
Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges


Abstract
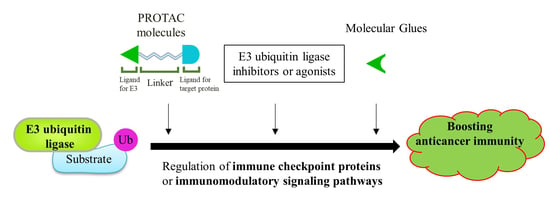
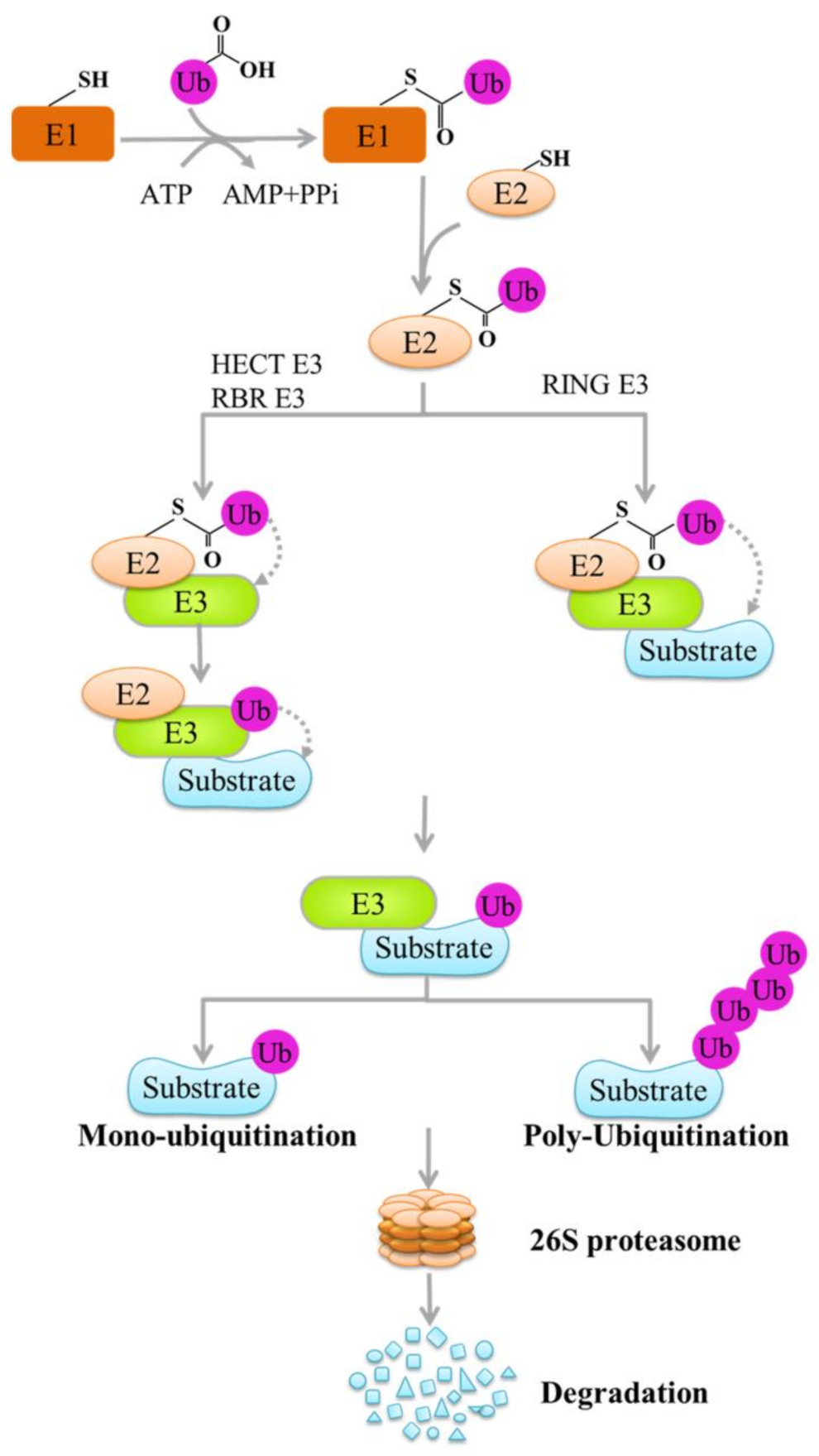
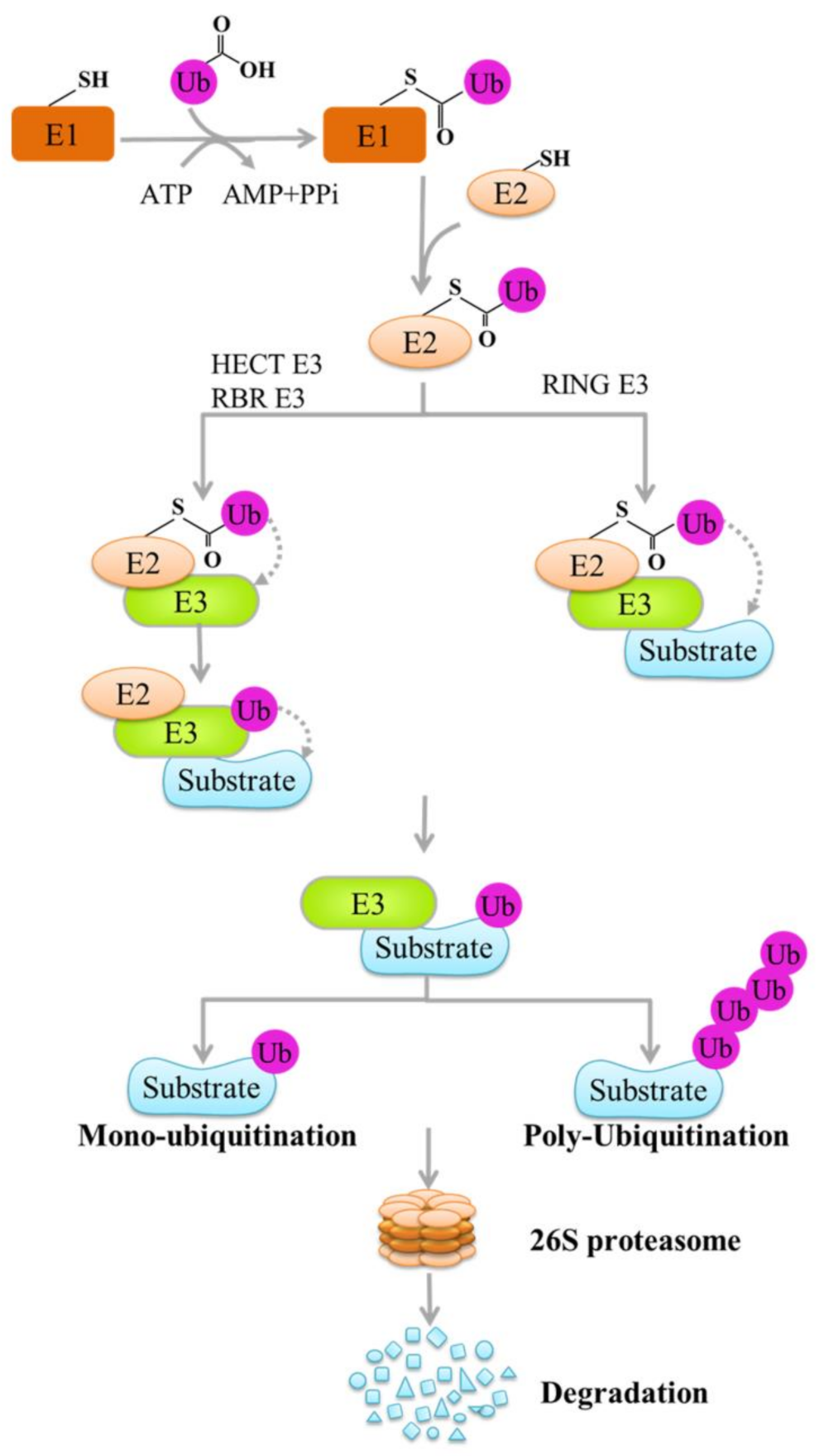
:
1. Introduction
2. E3 Ligases and Tumor Immune Checkpoints
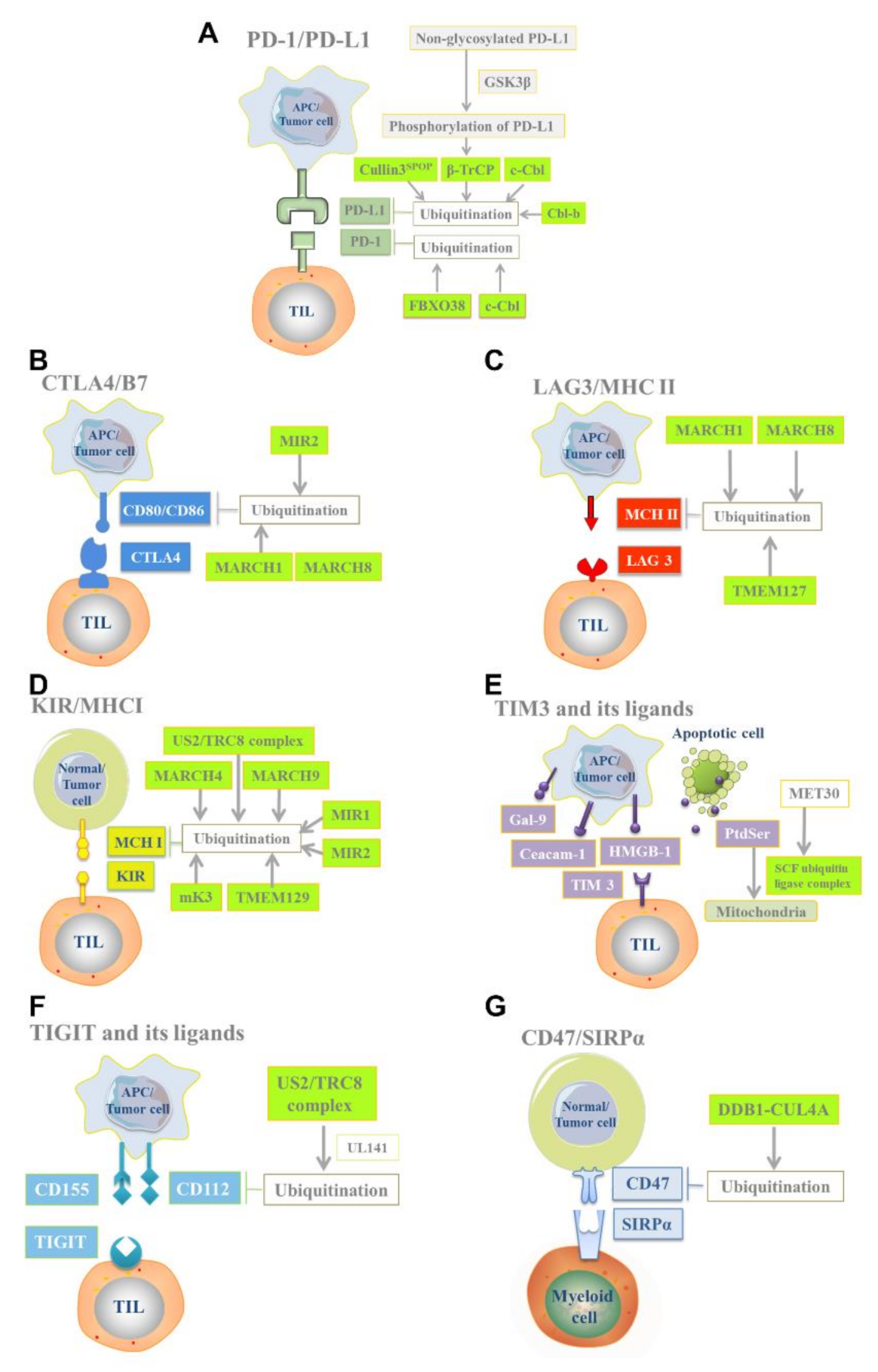
2.1. PD-1/Programmed Cell Death Protein Ligand 1 (PD-L1)
2.2. CTLA4/B7
2.3. Lymphocyte Activation Gene-3 (LAG-3)/Major Histocompatibility Complex-II (MHC-II)
2.4. Killer Cell Immunoglobulin-like Receptor (KIR)/MHC-I
2.5. T-Cell Immunoglobulin and Mucin Domain-3 (TIM-3)
2.6. T-Cell Ig and ITIM Domain (TIGIT) and Its Ligands CD155 and CD112
2.7. CD47/Signal Regulatory Protein α (SIRPα)
3. E3 Ligases and Immunomodulatory Pathways
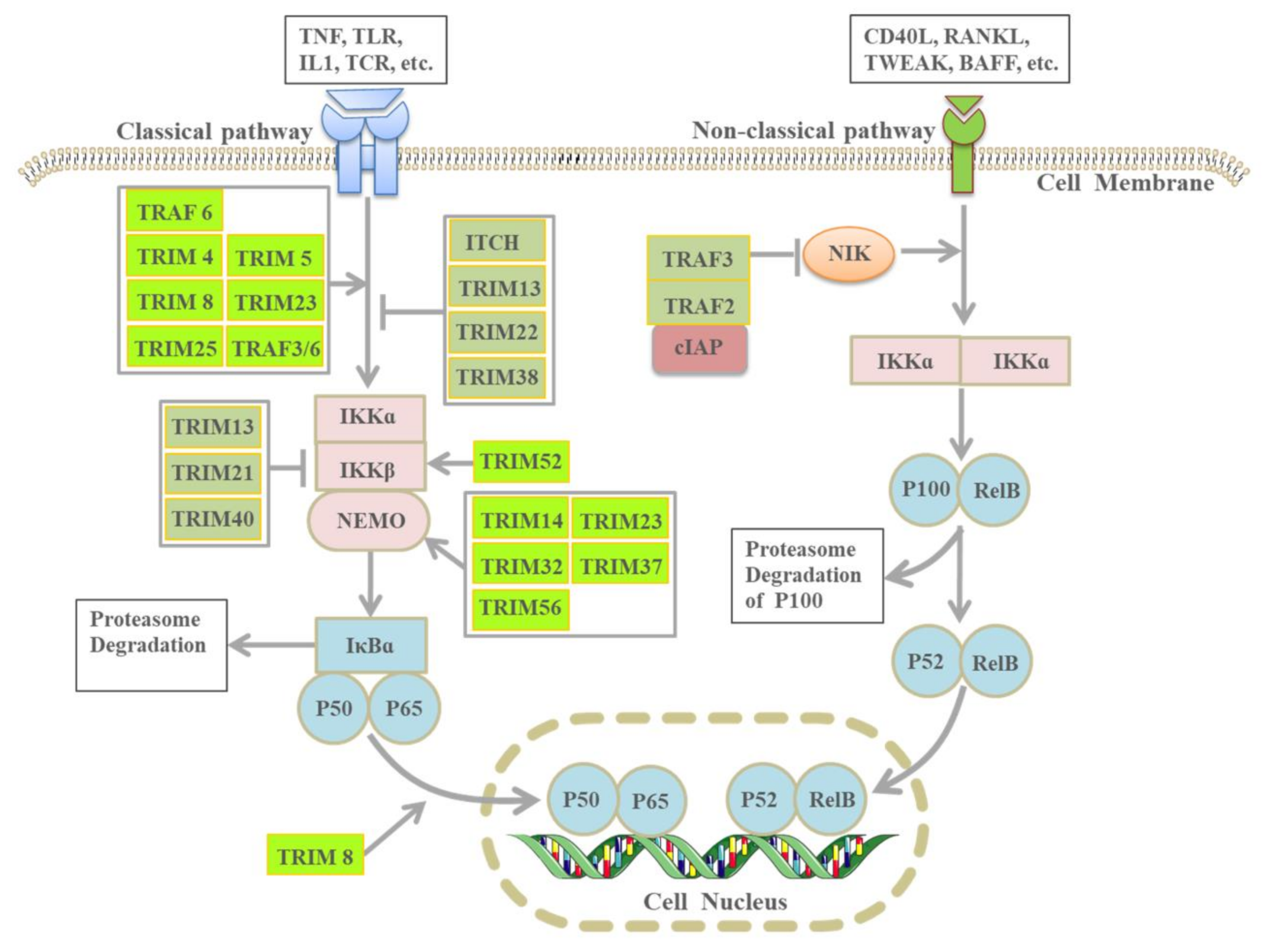
3.1. NF-κB Signaling Pathway
3.2. JAK-STAT Signaling Pathway
3.3. Stimulator of Interferon Gene (STING) Signaling Pathway
4. Therapeutic Targeting of E3 Ligases in Cancer Immunotherapy
4.1. E3 Ligase Inhibitors
4.1.1. RING-Type E3 Ligase Inhibitors
4.1.2. HECT-Type E3 Ligase Inhibitors
4.1.3. RBR-Type E3 Ligase Inhibitors
4.2. E3 Ligase Agonists
4.2.1. CRBN
4.2.2. β-TrCP
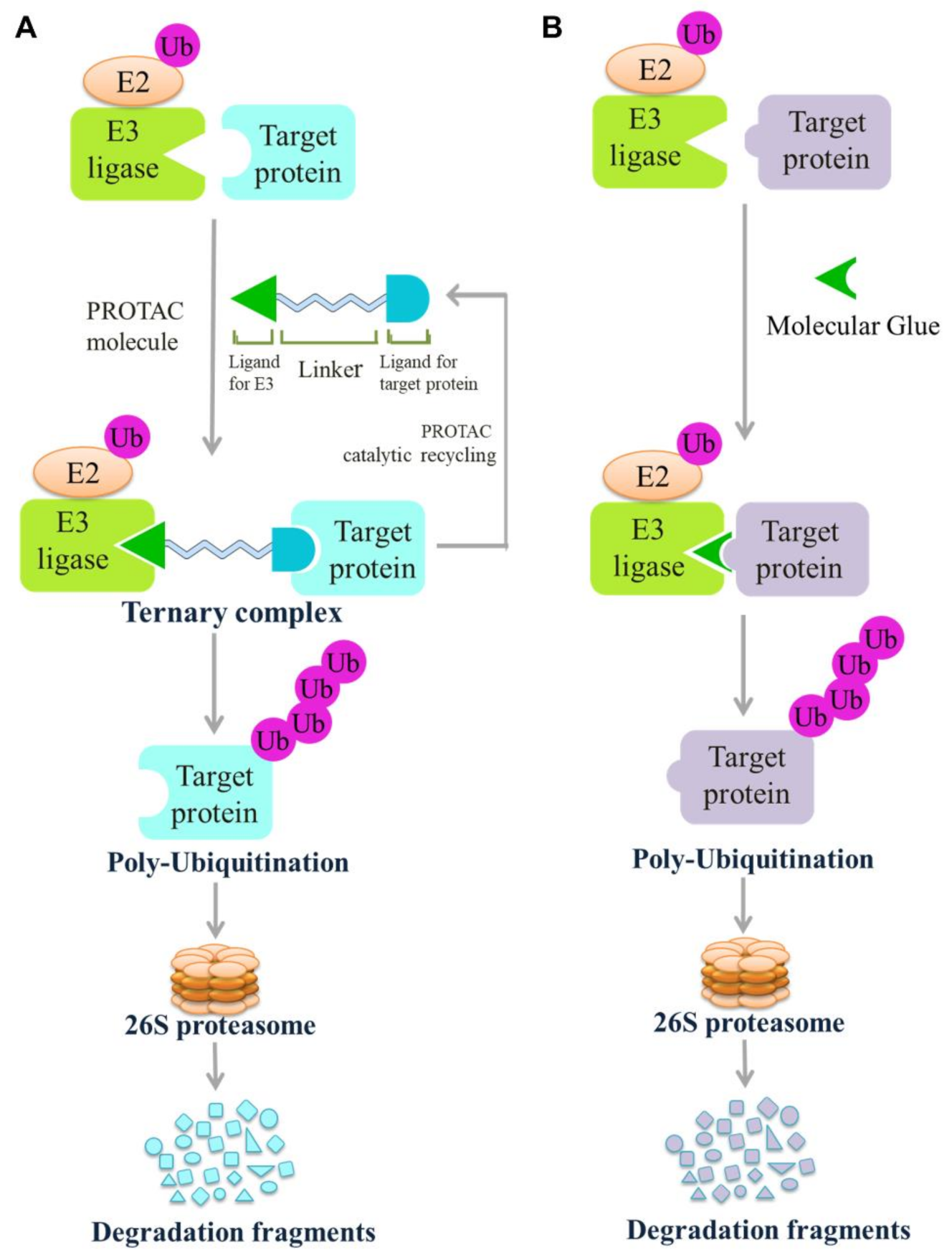
4.3. Other Drug Development Based on E3 Ligases: PROTACs and Molecular Glue
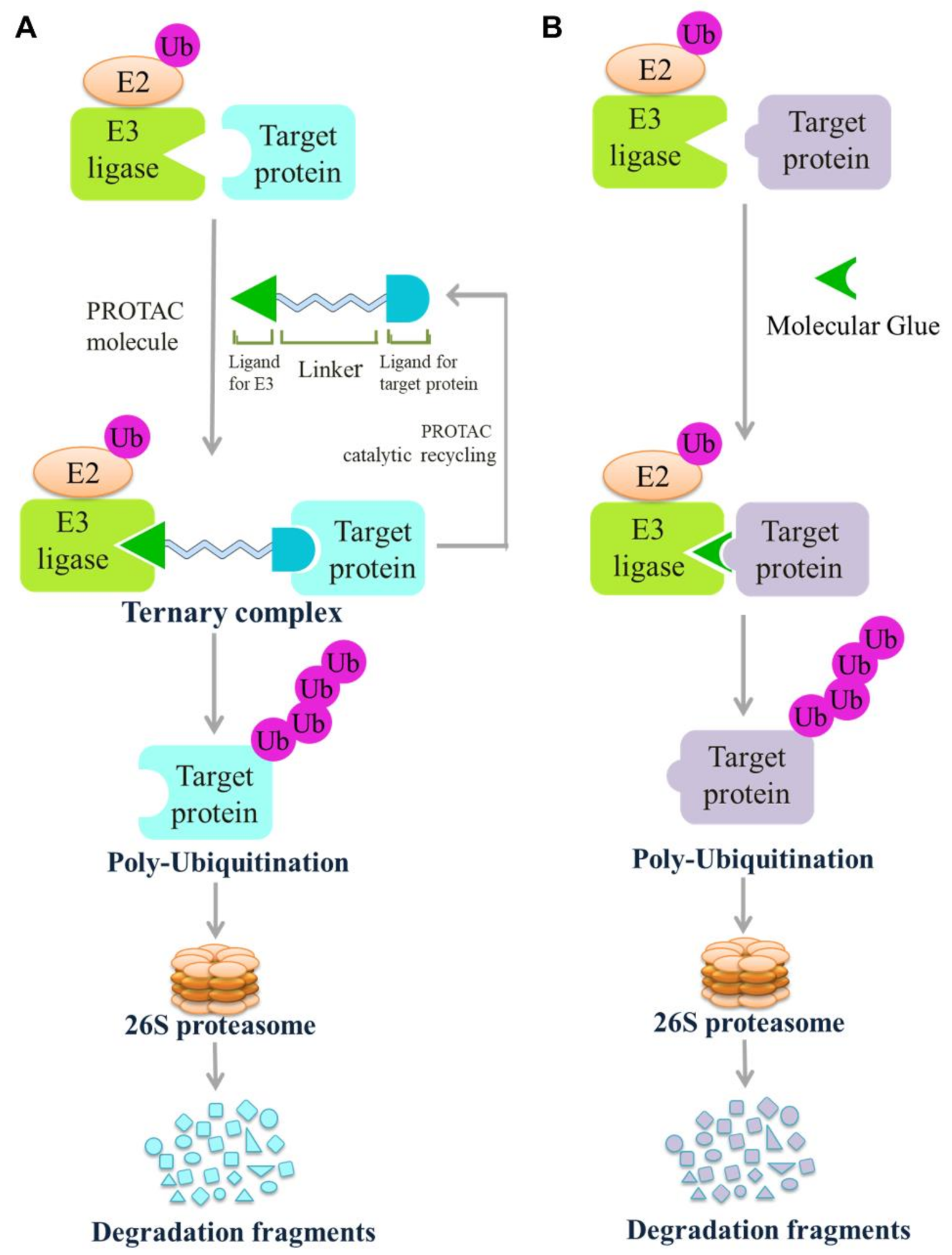
4.3.1. Proteolysis Targeting Chimeras (PROTACs)
4.3.2. Molecular Glue
