
The Role of T Cell Immunotherapy in Acute Myeloid Leukemia

Abstract
1. Introduction
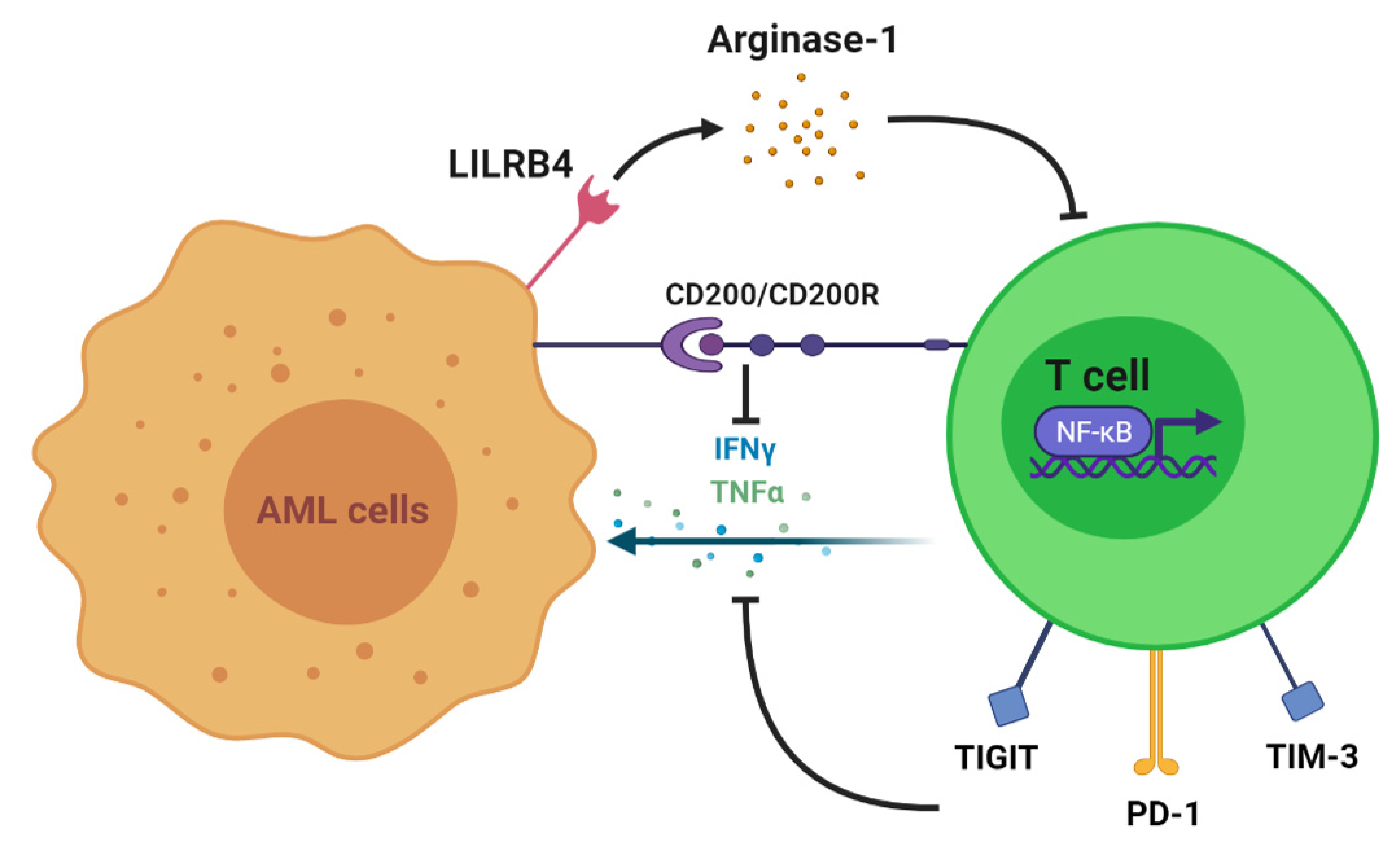
2. T Cell Alteration in AML
3. T Cell Antileukemia Effect
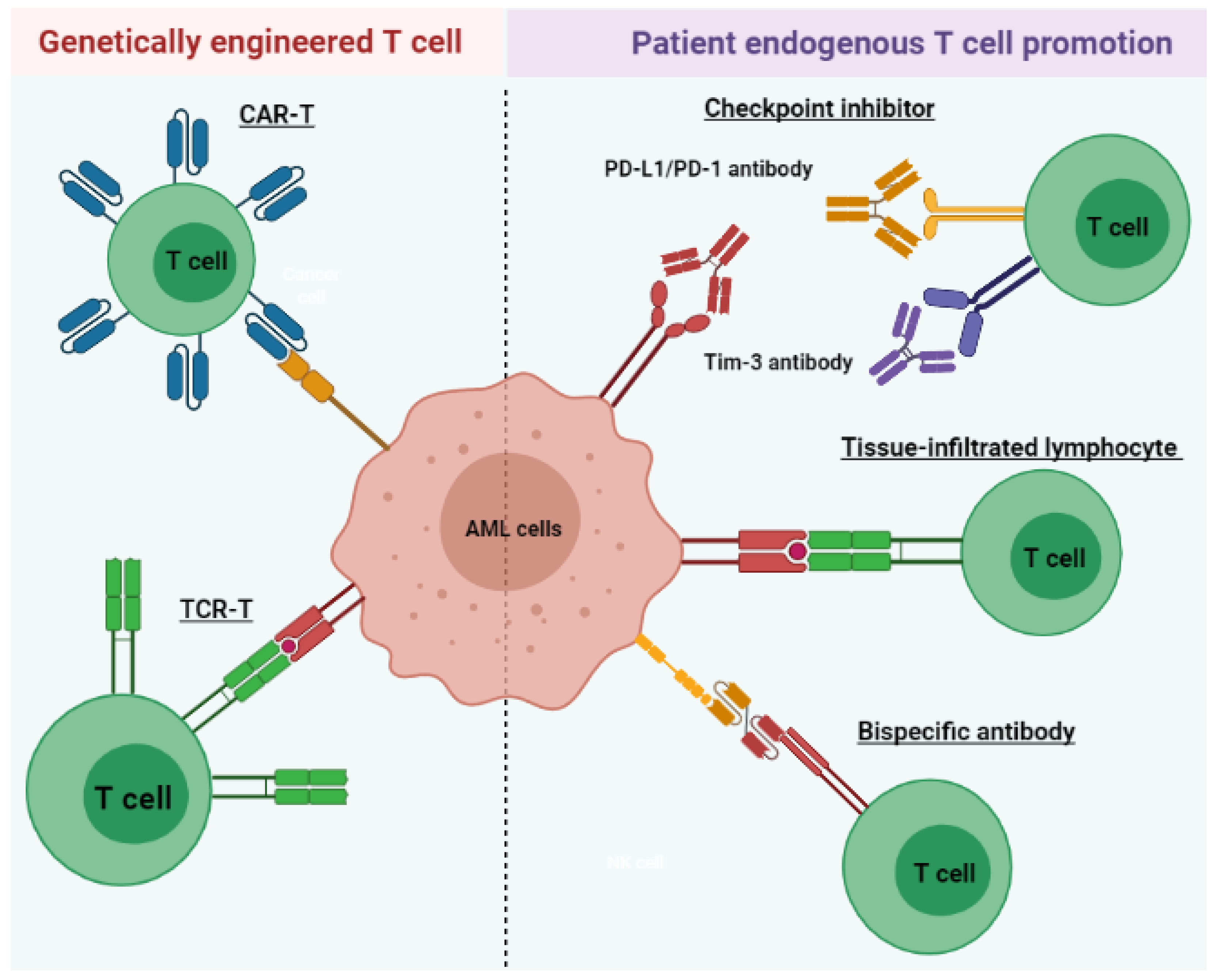
4. T Cell Immunotherapy
4.1. Tissue-Infiltrated Lymphocyte
4.2. T Cell-Recruiting Bispecific Antibody
4.3. Immune Checkpoint Inhibitor
4.4. Chimeric Antigen Receptor T (CAR-T) Cell Therapy
4.5. Tumor-Specific T Cell Receptor Gene-Transduced T Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Löwenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Cai, S.F.; Levine, R.L. Genetic and epigenetic determinants of AML pathogenesis. Semin. Hematol. 2019, 56, 84–89. [Google Scholar] [CrossRef]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med J. 2020, 103, 38–40. [Google Scholar]
- Kantarjian, H. Acute myeloid leukemia—Major progress over four decades and glimpses into the future. Am. J. Hematol. 2016, 91, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Wang, E.S. Novel therapies for AML: A round-up for clinicians. Expert Rev. Clin. Pharmacol. 2020, 13, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Chan, S.M.; Daver, N.G.; Jonas, B.A.; Pollyea, D.A. Optimizing survival outcomes with post-remission therapy in acute myeloid leukemia. Am. J. Hematol. 2019, 94, 803–811. [Google Scholar] [CrossRef]
- Mueller, B.U.; Seipel, K.; Bacher, U.; Pabst, T. Autologous Transplantation for Older Adults with AML. Cancers 2018, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Takami, A. Hematopoietic stem cell transplantation for acute myeloid leukemia. Int. J. Hematol. 2018, 107, 513–518. [Google Scholar] [CrossRef]
- Kassim, A.A.; Savani, B.N. Hematopoietic stem cell transplantation for acute myeloid leukemia: A review. Hematol. Oncol. Stem Cell Ther. 2017, 10, 245–251. [Google Scholar] [CrossRef]
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 17, 475–492. [Google Scholar] [CrossRef]
- Sweeney, C.; Vyas, P. The Graft-Versus-Leukemia Effect in AML. Front. Oncol. 2019, 9, 1217. [Google Scholar] [CrossRef] [PubMed]
