.I.; Yoo, M.A. Synergistic effects of dexamethasone and genistein on the expression of Cdk inhibitor p21WAF1/CIP1 in human hepatocellular and colorectal carcinoma cells. Int. J. Oncol. 2001, 18, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S.L. Acceleration of canalicular development in lungs of fetal mice exposed transplacentally to dexamethasone. Lab. Invest. 1977, 36, 395–401. [Google Scholar] [PubMed]
- Oshika, E.; Liu, S.; Ung, L.P.; Singh, G.; Shinozuka, H.; Michalopoulos, G.K.; Katyal, S.L. Glucocorticoid-induced effects on pattern formation and epithelial cell differentiation in early embryonic rat lungs. Pediatr. Res. 1998, 43, 305–314. [Google Scholar] [CrossRef]
- Bolt, R.J.; van Weissenbruch, M.M.; Lafeber, H.N.; Delemarre-van de Waal, H.A. Glucocorticoids and lung development in the fetus and preterm infant. Pediatr. Pulmonol. 2001, 32, 76–91. [Google Scholar] [CrossRef]
- Patki, M.; McFall, T.; Rosati, R.; Huang, Y.; Malysa, A.; Polin, L.; Fielder, A.; Wilson, M.R.; Lonardo, F.; Back, J.; et al. Chronic p27(Kip1) induction by dexamethasone causes senescence phenotype and permanent cell cycle blockade in lung adenocarcinoma cells over-expressing glucocorticoid receptor. Sci. Rep. 2018, 8, 16006. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Hu, J.; Basu, S.; Hay, J.; Reibman, J.; Yie, T.A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am. J. Respir. Cell Mol. Biol. 2002, 27, 320–328. [Google Scholar] [CrossRef]
- Lu, Y.S.; Lien, H.C.; Yeh, P.Y.; Kuo, S.H.; Chang, W.C.; Kuo, M.L.; Cheng, A.L. Glucocorticoid receptor expression in advanced non-small cell lung cancer: Clinicopathological correlation and in vitro effect of glucocorticoid on cell growth and chemosensitivity. Lung Cancer 2006, 53, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, U.; Hofmann, J.; Schilli, M.; Wegmann, B.; Klotz, U.; Wedel, S.; Virmani, A.K.; Wollmer, E.; Branscheid, D.; Gazdar, A.F.; et al. Steroid-hormone receptors in cell lines and tumor biopsies of human lung cancer. Int. J. Cancer 1996, 67, 357–364. [Google Scholar] [CrossRef]
- Brown, R.W.; Chapman, K.E.; Murad, P.; Edwards, C.R.; Seckl, J.R. Purification of 11 beta-hydroxysteroid dehydrogenase type 2 from human placenta utilizing a novel affinity labelling technique. Biochem. J. 1996, 313, 997–1005. [Google Scholar] [CrossRef]
- Brown, R.W.; Diaz, R.; Robson, A.C.; Kotelevtsev, Y.V.; Mullins, J.J.; Kaufman, M.H.; Seckl, J.R. The ontogeny of 11 beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor gene expression reveal intricate control of glucocorticoid action in development. Endocrinology 1996, 137, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Liggins, G.C. Adrenocortical-related maturational events in the fetus. Am. J. Obstet. Gynecol. 1976, 126, 931–941. [Google Scholar] [CrossRef]
- Oliveira, M.; Bessa, J.M.; Mesquita, A.; Tavares, H.; Carvalho, A.; Silva, R.; Pego, J.M.; Cerqueira, J.J.; Palha, J.A.; Almeida, O.F.; et al. Induction of a hyperanxious state by antenatal dexamethasone: A case for less detrimental natural corticosteroids. Biol. Psychiatry 2006, 59, 844–852. [Google Scholar] [CrossRef]
- Oliveira, M.; Rodrigues, A.J.; Leao, P.; Cardona, D.; Pego, J.M.; Sousa, N. The bed nucleus of stria terminalis and the amygdala as targets of antenatal glucocorticoids: Implications for fear and anxiety responses. Psychopharmacology 2012, 220, 443–453. [Google Scholar] [CrossRef]
- Rodrigues, A.J.; Leao, P.; Pego, J.M.; Cardona, D.; Carvalho, M.M.; Oliveira, M.; Costa, B.M.; Carvalho, A.F.; Morgado, P.; Araujo, D.; et al. Mechanisms of initiation and reversal of drug-seeking behavior induced by prenatal exposure to glucocorticoids. Mol. Psychiatry 2012, 17, 1295–1305. [Google Scholar] [CrossRef]
- Gould, E.; Tanapat, P. Stress and hippocampal neurogenesis. Biol. Psychiatry 1999, 46, 1472–1479. [Google Scholar] [CrossRef]
- Sapolsky, R.M.; Krey, L.C.; McEwen, B.S. Prolonged glucocorticoid exposure reduces hippocampal neuron number: Implications for aging. J. Neurosci. 1985, 5, 1222–1227. [Google Scholar] [CrossRef]
- Reagan, L.P.; McEwen, B.S. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J. Chem. Neuroanat. 1997, 13, 149–167. [Google Scholar] [CrossRef]