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.A. Ubiquitination: Friend and foe in cancer. Int. J. Biochem. Cell Biol. 2018, 101, 80–93. [Google Scholar] [CrossRef]
- LaPlante, G.; Zhang, W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers 2021, 13, 3079. [Google Scholar] [CrossRef]
- Morrow, J.K.; Lin, H.-K.; Sun, S.-C.; Zhang, S. Targeting ubiquitination for cancer therapies. Futur. Med. Chem. 2015, 7, 2333–2350. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Li, S.; Shu, H.-B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Duan, C.; Zhang, C. E3 Ubiquitin Ligase in Anticancer Drugdsla Resistance: Recent Advances and Future Potential. Front. Pharmacol. 2021, 12, 645864. [Google Scholar] [CrossRef]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, K.; Oikawa, D.; Iio, K.; Obika, S.; Hori, Y.; Urashima, T.; Ayukawa, K.; Tokunaga, F. Small-molecule inhibitors of linear ubiquitin chain assembly complex (LUBAC), HOIPINs, suppress NF-κB signaling. Biochem. Biophys. Res. Commun. 2019, 509, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Baietti, M.F.; Sablina, A.A. Cracking the Monoubiquitin Code of Genetic Diseases. Int. J. Mol. Sci. 2020, 21, 3036. [Google Scholar] [CrossRef]
