
Mitochondrial Phenotypes in Parkinson’s Diseases—A Focus on Human iPSC-Derived Dopaminergic Neurons

 , and
, and
Abstract
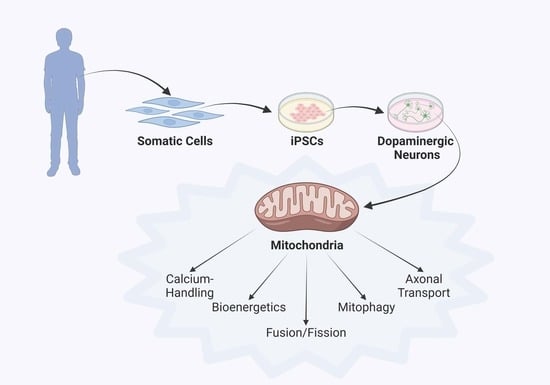
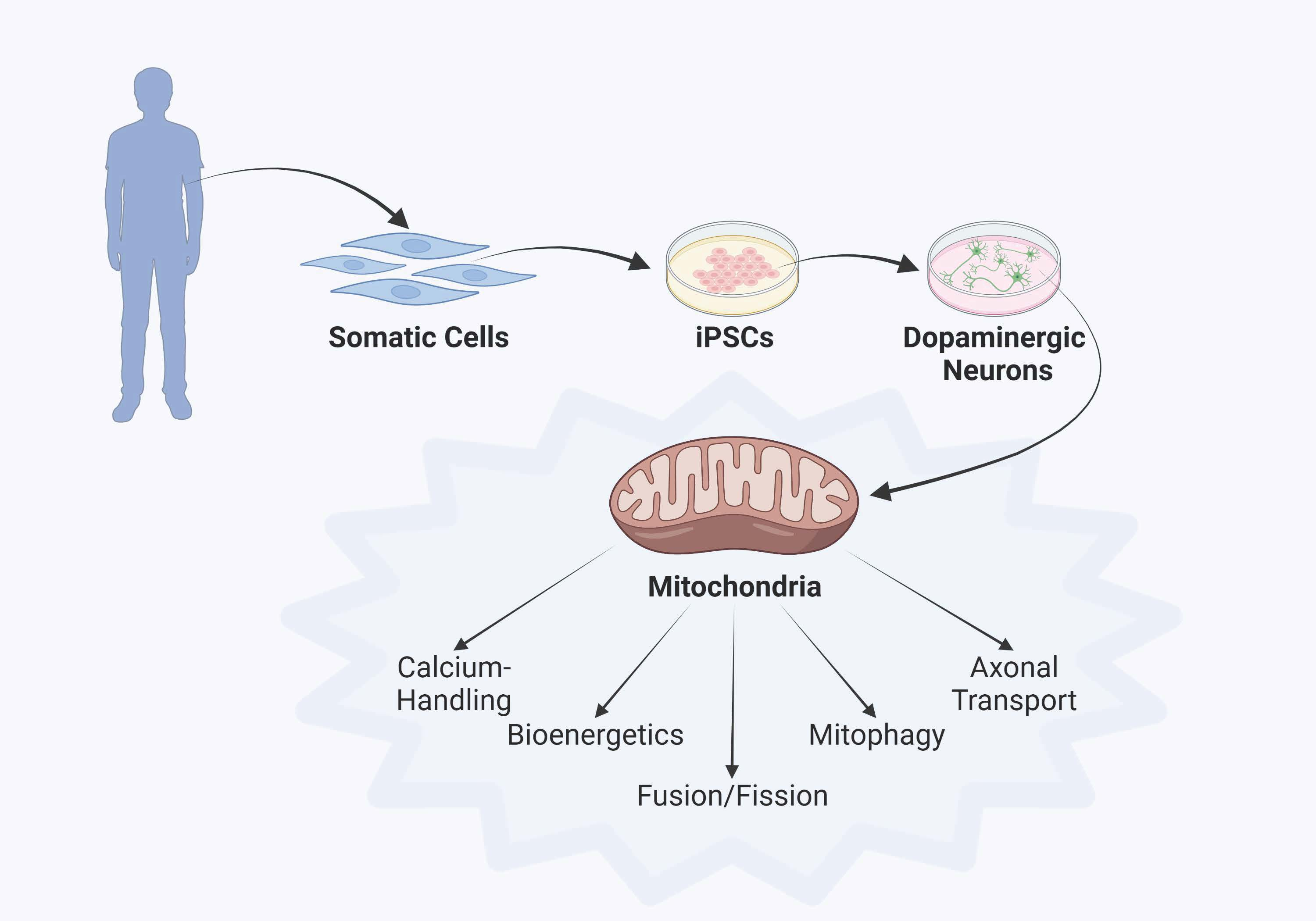
:
1. Introduction
2. Selective Neuron Vulnerability
3. Parkinson’s Disease Modeling Using Human iPSC Technology
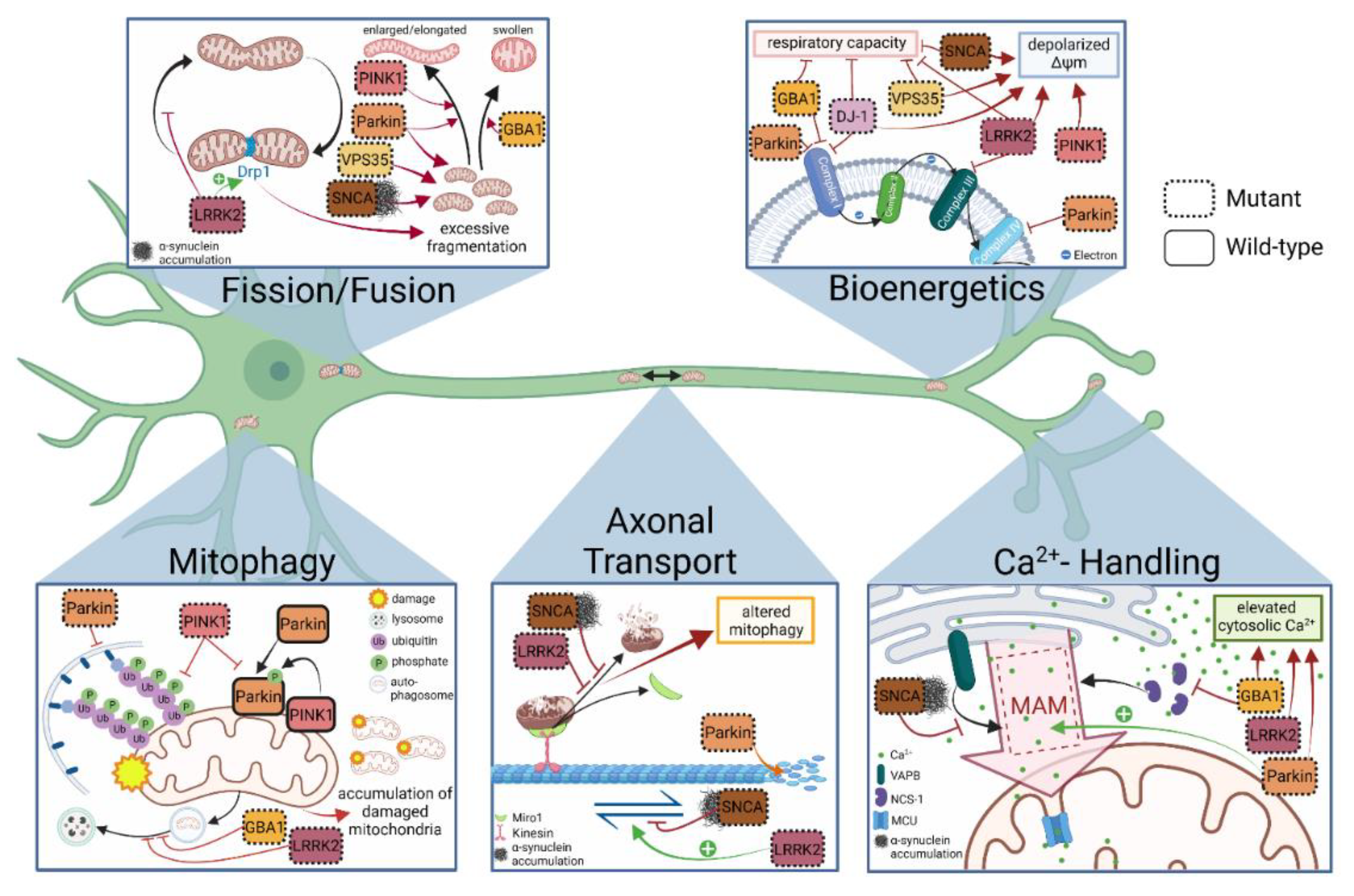
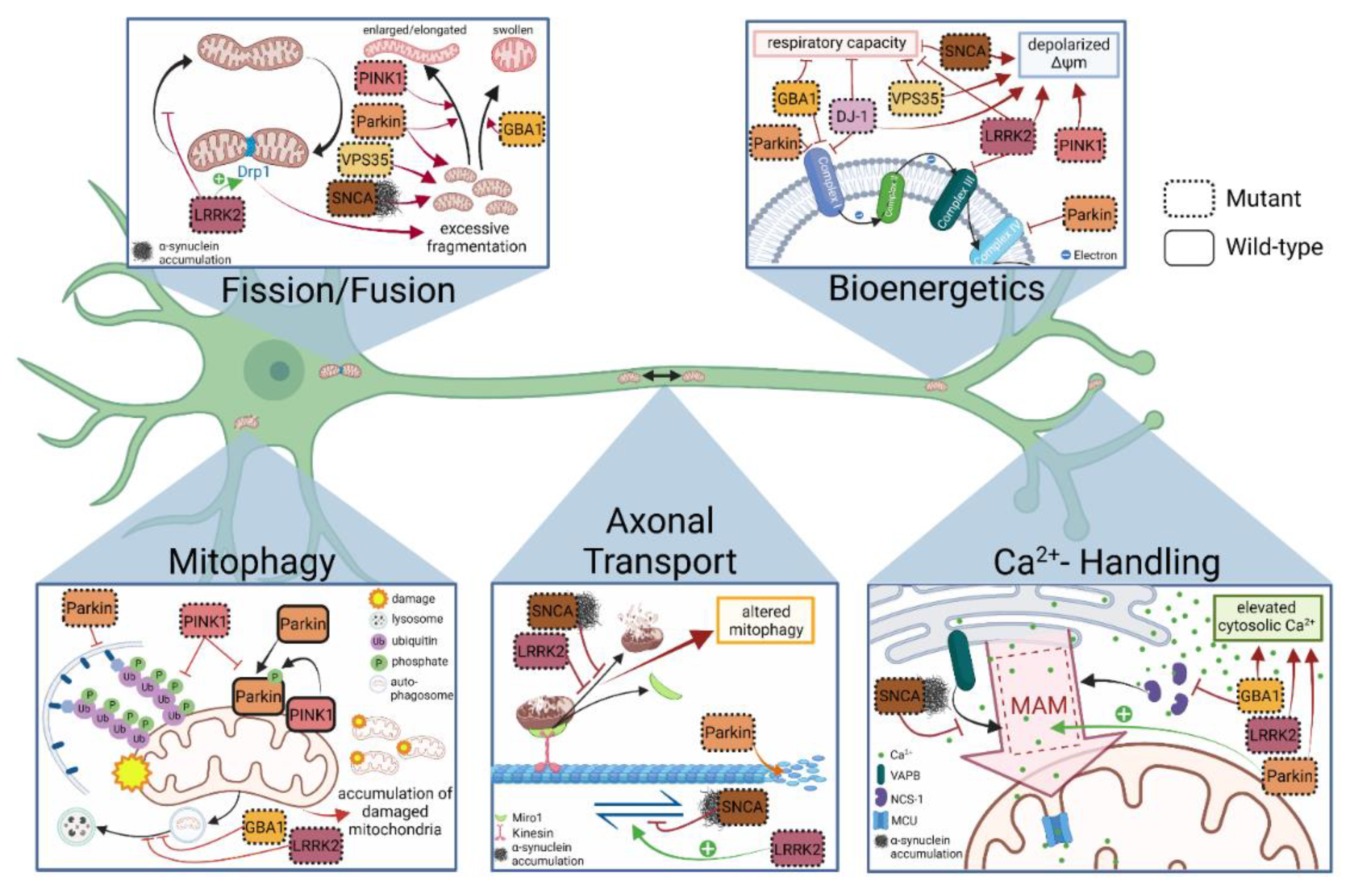
4. Discoveries of Mitochondria-Specific Phenotypes in iPSC Models of PD
4.1. Mitochondrial Respiration and Membrane Integrity
4.2. Mitochondrial Fusion, Fission and Morphology
4.3. Axonal Transport of Mitochondria
4.4. Mitophagy
4.5. Mitochondrial Calcium Handling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. 2015, 593, 3417–3429. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nature Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Et Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular models for Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 121–130. [Google Scholar] [CrossRef]
- Jagmag, S.A.; Tripathi, N.; Shukla, S.D.; Maiti, S.; Khurana, S. Evaluation of Models of Parkinson’s Disease. Front. Neurosci. 2016, 9, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and Parkinson’s disease. New Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Schrag, A.; Horsfall, L.; Walters, K.; Noyce, A.; Petersen, I. Prediagnostic presentations of Parkinson’s disease in primary care: A case-control study. Lancet Neurol. 2015, 14, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. New Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, T. Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov. Disord. 2012, 27, 617–626. [Google Scholar] [CrossRef]
- Alberico, S.L.; Cassell, M.D.; Narayanan, N.S. The Vulnerable Ventral Tegmental Area in Parkinson’s Disease. Basal Ganglia 2015, 5, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K. Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging 2004, 25, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, J.; Hess, S.; Roy, S.; Beccano-Kelly, D.; Wiederspohn, N.; Duda, J.; Simons, C.; Patil, K.; Gaifullina, A.; Mannal, N.; et al. Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson’s disease. Nat. Commun. 2019, 10, 5094. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Unternaehrer, J.J.; Daley, G.Q. Induced pluripotent stem cells for modelling human diseases. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 2274–2285. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, M. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol. 2020, 16, 465–480. [Google Scholar] [CrossRef]