- Radujkovic, A.; Guglielmi, C.; Bergantini, S.; Iacobelli, S.; van Biezen, A.; Milojkovic, D.; Gratwohl, A.; Schattenberg, A.V.; Verdonck, L.F.; Niederwieser, D.W.; et al. Donor Lymphocyte Infusions for Chronic Myeloid Leukemia Relapsing after Allogeneic Stem Cell Transplantation: May We Predict Graft-versus-Leukemia Without Graft-versus-Host Disease? Biol. Blood Marrow Transplant. 2015, 21, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Orti, G.; Barba, P.; Fox, L.; Salamero, O.; Bosch, F.; Valcarcel, D. Donor lymphocyte infusions in AML and MDS: Enhancing the graft-versus-leukemia effect. Exp. Hematol. 2017, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.; Kolli, S.S.; Strowd, L.C. Review of Graft-Versus-Host Disease. Dermatol. Clin. 2019, 37, 569–582. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Orleans-Lindsay, J.K.; Barber, L.D.; Prentice, H.G.; Lowdell, M.W. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function--implications for the adoptive immunotherapy of leukaemia. Clin. Exp. Immunol. 2001, 126, 403–411. [Google Scholar] [CrossRef]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef]
- Deng, M.; Gui, X.; Kim, J.; Xie, L.; Chen, W.; Li, Z.; He, L.; Chen, Y.; Chen, H.; Luo, W.; et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018, 562, 605–609. [Google Scholar] [CrossRef]
- Coles, S.J.; Hills, R.K.; Wang, E.C.Y.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia 2012, 26, 2146–2148. [Google Scholar] [CrossRef]
- Ustun, C.; Miller, J.S.; Munn, D.H.; Weisdorf, D.J.; Blazar, B.R. Regulatory T cells in acute myelogenous leukemia: Is it time for immunomodulation? Blood 2011, 118, 5084–5095. [Google Scholar] [CrossRef]
- Zhou, Q.; Bucher, C.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Munn, D.H.; Levine, B.L.; Riddle, M.; June, C.H.; Vallera, D.A.; et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood 2009, 114, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated frequencies of CD4⁺ CD25⁺ CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yu, S.; Liu, L.; Xue, F.; Yuan, C.; Wang, M.; Ji, C.; Ma, D. The Profile of T Helper Subsets in Bone Marrow Microenvironment Is Distinct for Different Stages of Acute Myeloid Leukemia Patients and Chemotherapy Partly Ameliorates These Variations. PLoS ONE 2015, 10, e0131761. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef]
- Tan, J.; Chen, S.; Lu, Y.; Yao, D.; Xu, L.; Zhang, Y.; Yang, L.; Chen, J.; Lai, J.; Yu, Z.; et al. Higher PD-1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia. Chin. J. Cancer Res. 2017, 29, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef]
- Wang, M.; Bu, J.; Zhou, M.; Sido, J.; Lin, Y.; Liu, G.; Lin, Q.; Xu, X.; Leavenworth, J.W.; Shen, E. CD8(+)T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin. Immunol. 2018, 190, 64–73. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888. [Google Scholar]
- Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Zhu, L.; Zeng, H.; Schell, T.D.; Zheng, H. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015, 5, e330. [Google Scholar] [CrossRef]
- Radpour, R.; Riether, C.; Simillion, C.; Höpner, S.; Bruggmann, R.; Ochsenbein, A.F. CD8(+) T cells expand stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. Leukemia 2019, 33, 2379–2392. [Google Scholar] [CrossRef]
- Barbui, A.M.; Borleri, G.; Conti, E.; Ciocca, A.; Salvi, A.; Micò, C.; Introna, M.; Rambaldi, A. Clinical grade expansion of CD45RA, CD45RO, and CD62L-positive T-cell lines from HLA-compatible donors: High cytotoxic potential against AML and ALL cells. Exp. Hematol. 2006, 34, 475–485. [Google Scholar] [CrossRef]
- Lamble, A.J.; Lind, E.F. Targeting the Immune Microenvironment in Acute Myeloid Leukemia: A Focus on T Cell Immunity. Front. Oncol. 2018, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Heidel, F.; Tenzer, S.; Radsak, M.P.; Solem, F.K.; Britten, C.M.; Huber, C.; Fischer, T.; Wölfel, T. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 2007, 109, 2985–2988. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Ono, Y.; Hofmann, S.; Schmitt, A.; Mehring, E.; Götz, M.; Guillaume, P.; Döhner, K.; Mytilineos, J.; Döhner, H.; et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood 2012, 120, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.K.; Lane, T.A.; Ball, E.D. Generation of T-cell lines to autologous acute myeloid leukemia cells by competitive limiting dilution culture of acute myeloid leukemia mononuclear cells. Exp. Hematol. 2008, 36, 486–494. [Google Scholar] [CrossRef]
- Eljaafari, A.; Farre, A.; Duperrier, K.; Even, J.; Vie, H.; Michallet, M.; Souillet, G.; Catherine Freidel, A.; Gebuhrer, L.; Rigal, D. Generation of helper and cytotoxic CD4+T cell clones specific for the minor histocompatibility antigen H-Y, after in vitro priming of human T cells by HLA-identical monocyte-derived dendritic cells. Transplantation 2001, 71, 1449–1455. [Google Scholar] [CrossRef]
- Gertner-Dardenne, J.; Fauriat, C.; Vey, N.; Olive, D. Immunotherapy of acute myeloid leukemia based on γδ T cells. Oncoimmunology 2012, 1, 1614–1616. [Google Scholar] [CrossRef][Green Version]