- Lupien, S.J.; de Leon, M.; de Santi, S.; Convit, A.; Tarshish, C.; Nair, N.P.; Thakur, M.; McEwen, B.S.; Hauger, R.L.; Meaney, M.J. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1998, 1, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Simeoli, C.; de Martino, M.C.; Cozzolino, A.; de Leo, M.; Iacuaniello, D.; Pivonello, C.; Negri, M.; Pellecchia, M.T.; Iasevoli, F.; et al. Neuropsychiatric disorders in Cushing’s syndrome. Front. Neurosci. 2015, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [PubMed]
- Dodt, C.; Dittmann, J.; Hruby, J.; Spath-Schwalbe, E.; Born, J.; Schuttler, R.; Fehm, H.L. Different regulation of adrenocorticotropin and cortisol secretion in young, mentally healthy elderly and patients with senile dementia of Alzheimer’s type. J. Clin. Endocrinol. Metab. 1991, 72, 272–276. [Google Scholar] [CrossRef] [PubMed]
- MacLullich, A.M.; Ferguson, K.J.; Wardlaw, J.M.; Starr, J.M.; Deary, I.J.; Seckl, J.R. Smaller left anterior cingulate cortex volumes are associated with impaired hypothalamic-pituitary-adrenal axis regulation in healthy elderly men. J. Clin. Endocrinol. Metab. 2006, 91, 1591–1594. [Google Scholar] [CrossRef]
- Sandi, C.; Venero, C.; Guaza, C. Novelty-related rapid locomotor effects of corticosterone in rats. Eur. J. Neurosci. 1996, 8, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; Fiocco, A.; Wan, N.; Maheu, F.; Lord, C.; Schramek, T.; Tu, M.T. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology 2005, 30, 225–242. [Google Scholar] [CrossRef]
- Pivonello, R.; Isidori, A.M.; de Martino, M.C.; Newell-Price, J.; Biller, B.M.; Colao, A. Complications of Cushing’s syndrome: State of the art. Lancet Diabetes Endocrinol. 2016, 4, 611–629. [Google Scholar] [CrossRef]
- Weinstein, R.S. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol. Metab. Clin. North Am. 2012, 41, 595–611. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Akhter, M.P.; Gao, X.; Wang, X.Y.; Wang, X.B.; Zhao, G.; Wei, X.; Wu, H.J.; Chen, H.; Wang, D.; et al. Glucocorticoid-induced delayed fracture healing and impaired bone biomechanical properties in mice. Clin. Interv. Aging 2018, 13, 1465–1474. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Wang, T.; Yang, L.; Liang, Z.; Wang, L.; Su, F.; Wang, X.; You, X.; He, C. Targeting cellular senescence prevents glucocorticoid-induced bone loss through modulation of the DPP4-GLP-1 axis. Signal Transduct. Target. Ther. 2021, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Auer, M.K.; Paizoni, L.; Hofbauer, L.C.; Rauner, M.; Chen, Y.; Schmidt, H.; Huebner, A.; Bidlingmaier, M.; Reisch, N. Effects of androgen excess and glucocorticoid exposure on bone health in adult patients with 21-hydroxylase deficiency. J. Steroid Biochem. Mol. Biol. 2020, 204, 105734. [Google Scholar] [CrossRef]
- Zapata, E.; Ventura, J.L.; de la Cruz, K.; Rodriguez, E.; Damian, P.; Masso, F.; Montano, L.F.; Lopez-Marure, R. Dehydroepiandrosterone inhibits the proliferation of human umbilical vein endothelial cells by enhancing the expression of p53 and p21, restricting the phosphorylation of retinoblastoma protein, and is androgen- and estrogen-receptor independent. FEBS J. 2005, 272, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Hoshiga, M.; Negoro, N.; Fukui, R.; Nakakoji, T.; Kohbayashi, E.; Shibata, N.; Furutama, D.; Ishihara, T.; Hanafusa, T.; et al. Adrenal androgen dehydroepiandrosterone sulfate inhibits vascular remodeling following arterial injury. Atherosclerosis 2009, 206, 77–85. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Li, F.; Liu, B.; Wang, Y.; Wang, Y.; Zhou, H. Cellular Senescence in Adrenocortical Biology and Its Disorders. Cells 2021, 10, 3474. https://doi.org/10.3390/cells10123474
Gao X, Li F, Liu B, Wang Y, Wang Y, Zhou H. Cellular Senescence in Adrenocortical Biology and Its Disorders. Cells. 2021; 10(12):3474. https://doi.org/10.3390/cells10123474
Chicago/Turabian StyleGao, Xin, Faping Li, Bin Liu, Yuxiong Wang, Yishu Wang, and Honglan Zhou. 2021. "Cellular Senescence in Adrenocortical Biology and Its Disorders" Cells 10, no. 12: 3474. https://doi.org/10.3390/cells10123474
APA StyleGao, X., Li, F., Liu, B., Wang, Y., Wang, Y., & Zhou, H. (2021). Cellular Senescence in Adrenocortical Biology and Its Disorders. Cells, 10(12), 3474. https://doi.org/10.3390/cells10123474