- Soldner, F.; Jaenisch, R. Medicine. iPSC disease modeling. Science 2012, 338, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Hartfield, E.M.; Yamasaki-Mann, M.; Ribeiro Fernandes, H.J.; Vowles, J.; James, W.S.; Cowley, S.A.; Wade-Martins, R. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS ONE 2014, 9, e87388. [Google Scholar] [CrossRef]
- Xia, N.; Zhang, P.; Fang, F.; Wang, Z.; Rothstein, M.; Angulo, B.; Chiang, R.; Taylor, J.; Reijo Pera, R.A. Transcriptional comparison of human induced and primary midbrain dopaminergic neurons. Sci. Rep. 2016, 6, 20270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, D.; Ketteler, R.; Gissen, P.; Devine, M.J. Using stem cell-derived neurons in drug screening for neurological diseases. Neurobiol Aging 2019, 78, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Sasai, Y. Next-generation regenerative medicine: Organogenesis from stem cells in 3D culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; Volpi, L.; Pasquali, L.; Petrozzi, L.; Siciliano, G. Astrocyte-neuron interactions in neurological disorders. J. Biol Phys. 2009, 35, 317–336. [Google Scholar] [CrossRef] [Green Version]
- Krencik, R.; Ullian, E.M. A cellular star atlas: Using astrocytes from human pluripotent stem cells for disease studies. Front. Cell. Neurosci. 2013, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and alpha-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Kolodkin, N.A.; Sharma, R.P.; Colangelo, A.M.; Ignatenko, A.; Martorana, F.; Jennen, D.; Briedé, J.J.; Brady, N.; Barberis, M.; Mondeel, T.D.G.A.; et al. ROS networks: Designs, aging, Parkinson’s disease and precision therapies. NPJ Syst. Biol. Appl. 2020, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Dadgar, J.; Trevor, A.; Singer, T.P. Energy-driven uptake of N-methyl-4-phenylpyridine by brain mitochondria mediates the neurotoxicity of MPTP. Life Sci. 1986, 39, 581–588. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, Y.; Sone, N.; Suzuki, K.; Saitoh, T. Studies on the toxicity of 1-methyl-4-phenylpyridinium ion (MPP+) against mitochondria of mouse brain. J. Neurol. Sci. 1988, 86, 97–110. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Salama, M.; Arias-Carrión, O. Natural toxins implicated in the development of Parkinson’s disease. Ther. Adv. Neurol. Disord. 2011, 4, 361–373. [Google Scholar] [CrossRef]
