
Senescence Alterations in Pulmonary Hypertension

Abstract
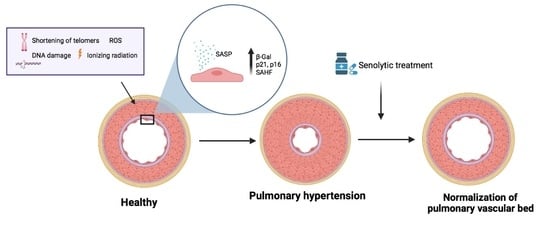
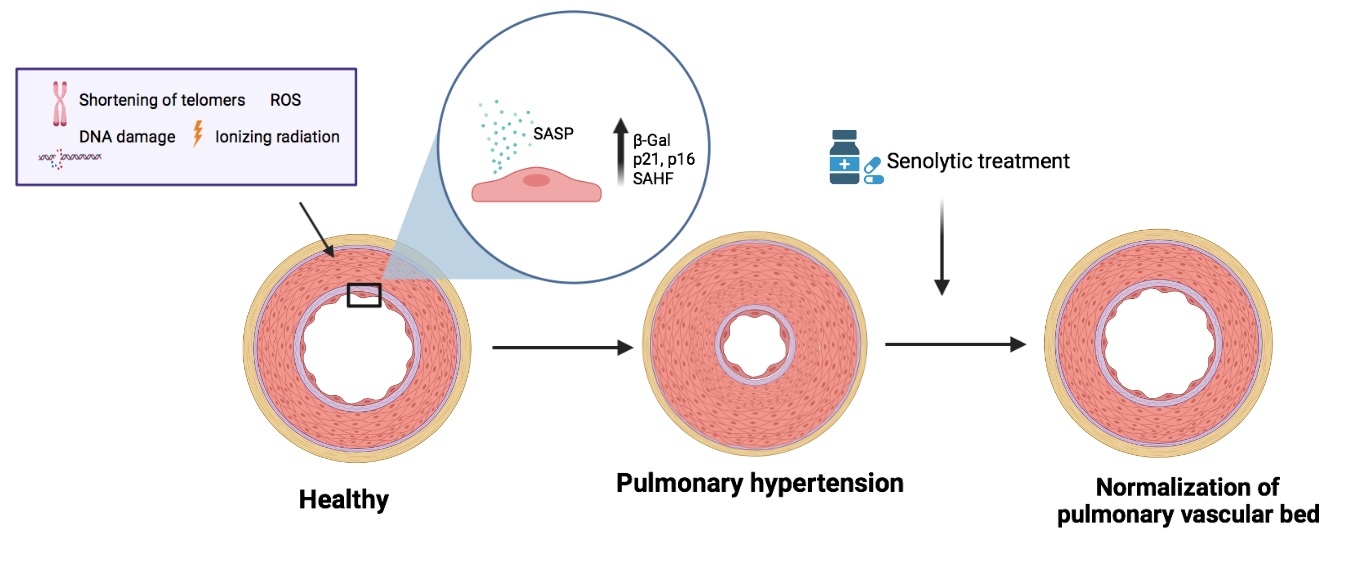
1. Introduction
1.1. Definition
1.2. Types of Senescence
1.3. Senescence Markers
1.4. Vessels and Senescence
2. Pulmonary Hypertension and Senescence
2.1. Definition and Classification
2.2. Expression and Distribution of Senescent Markers in Pulmonary Hypertension
2.3. Signaling Pathways Involved in Cellular Senescence and in PH
2.3.1. Growth Factors Imbalance Lead to Senescence and PH
2.3.2. Cytokines as Activators of Senescence in PH
2.3.3. Imbalance in Mediators of Vascular Tone Leads to Senescence in PH
2.3.4. Osteopontin and Senescence in PH
3. Senotherapy in PH
3.1. Senolytic Therapy
3.2. Senostatic Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Drug | Mechanism of Action | Groups of PH | Data of Trials in PH |
|---|---|---|---|---|
| Senolytics | Dasatinib | Inhibits BCR/ABL kinase. Targets multiple antiapoptotic pathways. | Clinical study | Incident cases of precapillary PH have been reported in patients who have chronic myelogenous leukemia [117] AM. |
| Pre-clinical PH models | Dasatinib exaggerates the response to MCT and hypoxia [118] AM. | |||
| Navitoclax (ABT263) | Bcl2 inhibitor. | Pre-clinical PH models | ABT263 reduced SASP elevation, vessel remodeling, and consequent hemodynamic manifestation in hypoxic mice and hypoxic IL-6 transgenic mice [119] AM. | |
| FOXO4-DRI | Stimulates p53-mediated apoptosis of senescent cells. | No data in PH. | ||
| HSP90 inhibitors * | Induces apoptosis in senescent cells. | Pre-clinical PH models | Hsp90 inhibitors reduce mPAP, RVSP, and vascular remodeling and right ventricular hypertrophy in MCT rats [121,122] AM. | |
| UBX0101 | Stimulates p53-mediated apoptosis of senescent cells. | No data in PH. | ||
| Senostatic | Rapamycin * | mTOR inhibitor | Clinical study: Severe PAH | Phase I (NCT02587325) |
| Pre-clinical PH models | Rapamycin attenuates pulmonary vascular remodeling and right ventricular hypertrophy in MCT rats and hypoxic mice [122,123,124,125]. | |||
| Everolimus * | Clinical study: PAH and chronic thromboembolic PH | Everolimus decreased iPVR and increases 6MWD [128] AM. | ||
| Metformin * | NF-κB inhibitor; suppression of the SASP. | Clinical study: CHD-PH | Metformin with bosetan provides improvements in important outcomes [136] AM. | |
| Clinical Study: PAH | Phase II (NCT03617458) Phase II (NCT01352026) Recruiting (NCT01884051). | |||
| Clinical study: HFpEF-PH | Active, not recruiting (NCT03629340). | |||
| Pre-clinical PH models | Metformin improved hemodynamic parameters and right ventricle hypertrophy in well-established models of severe PH. | |||
| Selonsertib * (GS-444217) | ASK1 inhibition suppression of the SASP. | Pre-clinical PH models | GS-444217 reduced pulmonary arterial pressure and reduced RV hypertrophy in MCT and Sugen/hypoxia models [138] AM. | |
| Clinical study: PAH | Completed (NCT02234141). Selonsertib had no significant effect [137] AM. | |||
| SB-203580 * | P38 MAPK inhibitor | Pre-clinical PH models | SB203580 caused decreased right ventricle systolic pressure, superoxide anion production, and the production of tissue and circulating IL-6 in hypoxic rats [139,140] AM. | |
| PH-797804 * | Pre-clinical PH models | Chronic hypoxic and MCT-induced PH was reversed with PH-797804. The production of tissue and circulating IL-6 was reduced too [137] AM. | ||
| FR167653 * | Pre-clinical PH models | FR167653 attenuates vascular proliferation and reduces mean pulmonary artery pressure in MCT rats [142] AM. | ||
| Curcumin | Antioxidant therapy | Pre-clinical PH models | Curcumin administration was associated with reduced right ventricular wall thickness and a decreased right ventricle weight/body weight ratio [145] AM. | |
| NAC | Pre-clinical PH models | NAC reduced right ventricular hypertrophy index, mean pulmonary artery pressure, PVR and pulmonary inflammation [147] AM. | ||
| Clinical study: PAH | Recruiting (NCT04081012) | |||
| Tocilizumab | IL-6Rα inhibitor | Clinical study: PAH | Phase II (NCT02676947) | |
| Anakinra | IL-1α receptor antagonist | Clinical study: PAH | Complete (NCT03057028) | |
| Pre-clinical PH models | Experimental animal data suggesting anakinra protects against development of PAH [150,151] AM. | |||
| Etanercept | TNFα inhibitor | Pre-clinical PH models | Etanercept prevents and reverses MCT-PH in rats and endotoxin-PH in pigs5) [153,154,155]. |