- Baur, R.; Rape, M. Getting Close: Insight into the Structure and Function of K11/K48-Branched Ubiquitin Chains. Structure 2020, 28, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Sun, S.-C. Targeting ubiquitin signaling for cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases-emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs228072. [Google Scholar] [CrossRef]
- Berndsen, C.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Uchida, C.; Kitagawa, M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr. Cancer Drug Targets 2016, 16, 157–174. [Google Scholar] [CrossRef]
- Cable, J.; Greenbaum, B.; Pe’Er, D.; Bollard, C.M.; Bruni, S.; Griffin, M.E.; Allison, J.P.; Wu, C.J.; Subudhi, S.K.; Mardis, E.R.; et al. Frontiers in cancer immunotherapy—A symposium report. Ann. N. Y. Acad. Sci. 2021, 1489, 30–47. [Google Scholar] [CrossRef]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Serman, T.M.; Gack, M.U. FBXO38 Drives PD-1 to Destruction. Trends Immunol. 2019, 40, 81–83. [Google Scholar] [CrossRef]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Ghahremanloo, A.; Soltani, A.; Modaresi, S.M.S.; Hashemy, S.I. Recent advances in the clinical development of immune checkpoint blockade therapy. Cell. Oncol. 2019, 42, 609–626. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y. Biomarkers for Immune Checkpoint Inhibitor-Mediated Tumor Response and Adverse Events. Front. Med. 2019, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Lan, J.; Yang, J. Mechanisms of Resistance to Checkpoint Blockade Therapy. Adv. Exp. Med. Biol. 2020, 1248, 83–117. [Google Scholar] [CrossRef] [PubMed]