- Gertner-Dardenne, J.; Castellano, R.; Mamessier, E.; Garbit, S.; Kochbati, E.; Etienne, A.; Charbonnier, A.; Collette, Y.; Vey, N.; Olive, D. Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts. J. Immunol. 2012, 188, 4701–4708. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef]
- Lin, B.; Du, L.; Li, H.; Zhu, X.; Cui, L.; Li, X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed. Pharmacother. 2020, 132, 110873. [Google Scholar] [CrossRef]
- Li, Z.; Philip, M.; Ferrell, P.B. Alterations of T-cell-mediated immunity in acute myeloid leukemia. Oncogene 2020, 39, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, J.; Liu, J.; Yao, J.; He, Y.; Li, X.; Yu, J.; Yang, J.; Liu, Z.; Huang, S. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur. J. Haematol. 2005, 75, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, Z.; Zhang, Z.; Li, Y.; Fan, S.; Zhao, Y.; Liu, Z.; Ye, X.; Zhang, F.; Yu, Y.; et al. Assessment of the presence and anti-tumor potential of tumor-infiltrating lymphocytes in patients with acute myeloid leukemia. Cancer Manag. Res. 2019, 11, 3187–3196. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Sánchez-Abarca, L.I.; Domingo, E.; Ramos, T.L.; Bejarano-García, J.A.; Gonzalez-Campos, J.A.; Caballero-Velázquez, T.; Pérez-Simón, J.A. Selection of Tumor-Specific Cytotoxic T Lymphocytes in Acute Myeloid Leukemia Patients Through the Identification of T-Cells Capable to Establish Stable Interactions With the Leukemic Cells: “Doublet Technology”. Front. Immunol. 2018, 9, 1971. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841. [Google Scholar] [CrossRef]
- Kayser, S.; Levis, M.J. Advances in targeted therapy for acute myeloid leukaemia. Br. J. Haematol. 2018, 180, 484–500. [Google Scholar] [CrossRef]
- Stanchina, M.; Soong, D.; Zheng-Lin, B.; Watts, J.M.; Taylor, J. Advances in Acute Myeloid Leukemia: Recently Approved Therapies and Drugs in Development. Cancers 2020, 12, 3225. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Teague, R.M.; Kline, J. Immune evasion in acute myeloid leukemia: Current concepts and future directions. J. Immunother. Cancer 2013, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.C.; Stein, A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101224. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Ravandi, F. The potential role of Bi-specific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101218. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2:2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2019, 134, 834. [Google Scholar] [CrossRef]