4. Potential Application of Senolytics Therapies Used in Lung and Cardiac Disease and Perspectives on PH
5. The Pitfalls of Senescence in Pulmonary Hypertension
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory Phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox Signal. 2009, 11, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Banerjee, P.; Hamilton, D.J.; Le, N.-T.; Abe, J.-I. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020, 37, 101614. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef]
- Toussaint, O.; Medrano, E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to detect biomarkers of cellular senescence: The senescence-associated beta-galactosidase assay. Biol. Aging 2007, 371, 21–31. [Google Scholar]
- Sikora, E.; Bielak-Żmijewska, A.; Mosieniak, G. What is and what is not cell senescence. Postepy Biochem. 2018, 64, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nuñez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for highmobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kreiling, J.A.; Tamamori-Adachi, M.; Sexton, A.N.; Jeyapalan, J.C.; Munoz-Najar, U.; Peterson, A.L.; Manivannan, J.; Rogers, E.S.; Pchelintsev, N.A.; Adams, P.D.; et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 2010, 10, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.L.; McBryan, T.; Enders, G.H.; Johnson, F.B.; Zhang, R.; Adams, P.D. Senescent mouse cells fail to overtly regulate the HIRA histone chaperone and do not form robust Senescence Associated Heterochromatin Foci. Cell Div. 2010, 5, 16. [Google Scholar] [CrossRef]
- Košař, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16 ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; d’Ario, G.; Montani, E.; Mercurio, C.; Hahn, W.C.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. Inf. 2011, 13, 292–302. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. γH2AX as a molecular marker of aging and disease. Epigenetics 2010, 5, 129–136. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Sordet, O.; Dickey, J.S.; Gouliaeva, K.; Tabb, B.; Lawrence, S.; Kinders, R.J.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX detection in Peripheral blood Lymphocytes, Splenocytes, bone marrow, Xenografts, and Skin. Springer Protoc. Handb. 2010, 682, 249–270. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX Phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Balajee, A.S.; Geard, C.R. Replication protein A and gamma-H2AX foci assembly is triggered by cellular response to DNA double-strand breaks. Exp. Cell Res. 2004, 300, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in Human Atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef]
- Tian, X.-L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Van der Feen, D.E.; Berger, R.M.F.; Bartelds, B. Converging paths of pulmonary arterial hypertension and cellular senescence. Am. J. Respir. Cell Mol. Biol. 2019, 61, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Okumura, N.; Adachi, S.; Murohara, T. Pulmonary hypertension: Diagnosis, management, and treatment. Nagoya J. Med. Sci. 2019, 81, 19–30. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart. J. 2016, 37, 67–119. [Google Scholar]
- van der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.-R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Trans. Med. 2020, 12, 554. [Google Scholar] [CrossRef]
- Sugimoto, K.; Yokokawa, T.; Misaka, T.; Nakazato, K.; Ishida, T.; Takeishi, Y. Senescence marker protein 30 deficiency exacerbates pulmonary hypertension in hypoxia-exposed mice. Int. Hear. J. 2019, 60, 1430–1434. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, K.; Zheng, Q.; Zhang, C.; Tang, H.; Babicheva, A.; Jiang, Q.; Li, M.; Chen, Y.; Carr, S.G.; et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L216–L228. [Google Scholar] [CrossRef]
- Wakasugi, T.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Katsuumi, G.; Nakao, M.; Hoyano, M.; Kashimura, T.; et al. Role of smooth muscle cell p53 in pulmonary arterial hypertension. PLoS ONE 2019, 14, e0212889. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.-I.; et al. BMPR2 preserves mitochondrial function and DNA during Reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef]