- Lyle, C.; Richards, S.; Yasuda, K.; Napoleon, M.A.; Walker, J.; Arinze, N.; Belghasem, M.; Vellard, I.; Yin, W.; Ravid, J.D.; et al. c-Cbl targets PD-1 in immune cells for proteasomal degradation and modulates colorectal tumor growth. Sci. Rep. 2019, 9, 20257. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, L.; Che, X.; Li, C.; Xu, L.; Hou, K.; Fan, Y.; Wen, T.; Qu, X.; Liu, Y. E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. 2018, 592, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Xu, J. Regulation of Cancer Immune Checkpoint: Mono- and Poly-Ubiquitination: Tags for Fate. Adv. Exp. Med. Biol. 2020, 1248, 295–324. [Google Scholar] [CrossRef]
- Dermani, F.K.; Samadi, P.; Rahmani, G.; Kohlan, A.K.; Najafi, R. PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J. Cell. Physiol. 2019, 234, 1313–1325. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Anstadt, E.J.; Clark, R.B. Cbl-b Deficiency Mediates Resistance to Programed Death-Ligand 1/Programed Death-1 Regulation. Front. Immunol. 2017, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Peer, S.; Baier, G.; Gruber, T. Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget 2017, 8, 41841–41853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar] [CrossRef]