- Ming, F.; Tan, J.; Qin, L.; Zhang, H.; Tang, J.; Tan, X.; Wang, C. The PARK2 Mutation Associated with Parkinson’s Disease Enhances the Vulnerability of Peripheral Blood Lymphocytes to Paraquat. BioMed. Res. Int. 2020, 2020, 4658109. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.-c.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [Green Version]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef] [Green Version]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grünewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425. [Google Scholar] [CrossRef] [Green Version]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogetofte, H.; Jensen, P.; Ryding, M.; Schmidt, S.I.; Okarmus, J.; Ritter, L.; Worm, C.S.; Hohnholt, M.C.; Azevedo, C.; Roybon, L.; et al. PARK2 Mutation Causes Metabolic Disturbances and Impaired Survival of Human iPSC-Derived Neurons. Front. Cell Neurosci. 2019, 13, 297. [Google Scholar] [CrossRef] [Green Version]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, P.; Allaman, I.; Rothenfusser, K.; Fiumelli, H.; Marquet, P.; Magistretti, P.J. L-Lactate protects neurons against excitotoxicity: Implication of an ATP-mediated signaling cascade. Sci. Rep. 2016, 6, 21250. [Google Scholar] [CrossRef] [Green Version]
- Hanss, Z.; Larsen, S.B.; Antony, P.; Mencke, P.; Massart, F.; Jarazo, J.; Schwamborn, J.C.; Barbuti, P.A.; Mellick, G.D.; Krüger, R. Mitochondrial and Clearance Impairment in p.D620N VPS35 Patient-Derived Neurons. Mov. Disord. 2021, 36, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141–190. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Arias-Fuenzalida, J.; Jarazo, J.; Qing, X.; Walter, J.; Gomez-Giro, G.; Nickels, S.L.; Zaehres, H.; Scholer, H.R.; Schwamborn, J.C. FACS-Assisted CRISPR-Cas9 Genome Editing Facilitates Parkinson’s Disease Modeling. Stem Cell Rep. 2017, 9, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Flierl, A.; Oliveira, L.M.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS ONE 2014, 9, e112413. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Ryan, B.J.; Lourenco-Venda, L.L.; Crabtree, M.J.; Hale, A.B.; Channon, K.M.; Wade-Martins, R. alpha-Synuclein and mitochondrial bioenergetics regulate tetrahydrobiopterin levels in a human dopaminergic model of Parkinson disease. Free Radic. Biol. Med. 2014, 67, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033. [Google Scholar] [CrossRef] [Green Version]