- Ravi, Y.; Selvendiran, K.; Meduru, S.; Citro, L.; Naidu, S.; Khan, M.; Rivera, B.K.; Sai-Sudhakar, C.B.; Kuppusamy, P. Dysregulation of PTEN in cardiopulmonary vascular remodeling induced by pulmonary hypertension. Cell Biophys. 2013, 67, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Houssaïni, A.; Mouraret, N.; Marcos, E.; Amsellem, V.; Wan, F.; Dubois-Randé, J.L.; Derumeaux, G.; Boczkowski, J.; Motterlini, R.; et al. p21-dependent protective effects of a carbon monoxide–Releasing molecule-3 in pulmonary hypertension. Arter. Thromb. Vasc. Biol. 2014, 34, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Kraskauskas, D.; Alhussaini, A.; Gomez-Arroyo, J.; Voelkel, N.F.; Ishizaki, T. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L753–L761. [Google Scholar] [CrossRef] [PubMed]
- Saker, M.; Lipskaia, L.; Marcos, E.; Abid, S.; Parpaleix, A.; Houssaini, A.; Validire, P.; Girard, P.; Noureddine, H.; Boyer, L.; et al. Osteopontin, a key mediator expressed by senescent pulmonary vascular cells in pulmonary hypertension. Arter. Thromb. Vasc. Biol. 2016, 36, 1879–1890. [Google Scholar] [CrossRef]
- Roger, I.; Milara, J.; Montero, P.; Ribera, P.; Cortijo, J. IL-11 promotes pulmonary vascular remodeling and lung fibrosis through the activation of endothelial to mesenchymal transition. Eur. Respir. J. 2020, 56, 3378. [Google Scholar]
- Roger, I.; Milara, J.; Estornut, C.; Bayarri, A.; Garcia, A.; Cortijo, J. Role of IL-11 system in pulmonay hypertension. Pulm. Hypertens. 2020, 56, 1495. [Google Scholar]
- Roger, I.; Estornut, C.; Ballester, B.; Milara, J.; Cortijo, J. Role of IL-11 in vascular function of pulmonary fibrosis patients. Pulm. Hypertens. 2019, 54, PA1424. [Google Scholar]
- Zhang, D.; Wang, G.; Han, D.; Zhang, Y.; Xu, J.; Lu, J.; Li, S.; Xie, X.; Liu, L.; Dong, L.; et al. Activation of PPAR-γ ameliorates pulmonary arterial hypertension via inducing heme oxygenase-1 and p21WAF1: An in vivo study in rats. Life Sci. 2014, 98, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, H.; Gary-Bobo, G.; Alifano, M.; Marcos, E.; Saker, M.; Vienney, N.; Amsellem, V.; Maitre, B.; Chaouat, A.; Chouaid, C.; et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ. Res. 2011, 109, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.R.; Cool, C.D.; King, J.A.C.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef]
- Benza, R.L.; Williams, G.; Wu, C.; Shields, K.J.; Raina, A.; Murali, S.; Passineau, M.J. In situ expression of Bcl-2 in pulmonary artery endothelial cells associates with pulmonary arterial hypertension relative to heart failure with preserved ejection fraction. Pulm. Circ. 2016, 6, 551–556. [Google Scholar] [CrossRef]
- Ding, X.; Zhou, S.; Li, M.; Cao, C.; Wu, P.; Sun, L.; Fei, G.; Wang, R. Upregulation of SRF is associated with hypoxic pulmonary hypertension by promoting viability of smooth muscle cells via increasing expression of Bcl-2. J. Cell. Biochem. 2017, 118, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Harada, T.; Kikuzuki, R.; Yamawaki, H.; Hara, Y. Effects of telmisartan on right ventricular remodeling induced by monocrotaline in rats. J. Pharmacol. Sci. 2009, 111, 193–200. [Google Scholar] [CrossRef]
- Wetzl, V.; Tiede, S.L.; Faerber, L.; Weissmann, N.; Schermuly, R.T.; Ghofrani, H.A.; Gall, H. Plasma MMP2/TIMP4 ratio at follow-up assessment predicts disease progression of idiopathic pulmonary arterial hypertension. Nat. Cell Biol. Inf. 2017, 195, 489–496. [Google Scholar] [CrossRef]
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1995, 151, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A. Waxman AB. IL-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Agrotis, A.; Kalinina, N.; Bobik, A. Transforming growth factor-beta, cell signaling and cardiovascular disorders. Curr. Vasc. Pharmacol. 2005, 3, 55–61. [Google Scholar] [CrossRef]
- Lu, A.; Zuo, C.; He, Y.; Chen, G.; Piao, L.; Zhang, J.; XiaoChun, F.; Shen, Y.; Tang, J.; Kong, D.; et al. EP3 receptor deficiency attenuates pulmonary hypertension through suppression of Rho/TGF-β1 signaling. J. Clin. Investig. 2015, 125, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β–dependent mouse model of pulmonary hypertension and in systemic Sclerosis. Am. J. Respir. Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.V.; Ornitz, D.M.; Singh, G.K. Diagnosis and pathophysiological mechanisms of group 3 hypoxia-induced pulmonary hypertension. Curr. Treat. Options Cardiovasc. Med. 2019, 21, 16. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Chen, Z.-L.; Zhang, T.; Wei, C.; Shi, W.-Y. TGF-β and NF-κB signaling pathway crosstalk potentiates corneal epithelial senescence through an RNA stress response. Aging 2016, 8, 2337–2354. [Google Scholar] [CrossRef]