- Okada, R.; Kato, T.; Furusawa, A.; Inagaki, F.; Wakiyama, H.; Choyke, P.L.; Kobayashi, H. Local Depletion of Immune Checkpoint Ligand CTLA4 Expressing Cells in Tumor Beds Enhances Antitumor Host Immunity. Adv. Ther. 2021, 4, 2000269. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, Y.; Zheng, M.; Yuan, B.; Wang, Z.; Li, X.; Yin, J.; Ye, M.; Song, Y. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduct. Target. Ther. 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Gál, I.; Vermes, C.; Alegre, M.-L.; Chong, A.S.F.; Chen, L.; Shao, Q.; Adarichev, V.; Xu, X.; Koreny, T.; et al. Cutting edge: Cbl-b: One of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 2004, 173, 7135–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiting, C.C.; Su, L.L.; Lin, J.T.; Fathman, C.G. GRAIL: A unique mediator of CD4 T-lymphocyte unresponsiveness. FEBS J. 2011, 278, 47–58. [Google Scholar] [CrossRef]
- Stempin, C.C.; Marquez, D.R.; Ana, Y.; Cerban, F.M. GRAIL and Otubain-1 are Related to T Cell Hyporesponsiveness during Trypanosoma cruzi Infection. PLoS Negl. Trop. Dis. 2017, 11, e0005307. [Google Scholar] [CrossRef] [Green Version]
- Gibson, H.; Mishra, A.; Chan, D.V.; Hake, T.S.; Porcu, P.; Wong, H.K. Impaired Proteasome Function Activates GATA3 in T Cells and Upregulates CTLA-4: Relevance for Sézary Syndrome. J. Investig. Dermatol. 