- Vos, M.; Geens, A.; Böhm, C.; Deaulmerie, L.; Swerts, J.; Rossi, M.; Craessaerts, K.; Leites, E.P.; Seibler, P.; Rakovic, A.; et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 2017, 216, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Safiulina, D.; Kaasik, A. Energetic and Dynamic: How Mitochondria Meet Neuronal Energy Demands. PLoS Biol. 2014, 11, e1001755. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Impairing the mitochondrial fission and fusion balance: A new mechanism of neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 283–292. [Google Scholar] [CrossRef] [Green Version]
- MacAskill, A.F.; Kittler, J.T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010, 20, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Meeusen, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial fusion intermediates revealed in vitro. Science 2004, 305, 1747–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo. J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bus, C.; Zizmare, L.; Feldkaemper, M.; Geisler, S.; Zarani, M.; Schaedler, A.; Klose, F.; Admard, J.; Mageean, C.J.; Arena, G.; et al. Human Dopaminergic Neurons Lacking PINK1 Exhibit Disrupted Dopamine Metabolism Related to Vitamin B6 Co-Factors. iScience 2020, 23, 101797. [Google Scholar] [CrossRef] [PubMed]
- Aboud, A.A.; Tidball, A.M.; Kumar, K.K.; Neely, M.D.; Han, B.; Ess, K.C.; Hong, C.C.; Erikson, K.M.; Hedera, P.; Bowman, A.B. PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol. Dis. 2015, 73, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Yokota, M.; Kakuta, S.; Shiga, T.; Ishikawa, K.-i.; Okano, H.; Hattori, N.; Akamatsu, W.; Koike, M. Establishment of an in vitro model for analyzing mitochondrial ultrastructure in PRKN-mutated patient iPSC-derived dopaminergic neurons. Mol. Brain 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Qi, X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loss, O.; Stephenson, F.A. Developmental changes in trak-mediated mitochondrial transport in neurons. Mol. Cell Neurosci. 2017, 80, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- LaMonte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascaño, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 2002, 34, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Beaumont, K.G.; Mrksich, M.; Krainc, D. Micropatterning Facilitates the Long-Term Growth and Analysis of iPSC-Derived Individual Human Neurons and Neuronal Networks. Adv. Healthc. Mater. 