- Wu, J.; Niu, J.; Li, X.; Wang, X.; Guo, Z.; Zhang, F. TGF-β1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev. Biol. 2014, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Kretova, M.; Sabova, L.; Hodny, Z.; Bartek, J.; Kollarovic, G.; Nelson, B.D.; Hubackova, S.; Luciakova, K. TGF-β/NF1/Smad4-mediated suppression of ANT2 contributes to oxidative stress in cellular senescence. Cell Signal. 2014, 26, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, L.; Jia, P.; Zhao, J.; Liu, D.; Liu, B. Elevated plasma connective tissue growth factor levels in children with pulmonary arterial hypertension associated with congenital heart disease. Pediatr. Cardiol. 2015, 37, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Park, G.-T.; Rue, S.-W.; Sonn, J.-K.; Park, J.-W.; Lim, Y.-B.; Jung, J.-C.; Bae, Y.-S.; Lee, Y.-S. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor β-mediated signaling pathway. Biochem. Biophys. Res. Commun. 2004, 318, 819–825. [Google Scholar] [CrossRef]
- Jun, J.-I.; Lau, L.F. CCN2 induces cellular senescence in fibroblasts. J. Cell Commun. Signal. 2016, 11, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension: Composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Nickel, N.; Kümpers, P.; Olsson, K.M.; Westerkamp, V.; Golpon, H.; Hoeper, M.M. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. In Proceedings of the American Thoracic Society 2011 International Conference, Denver, CO, USA, 13–18 May 2011; pp. 2291–2300. [Google Scholar] [CrossRef]
- Laddha, A.P.; Kulkarni, Y.A. VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir. Med. 2019, 156, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 signaling underlies fibrosis, parenchymal dysfunction, and chronic inflammation of the airway. Exp. Mol. Med. 2020, 52, 1871–1878. [Google Scholar] [CrossRef]
- Kojima, H.; Kunimoto, H.; Inoue, T.; Nakajima, K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 2012, 11, 730–739. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer Oxf. Engl. 1990, 49, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zeng, F.; Han, L.; Wang, J.; Yin, Z.; Lv, L.; Guo, L.; Wang, D.; Xu, Y.; Zhou, H. The synergistic action of phosphate and interleukin-6 enhances senescence-associated calcification in vascular smooth muscle cells depending on p53. Mech. Ageing Dev. 2019, 182, 111124. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Inoue, T.; Kunimoto, H.; Nakajima, K. IL-6-STAT3 signaling and premature senescence. JAK-STAT 2013, 2, e25763. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Shannon, J.M.; Irvin, C.G.; Fagan, K.A.; Cool, C.; Augustin, A.; Mason, R.J. Overexpression of tumor necrosis factor-α produces an increase in lung volumes and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L39–L49. [Google Scholar] [CrossRef]
- Li, X.-Q.; Wang, H.-M.; Yang, C.-G.; Zhang, X.-H.; Han, D.-D.; Wang, H.-L. Fluoxetine inhibited extracellular matrix of pulmonary artery and inflammation of lungs in monocrotaline-treated rats. Acta Pharmacol. Sin. 2011, 32, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Beyne-Rauzy, O.; Recher, C.; Dastugue, N.; Demur, C.; Pottier, G.; Laurent, G.; Sabatier, L.; Mas, V.M.-D. Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene 2004, 23, 7507–7516. [Google Scholar] [CrossRef]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Konstantinou, A.; Kletsas, D. Long-term exposure to TNF-α leads human skin fibroblasts to a p38 MAPK- and ROS-mediated premature senescence. Biogerontology 2018, 19, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.-C.; Schermuly, R.; Ghofrani, A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Frazziano, G.; Champion, H.C.; Pagano, P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol. Circ. Physiol. 2012, 302, H2166–H2177. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef]
- Klinger, J.R.; Abman, S.H.; Gladwin, M.T. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am. J. Respir. Crit. Care. Med. 2013, 188, 639–646. [Google Scholar] [CrossRef]
- Hagan, G.; Pepke-Zaba, J. Pulmonary hypertension, nitric oxide and nitric oxide-releasing compounds. Expert Rev. Respir. Med. 2011, 5, 163–171. [Google Scholar] [CrossRef]
- Giaid, A.; Saleh, D. Reduced expression of Endothelial Nitric Oxide Synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Xue, C.; Johns, A.R. Endothelial Nitric Oxide Synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.A.; Springall, D.R.; Burke, M.; Pollock, J.; Mikhail, G.; Yacoub, M.H.; Polak, J.M. High expression of endothelial nitric oxide synthase in plexiform lesions of pulmonary hypertension. J. Pathol. 1998, 185, 313–318. [Google Scholar] [CrossRef]