2013, 133, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Bakke, O.; Morth, J.P. Overview of the membrane-associated RING-CH (MARCH) E3 ligase family. New Biotechnol. 2017, 38, 7–15. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Investig. 2001, 107, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscoy, L.; Sanchez, D.J.; Ganem, D. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 2001, 155, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Baravalle, G.; Park, H.; McSweeney, M.; Ohmura-Hoshino, M.; Matsuki, Y.; Ishido, S.; Shin, J.-S. Ubiquitination of CD86 Is a Key Mechanism in Regulating Antigen Presentation by Dendritic Cells. J. Immunol. 2011, 187, 2966–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Li, X.; Zhang, J. Progress of immune checkpoint LAG-3 in immunotherapy. Oncol. Lett. 2020, 20, 207. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in Regulatory T Cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echo, K.J.; Roche, P.A. Regulation of MHC Class II-Peptide Complex Expression by Ubiquitination. Front. Immunol. 2013, 4, 369. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [Green Version]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC Class II Engagement by Its Ligand LAG-3 (CD223) Contributes to Melanoma Resistance to Apoptosis. J. Immunol. 2011, 186, 5173–5183. [Google Scholar] [CrossRef] [Green Version]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.M.; Mintern, J.D.; Villadangos, J. Control of MHC II antigen presentation by ubiquitination. Curr. Opin. Immunol. 2013, 25, 109–114. [Google Scholar] [CrossRef]