- Sarde, A.; Eckard, S.; Mei, L.; Ruegg, C.; Chun, P.; Smith, V. Selectivity of T Cell Engager AMV564 Against Different Leukemic Blast Populations and Potential Application for Patient Selection. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥65 Years) AML Patients. Blood 2019, 134, 832. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Vincent, B.G.; Esparza, S.; Ivanova, A.; Moore, D.T.; Foster, M.C.; Coombs, C.C.; Jamieson, K.; Van Deventer, H.W.; Blanchard, L.; et al. Final Clinical Results of a Phase II Study of High Dose Cytarabine Followed By Pembrolizumab in Relapsed/Refractory AML. Blood 2019, 134, 831. [Google Scholar] [CrossRef]
- Ghosh, A.; Barba, P.; Perales, M.A. Checkpoint inhibitors in AML: Are we there yet? Br. J. Haematol. 2020, 188, 159–167. [Google Scholar] [CrossRef]
- Liao, D.; Wang, M.; Liao, Y.; Li, J.; Niu, T. A Review of Efficacy and Safety of Checkpoint Inhibitor for the Treatment of Acute Myeloid Leukemia. Front. Pharmacol. 2019, 10, 609. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Bauer, K.; Herrmann, H.; Gleixner, K.V.; Schulenburg, A.; Rabitsch, W.; Sperr, W.R.; Wolf, D. Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. Int. J. Mol. Sci. 2019, 20, 4233. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, M.A.; Brault, M.; Bleakley, M. T-Cell Receptor-Based Immunotherapy for Hematologic Malignancies. Cancer J. 2019, 25, 179–190. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Chapuis Aude, G.; Ragnarsson Gunnar, B.; Nguyen Hieu, N.; Chaney Colette, N.; Pufnock Jeffrey, S.; Schmitt Thomas, M.; Duerkopp, N.; Roberts Ilana, M.; Pogosov Galina, L.; Ho William, Y.; et al. Transferred WT1-Reactive CD8+ T Cells Can Mediate Antileukemic Activity and Persist in Post-Transplant Patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene−transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, H.; Jiang, B.; Li, J.; Lu, X.; Li, L.; Ruan, G.; Liu, Y.; Chen, S.; Huang, X. Expression patterns of WT1 and PRAME in acute myeloid leukemia patients and their usefulness for monitoring minimal residual disease. Leuk. Res. 2009, 33, 384–390. [Google Scholar] [CrossRef]
- Van Baren, N.; Chambost, H.; Ferrant, A.; Michaux, L.; Ikeda, H.; Millard, I.; Olive, D.; Boon, T.; Coulie, P.G. PRAME, a gene encoding an antigen recognized on a human melanoma by cytolytic T cells, is expressed in acute leukaemia cells. Br. J. Haematol. 1998, 102, 1376–1379. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name | Target CheckPoint/Antigen | Clinical Trial Start Time | Trial Number/Phase | Disease | Status |