- Almudéver, P.; Milara, J.; De Diego, A.; Serrano-Mollar, A.; Xaubet, A.; Perez-Vizcaino, F.; Cogolludo, A.; Cortijo, J. Role of tetrahydrobiopterin in pulmonary vascular remodelling associated with pulmonary fibrosis. Thorax 2013, 68, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Chang, E.; Glassford, A.J.; Cooke, J.P.; Chiu, C.P.; Tsao, P.S. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Gross, C.M.; Sharma, S.; Fineman, J.R.; Black, S.M. Reactive oxygen species in pulmonary vascular remodeling. Compr. Physiol. 2013, 3, 1011–1034. [Google Scholar] [CrossRef]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Guibert, C.; Dubois, M.; Courtois, A.; Marthan, R.; Savineau, J.-P.; Muller, B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Ther. Adv. Respir. Dis. 2013, 7, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahè, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G. NADPH Oxidases and mitochondria in vascular senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M.; Nickel, N.; Krämer, R.; Golpon, H.; Westerkamp, V.; Olsson, K.M.; Haller, H.; Hoeper, M. Osteopontin in patients with idiopathic pulmonary hypertension. Chest 2011, 139, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Yang, T.; Li, W.; Song, L.; Meng, X.; Gu, Q.; Xiong, C.; He, J. Transgelin as a potential target in the reversibility of pulmonary arterial hypertension secondary to congenital heart disease. J. Cell. Mol. Med. 2018, 22, 6249–6261. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xin, X.; Wang, L.; Wang, B.; Chen, L.; Liu, O.; Rowe, D.W.; Xu, M. Senolytics improve bone forming potential of bone marrow mesenchymal stem cells from aged mice. NPJ Regen. Med. 2021, 6, 1–5. [Google Scholar] [CrossRef]
- Montani, D.; Bergot, E.; Günther, S.; Savale, L.; Bergeron, A.; Bourdin, A.; Bouvaist, H.; Canuet, M.; Pison, C.; Macro, M.; et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012, 125, 2128–2137. [Google Scholar] [CrossRef]
- Guignabert, C.; Phan, C.; Seferian, A.; Huertas, A.; Tu, L.; Thuillet, R.; Sattler, C.; Le Hiress, M.; Tamura, Y.; Jutant, E.-M.; et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Investig. 2016, 126, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Culley, M.K.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Perk, D.; Negi, V.; Yu, Q.; Woodcock, C.-S.C.; Handen, A.; Speyer, G.; et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef]
- Wang, G.; Li, S.; Zhao, Z.-M.; Liu, S.-X.; Zhang, G.-X.; Yang, F.; Wang, Y.; Wu, F.; Zhao, X.-X.; Xu, Z.-Y. Inhibition of heat shock protein 90 improves pulmonary arteriole remodeling in pulmonary arterial hypertension. Oncotarget 2016, 7, 54263–54273. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Anti-ageing pipeline starts to mature. Nat. Rev. Drug Discov. 2018, 17, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circiulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef]
- Houssaini, A.; Abid, S.; Mouraret, N.; Wan, F.; Rideau, D.; Saker, M.; Marcos, E.; Tissot, C.-M.; Dubois-Randé, J.-L.; Amsellem, V.; et al. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 568–577. [Google Scholar] [CrossRef]
- Nishimura, T.; Faul, J.L.; Berry, G.J.; Veve, I.; Pearl, R.G.; Kao, P.N. 40-O-(2-Hydroxyethyl)-rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am. J. Respir. Crit. Care Med. 2001, 163, 498–502. [Google Scholar] [CrossRef][Green Version]
- Paddenberg, R.; Stieger, P.; Von Lilien, A.-L.; Faulhammer, P.; Goldenberg, A.; Tillmanns, H.H.; Kummer, W.; Braun-Dullaeus, R.C. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir. Res. 2007, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Seyfarth, H.-J.; Hammerschmidt, S.; Halank, M.; Neuhaus, P.; Wirtz, H.R. Everolimus in patients with severe pulmonary hypertension: A safety and efficacy pilot trial. Pulm. Circ. 2013, 3, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Ilgin, S.; Burukoglu, D.; Atli, O.; Sirmagul, B. Effects of everolimus in combination with sildenafil in monocrotaline-induced pulmonary hypertension in rats. Cardiovasc. Toxicol. 2012, 12, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Atlı, Ö.; Ilgın, S.; Ergun, B.; Burukoğlu, D.; Musmul, A.; Sırmagül, B. Matrix metalloproteinases are possible targets in monocrotaline-induced pulmonary hypertension: Investigation of anti-remodeling effects of alagebrium and everolimus. Anatol. J. Cardiol. 