2016, 5, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirinzi, T.; Madeo, G.; Martella, G.; Maltese, M.; Picconi, B.; Calabresi, P.; Pisani, A. Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov. Disord. 2016, 31, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Goldwurm, S.; Casagrande, F.; Pezzoli, G.; Cappelletti, G. Microtubule Destabilization Is Shared by Genetic and Idiopathic Parkinson’s Disease Patient Fibroblasts. PLoS ONE 2012, 7, e37467. [Google Scholar] [CrossRef]
- Cartelli, D.; Amadeo, A.; Calogero, A.M.; Casagrande, F.V.M.; De Gregorio, C.; Gioria, M.; Kuzumaki, N.; Costa, I.; Sassone, J.; Ciammola, A.; et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol. Aging 2018, 61, 66–74. [Google Scholar] [CrossRef] [PubMed]
- López-Doménech, G.; Higgs, N.F.; Vaccaro, V.; Roš, H.; Arancibia-Cárcamo, I.L.; MacAskill, A.F.; Kittler, J.T. Loss of Dendritic Complexity Precedes Neurodegeneration in a Mouse Model with Disrupted Mitochondrial Distribution in Mature Dendrites. Cell Rep. 2016, 17, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenguer-Escuder, C.; Grossmann, D.; Antony, P.; Arena, G.; Wasner, K.; Massart, F.; Jarazo, J.; Walter, J.; Schwamborn, J.C.; Grünewald, A.; et al. Impaired mitochondrial-endoplasmic reticulum interaction and mitophagy in Miro1-mutant neurons in Parkinson’s disease. Hum. Mol. Genet. 2020, 29, 1353–1364. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Shaltouki, A.; Hsieh, C.H.; Kim, M.J.; Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Li, L.; Vanhauwaert, R.; Nguyen, K.T.; Davis, M.D.; Bu, G.; Wszolek, Z.K.; Wang, X. Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models. Cell Metab. 2019, 30, 1131–1140 e1137. [Google Scholar] [CrossRef] [PubMed]
- Bharat, V.; Wang, X. Precision Neurology for Parkinson’s Disease: Coupling Miro1-Based Diagnosis with Drug Discovery. Mov. Disord. 2020, 35, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Grenier, K.; McLelland, G.L.; Fon, E.A. Parkin- and PINK1-Dependent Mitophagy in Neurons: Will the Real Pathway Please Stand Up? Front. Neurol. 2013, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, V.S.; Arnold, B.; Cassady, S.J.; Chu, C.T.; Burton, E.A.; Berman, S.B. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 2011, 20, 927–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Ishigaki, S.; Fujioka, Y.; Kimura, M.; Sobue, G.; Tanaka, K.; Matsuda, N. The principal PINK1 and Parkin cellular events triggered in response to dissipation of mitochondrial membrane potential occur in primary neurons. Genes Cells 2013, 18, 672–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grünewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.-K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [Green Version]
- Trombetta-Lima, M.; Sabogal-Guáqueta, A.M.; Dolga, A.M. Mitochondrial dysfunction in neurodegenerative diseases: A focus on iPSC-derived neuronal models. Cell Calcium. 2021, 94, 102362. [Google Scholar] [CrossRef]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef]
- Schwartzentruber, A.; Boschian, C.; Lopes, F.M.; Myszczynska, M.A.; New, E.J.; Beyrath, J.; Smeitink, J.; Ferraiuolo, L.; Mortiboys, H. Oxidative switch drives mitophagy defects in dopaminergic parkin mutant patient neurons. Sci. Rep. 2020, 10, 15485. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Ishikawa, K.I.; Inoshita, T.; Izawa, N.; Takanashi, M.; Sato, S.; Onodera, O.; Akamatsu, W.; Okano, H.; Imai, Y.; et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3172–3185. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol 2021, 31, 2140–2154.e2146. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef] [PubMed]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Ichas, F.; Mazat, J.P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1998, 1366, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim Biophys Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in dopaminergic ventral tegmental area neurons: Depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Schumacker, P.T. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol Chem 2013, 288, 10736–10741. [Google Scholar] [CrossRef] [Green Version]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Yin, X.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.; et al. Defects in mRNA Translation in LRRK2-Mutant hiPSC-Derived Dopaminergic Neurons Lead to Dysregulated Calcium Homeostasis. Cell Stem Cell 2020, 27, 633–645.e637. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Mitochondrial Phenotype | PD-Associated Gene | Mitochondrial Dysfunction in PD | References |
|---|---|---|---|
| Respiration and membrane integrity | Parkin | Deficits of ETC complex I activity | [67] |
| PINK1 | [83] | ||
| LRRK | [74] | ||
| GBA1 | [68] | ||
| LRRK2 | Deficits of ETC complex III activity | [69] | |