|---|---|---|---|---|---|
| T cell-recruiting bispecific antibody | CD3 and CD33 | 2015 | NCT02520427/Phase 1 | Relapsed/Refractory AML | Recruiting; estimated completion on 28 February 2023 |
| 2017 | NCT03224819/Phase 1 | AML | Active; estimated completion on 21 March 2022 | ||
| 2017 | NCT03144245/Phase 1 | AML | Completed on 5 November 2020; no result posted | ||
| 2019 | NCT03915379/Phase 1 | AML and myelodysplastic syndromes | Recruiting; estimated completion on 26 October 2022 | ||
| 2018 | NCT03516760//Phase 1 | Relapsed/Refractory AML | Active; estimated completion on 31 December 2020 | ||
| Immune checkpoint inhibitor | PD-1 | 2015 | NCT02397720/Phase 2 | AML | Recruiting; estimated completion on 30 April 2022 |
| 2016 | NCT02845297/Phase 2 | AML | Active; estimated completion in October 2021 | ||
| 2020 | NCT04214249/Phase 2 | AML | Not yet recruiting; estimated completion on 31 July 2024 | ||
| Chimeric antigen receptor T cell therapy | CD123 | 2015 | NCT02159495/Phase 1 | AML | Recruiting; estimated completion on 15 December 2021 |
| CD33/Lewis Y | 2013 | NCT01864902/Phase 1 and 2 | Relapsed/refractory AML | Unknown recruiting status; estimated completion in April 2017 | |
| Tumor-specific T cell receptor gene-transduced T cells | WT 1 | 2012 | NCT01640301/Phase 1 and 2 | Recurrent and secondary AML | Active; estimated completion on 1 September 2029 |
| 2002 | NCT00052520/Phase 1 and 2 | AML | Completed in June 2013; no result posted |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, F.; Sholy, C.; Wang, C.; Cao, M.; Kang, X. The Role of T Cell Immunotherapy in Acute Myeloid Leukemia. Cells 2021, 10, 3376. https://doi.org/10.3390/cells10123376
Hao F, Sholy C, Wang C, Cao M, Kang X. The Role of T Cell Immunotherapy in Acute Myeloid Leukemia. Cells. 2021; 10(12):3376. https://doi.org/10.3390/cells10123376
Chicago/Turabian StyleHao, Fang, Christine Sholy, Chen Wang, Min Cao, and Xunlei Kang. 2021. "The Role of T Cell Immunotherapy in Acute Myeloid Leukemia" Cells 10, no. 12: 3376. https://doi.org/10.3390/cells10123376
APA StyleHao, F., Sholy, C., Wang, C., Cao, M., & Kang, X. (2021). The Role of T Cell Immunotherapy in Acute Myeloid Leukemia. Cells, 10(12), 3376. https://doi.org/10.3390/cells10123376