2017, 17, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Agard, C.; Rolli-Derkinderen, M.; Dumas-De-La-Roque, E.; Rio, M.; Sagan, C.; Savineau, J.; Loirand, G.; Pacaud, P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 2009, 158, 1285–1294. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Zhu, J.; Li, H.; Zhang, J.; Yang, G.; Sun, Z. Metformin prevents progression of experimental pulmonary hypertension via inhibition of autophagy and activation of adenosine monophosphate-activated protein kinase. J. Vasc. Res. 2019, 56, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, Y.; Yu, F.; Xu, Y.; Yanli, L.; Liu, N. Long non-coding RNA and mRNA profile analysis of metformin to reverse the pulmonary hypertension vascular remodeling induced by monocrotaline. Biomed. Pharmacother. 2019, 115, 108933. [Google Scholar] [CrossRef] [PubMed]
- Mulkareddy, V.; Simon, M.A. Metformin in pulmonary hypertension in left heart disease. Front. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.; Niswender, K.; Burke, K.; Fan, R.; Mallugari, R.; Newman, J.; Pugh, M.; Robbins, I.; Thompson, C.; Wheeler, L.; et al. Clinical trial of metformin in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 5588. [Google Scholar]
- Liao, S.; Li, N.; Hui, Z.; McLachlan, C.S.; Zhang, Y. Metformin added to bosentan therapy in patients with pulmonary arterial hypertension associated with congenital heart defects: A pilot study. ERJ Open Res. 2018, 4, 00060–02018. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Feldman, J.; McLaughlin, V.V.; Rischard, F.; Lange, T.J.; White, R.J.; Peacock, A.J.; Gerhardt, F.; Ebrahimi, R.; Brooks, G.; et al. Selonsertib in adults with pulmonary arterial hypertension (ARROW): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Budas, G.R.; Boehm, M.; Kojonazarov, B.; Viswanathan, G.; Tian, X.; Veeroju, S.; Novoyatleva, T.; Grimminger, F.; Hinojosa-Kirschenbaum, F.; Ghofrani, A.; et al. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Church, A.C.; Martin, D.H.; Wadsworth, R.; Bryson, G.; Fisher, A.J.; Welsh, D.J.; Peacock, A.J. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: A potential novel anti-inflammatory strategy in pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L333–L347. [Google Scholar] [CrossRef] [PubMed]
- Weerackody, R.P.; Welsh, D.J.; Wadsworth, R.M.; Peacock, A.J. Inhibition of p38 MAPK reverses hypoxia-induced pulmonary artery endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2009, 296, H1312–H1320. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.; Behr, T.; Galuppo, P.; Tas, P.; Angermann, C.E.; Fraccarollo, D.; Hu, K.; Ertl, G.; Bauersachs, J. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc. Res. 2004, 63, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shimpo, H.; Shimamoto, A.; Chong, A.J.; Hampton, C.R.; Spring, D.J.; Yada, M.; Takao, M.; Onoda, K.; Yada, I.; et al. Specific inhibition of p38 mitogen-activated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J. Thorac. Cardiovasc. Surg. 2004, 128, 850–859. [Google Scholar] [CrossRef]
- MacNee, W.; Allan, R.J.; Jones, I.; De Salvo, M.C.; Tan, L.F. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: A randomised clinical trial. Thorax 2013, 68, 738–745. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, X.; Hu, G.; Xu, C.; Jiang, H. Curcumin attenuates hydrogen peroxide-induced premature senescence via the activation of SIRT1 in human umbilical vein endothelial cells. Biol. Pharm. Bull. 2015, 38, 1134–1141. [Google Scholar] [CrossRef]
- Rice, K.M.; Manne, N.D.P.K.; Kolli, M.B.; Wehner, P.S.; Dornon, L.; Arvapalli, R.; Selvaraj, V.; Kumar, A.; Blough, E.R. Curcumin nanoparticles attenuate cardiac remodeling due to pulmonary arterial hypertension. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1909–1916. [Google Scholar] [CrossRef]
- Redout, E.M.; van der Toorn, A.; Zuidwijk, M.J.; van de Kolk, C.W.A.; van Echteld, C.J.A.; Musters, R.J.P.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Heart. Circ. Physiol. 2010, 298, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Song, X.; Lin, C.; Ji, W. Interventions and mechanisms of N-acetylcysteine on monocrotaline-induced pulmonary arterial hypertension. Exp. Ther. Med. 2018, 15, 5503–5509. [Google Scholar] [CrossRef] [PubMed]
- Transform-UK: A Phase 2 Trial of Tocilizumab in Pulmonary Arterial Hypertension. Available online: https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2018.197.1_MeetingAbstracts.A7804 (accessed on 13 October 2021).