| Parkin | Suppression of ETC complex IV proteins | [70] | |
| SNCA | Reduced respiratory capacity | [77,78,79] | |
| DJ-1 | [34] | ||
| LRRK2 | [69] | ||
| VPS35 | [73] | ||
| GBA1 | [68] | ||
| SNCA | Reduced membrane potential | [77,81] | |
| PINK1 | [82,83] | ||
| LRRK2 | [75] | ||
| VPS35 | [73] | ||
| DJ-1 | Mitochondrial oxidant stress | [34] | |
| Fusion, fission and morphology | SNCA | Abnormal mitochondrial morphology | [105] |
| Parkin | [43,70,98,100,101] | ||
| PINK1 | [43] | ||
| LRRK2 | [75,102] | ||
| VPS35 | [73] | ||
| GBA1 | [68] | ||
| Axonal mitochondrial transport | SNCA | Altered mitochondrial motility | [118,120] |
| LRRK2 | [69,74,119] | ||
| Parkin | Microtubule fragmentation | [115] | |
| Mitophagy | Parkin | Impaired mitophagy/mitophagic flux | [153,154] |
| PINK1 | [99] | ||
| PINK1 | Reduction in kinase activity | [155,156] | |
| LRRK2 | Malfunction in autophagosome transport | [157] | |
| SNCA | Delayed mitophagy through altered Miro1 kinetics | [120] | |
| LRRK2 | [119] | ||
| PINK1 | Impaired Parkin recruitment | [150] | |
| GBA1 | Diminished mitochondrial-lysosomal colocalization | [68] | |
| Calcium handling | LRRK2 | Increased cytosolic Ca2+ level | [32] |
| GBA1 | [176] | ||
| Parkin | Increased association between ER and mitochondria | [175] | |
| SNCA | Disrupted MAM tethering | [177] | |
| GBA1 | Reduced neuronal calcium sensor-1 (NCS-1) level | [178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heger, L.M.; Wise, R.M.; Hees, J.T.; Harbauer, A.B.; Burbulla, L.F. Mitochondrial Phenotypes in Parkinson’s Diseases—A Focus on Human iPSC-Derived Dopaminergic Neurons. Cells 2021, 10, 3436. https://doi.org/10.3390/cells10123436
Heger LM, Wise RM, Hees JT, Harbauer AB, Burbulla LF. Mitochondrial Phenotypes in Parkinson’s Diseases—A Focus on Human iPSC-Derived Dopaminergic Neurons. Cells. 2021; 10(12):3436. https://doi.org/10.3390/cells10123436
Chicago/Turabian StyleHeger, Leonie M., Rachel M. Wise, J. Tabitha Hees, Angelika B. Harbauer, and Lena F. Burbulla. 2021. "Mitochondrial Phenotypes in Parkinson’s Diseases—A Focus on Human iPSC-Derived Dopaminergic Neurons" Cells 10, no. 12: 3436. https://doi.org/10.3390/cells10123436
APA StyleHeger, L. M., Wise, R. M., Hees, J. T., Harbauer, A. B., & Burbulla, L. F. (2021). Mitochondrial Phenotypes in Parkinson’s Diseases—A Focus on Human iPSC-Derived Dopaminergic Neurons. Cells, 10(12), 3436. https://doi.org/10.3390/cells10123436