- Mertens, M.; Singh, J.A. Anakinra for Rheumatoid Arthritis: A systematic review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Parpaleix, A.; Amsellem, V.; Houssaini, A.; Abid, S.; Breau, M.; Marcos, E.; Sawaki, D.; Delcroix, M.; Quarck, R.; Adnot, S.; et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur. Respir. J. 2016, 48, 470–483. [Google Scholar] [CrossRef]
- Trankle, C.R.; Canada, J.M.; Kadariya, D.; Markley, R.; De Chazal, H.M.; Pinson, J.; Fox, A.; Van Tassell, B.W.; Abbate, A.; Grinnan, D. IL-1 blockade reduces inflammation in pulmonary arterial hypertension and right ventricular failure: A single-arm, open-label, phase IB/II pilot study. Am. J. Respir. Crit. Care Med. 2019, 199, 381–384. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Lu, J.; Li, M.-T.; Wang, Q.; Zeng, X.-F. Preventive and remedial application of etanercept attenuate monocrotaline-induced pulmonary arterial hypertension. Int. J. Rheum. Dis. 2014, 19, 192–198. [Google Scholar] [CrossRef]
- Wang, Q.; Zuo, X.; Wang, Y.; Xie, W.; Wang, H.; Zhang, M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-α antagonists via the suppression of TNF-α expression and NF-κB pathway in rats. Vasc. Pharmacol. 2013, 58, 71–77. [Google Scholar] [CrossRef]
- Mutschler, D.; Wikström, G.; Lind, L.; Larsson, A.; Lagrange, A.; Eriksson, M. Etanercept reduces late endotoxin-induced pulmonary hypertension in the pig. J. Interf. Cytokine Res. 2006, 26, 661–667. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, W.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.; Ogrodnik, M.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Houssaini, A.; Breau, M.; Kebe, K.; Abid, S.; Marcos, E.; Lipskaia, L.; Rideau, D.; Parpaleix, A.; Huang, J.; Amsellem, V.; et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Andrianifahanana, M.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Blenis, J.; Leof, E.B. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009, 69, 84–93. [Google Scholar] [CrossRef]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16 high senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020, 32, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Annual World Congress of the Pulmonary Vascular Research Institute. 2020. Available online: https://pvrinstitute.org/en/professionals/news/2019/3/15/2020-annual-world-congress-announcement/ (accessed on 1 December 2021).




| Senescence Marker | Expected Change | Senescent Cell Hallmark |
|---|---|---|
| Telomere | Shortening of telomere length | Telomere shortening |
| β-galactosidase | Enzyme activity depends on pH (in senescent cells at pH6 and in normal cells at pH 4) | Increased lysosomal compartment and activity |
| Heterochromatin foci | Over-expression of heterochromatin proteins such as H3K9Me2/3, macroH2A, and HP1 | Chromatin reorganization (SAHFs formation) |
| Histone γH2AX | Upregulated | DNA damage |
| 53BPI | Overexpression | DNA damage |
| Bcl-2 | Overexpression | Apoptosis resistance (DNA damage) |
| Lamin B1 | Downexpression | Morphological changes (Nuclear membrane) |
| P53 | Overexpression of p53 | Cell cycle arrest (Activation of p53-p21 axis) |
| P21 | Overexpression of p21 | Cell cycle arrest (Activation of p53-p21 axis) |
| P16 | Overexpression of p16INK4a | Cell cycle arrest (Activation of p16-pRB axis) |
| pRb | Overexpression of pRb | Cell cycle arrest (Activation of p16-pRB axis) |
| Ki67 | Downregulation | Cell cycle arrest (Lack of proliferation) |
| EdU/BrdU | Lack of edU/BrdU incorporation | Cell cycle arrest (Lack of proliferation) |
| MMP2 | Upregulated | Senescence-associated secretory phenotype |
| IL6 | Upregulated | Senescence-associated secretory phenotype |
| 1. Pulmonary Arterial Hypertension |
| 1.1. Idiopathic 1.2. Heritable 1.2.1 BMPR2 mutation 1.2.2 Other mutations 1.3. Drugs and toxins induced 1.4. Associated with: 1.4.1. Connective tissue disease 1.4.2. Human immunodeficiency virus (HIV) infection 1.4.3. Portal hypertension 1.4.4. Congenital heart disease 1.4.5. Schistosomiasis 1.5 PAH long-term responders to calcium channel blockers 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement 1.7 Persistent PH of the newborn syndrome |
| 2. PH due to Left Heart Disease |
| 2.1. PH due to heart failure with preserved LVEF 2.2. PH due to heart failure with reduced LVEF 2.3. Valvular heart disease 2.4. Congenital/acquired cardiovascular conditions leading to post-capillary PH |
| 3. PH due to Lung Disease and/or Hypoxia |
| 3.1. Obstructive lung disease 3.2. Restrictive lung disease 3.3. Other lung disease with mixed restrictive/obstructive pattern 3.4. Hypoxia without lung disease 3.5 Developmental lung disorders |
| 4. PH due to Pulmonary Artery Obstructions |
| 4.1. Chronic thromboembolic PH 4.2. Other pulmonary arteries OBSTRUCTIONS 4.2.1. Sarcoma or angiosarcoma 4.2.2. Other malignant tumors Renal carcinoma Uterine carcinoma Germ cell tumors of the testis 4.2.3 Non-malignant tumors Uterine leiomyoma 4.2.3. Arteritis without connective tissue disease 4.2.4. Congenital pulmonary arteries stenosis 4.2.5. Parasites Hydatidosis |
| 5. PH with Unclear and/or Multifactorial Mechanisms |
| 5.1. Hematological disorders: chronic hemolytic anemia, myeloproliferative disorders 5.2. Systemic and metabolic disorders: sarcoidosis, pulmonary Langerhans cell histiocutosis, Gaucher disease, neurofibromatosis. 5.3. Others: chronic renal failure with or without hemodialysis, fibrosing mediastinitis. 5.4. Complex congenital heart disease |
| Senescence Marker | PH, PH Animal Model and/or Cell Type | Senescence Marker Distribution | Senescence Marker mRNA and Protein Expression | Reference |
|---|---|---|---|---|
| P53 | PAECs from HPH mice | Increased protein expression | [43] | |
| PAECs from rats with MCT | Increased protein expression | [43] | ||
| PASMCs from HPH mice | Decreased protein expression | [43,44] | ||
| PASMCs from rats with MCT | Decreased protein expression | [43] | ||
| PASMCs and PAECs from patients with iPAH | Increased protein expression in iPAH-PAECs and reduced p53 expression in iPAH-PASMCs | [43,45] | ||
| P21 | Mice with HPH and rats treated with MCT | Lung tissue, endothelial cells | Increased mRNA and protein expression in lung tissue | [41,47,49,50,51,52,53] |
| P16 | Mice with HPH | Mainly located in the adventitia | Increased protein expression in older hypoxic mice | [41,49] |
| Bcl2 | Mice with HPH | Pulmonary arterial walls | Increased protein expression | [58] |
| PASMCs from HPH mice | Increased mRNA and protein expression | [58] | ||
| MMP2 | Rats treated with MCT | Endothelium and adventitia and in right ventricle | [41,59] | |
| IL-6 | Rats treated with MCT | Adventitia and diffuse staining in media/neointima | [41] |
| Senescence Marker | Group of PH | Senescence Marker Distribution | Senescence Marker mRNA and Protein Expression | Reference |
|---|---|---|---|---|
| P21 | PAH, CHD-PH and COPD-PH | Plexiform lesions | [41,54] | |
| P16 | Severe iPAH and COPD-PH | Plexiform lesions in pulmonary artery | [49,54,55] | |
| Bcl2 | HFpEF | Low values of endothelial Bcl2 index | [57] | |
| PAH | High values of endothelial Bcl2 index and Bcl2 in lung tissue | [56,57] | ||
| Survivin | PAH | Luminal cells of severe lesions | [41] | |
| MMP2 | iPAH | Increased protein expression in serum and urine | [60] | |
| IL-6 | iPAH | Increased protein expression in serum and lungs | [61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roger, I.; Milara, J.; Belhadj, N.; Cortijo, J. Senescence Alterations in Pulmonary Hypertension. Cells 2021, 10, 3456. https://doi.org/10.3390/cells10123456
Roger I, Milara J, Belhadj N, Cortijo J. Senescence Alterations in Pulmonary Hypertension. Cells. 2021; 10(12):3456. https://doi.org/10.3390/cells10123456
Chicago/Turabian StyleRoger, Inés, Javier Milara, Nada Belhadj, and Julio Cortijo. 2021. "Senescence Alterations in Pulmonary Hypertension" Cells 10, no. 12: 3456. https://doi.org/10.3390/cells10123456
APA StyleRoger, I., Milara, J., Belhadj, N., & Cortijo, J. (2021). Senescence Alterations in Pulmonary Hypertension. Cells, 10(12), 3456. https://doi.org/10.3390/cells10123456