- Shin, J.-S.; Ebersold, M.; Pypaert, M.; Delamarre, L.; Hartley, A.; Mellman, I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 2006, 444, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Liu, H.; Healey, G.; Vuong, V.; Ishido, S.; Herold, M.; Villadangos, J.A.; Mintern, J.D. MARCH1-mediated ubiquitination of MHC II impacts the MHC I antigen presentation pathway. PLoS ONE 2018, 13, e0200540. [Google Scholar] [CrossRef] [Green Version]
- De Gassart, A.; Camosseto, V.; Thibodeau, J.; Ceppi, M.; Catalan, N.; Pierre, P.; Gatti, E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 3491–3496. [Google Scholar] [CrossRef] [Green Version]
- Ishido, S.; Kajikawa, M. MHC class II fine tuning by ubiquitination: Lesson from MARCHs. Immunogenetics 2019, 71, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Hoshino, M.; Matsuki, Y.; Aoki, M.; Goto, E.; Mito, M.; Uematsu, M.; Kakiuchi, T.; Hotta, H.; Ishido, S. Inhibition of MHC Class II Expression and Immune Responses by c-MIR. J. Immunol. 2006, 177, 341–354. [Google Scholar] [CrossRef]
- Liu, H.; Jain, R.; Guan, J.; Vuong, V.; Ishido, S.; La Gruta, N.; Gray, D.; Villadangos, J.A.; Mintern, J.D. Ubiquitin ligase MARCH 8 cooperates with CD83 to control surface MHC II expression in thymic epithelium and CD4 T cell selection. J. Exp. Med. 2016, 213, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Von Rohrscheidt, J.; Petrozziello, E.; Nedjic, J.; Federle, C.; Krzyzak, L.; Ploegh, H.L.; Ishido, S.; Steinkasserer, A.; Klein, L. Thymic CD4 T cell selection requires attenuation of March8-mediated MHCII turnover in cortical epithelial cells through CD83. J. Exp. Med. 2016, 213, 1685–1694. [Google Scholar] [CrossRef]
- Bayer-Santos, E.; Durkin, C.H.; Rigano, L.A.; Kupz, A.; Alix, E.; Černý, O.; Jennings, E.; Liu, M.; Ryan, A.S.; Lapaque, N.; et al. The Salmonella Effector SteD Mediates MARCH8-Dependent Ubiquitination of MHC II Molecules and Inhibits T Cell Activation. Cell Host Microbe 2016, 20, 584–595. [Google Scholar] [CrossRef]
- Gatti, E. Monitoring MHC Ubiquitination by MARCH Ubiquitin Ligases. Methods Mol. Biol. 2019, 1988, 259–270. [Google Scholar] [CrossRef]
- Van Niel, G.; Wubbolts, R.; Broeke, T.T.; Buschow, S.; Ossendorp, F.A.; Melief, C.J.; Raposo, G.; van Balkom, B.W.; Stoorvogel, W. Dendritic Cells Regulate Exposure of MHC Class II at Their Plasma Membrane by Oligoubiquitination. Immunity 2006, 25, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Wu, N.; Baravalle, G.; Cohn, B.; Ma, J.; Lo, B.; Mellman, I.; Ishido, S.; Anderson, M.; Shin, J.-S. MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J. Exp. Med. 2013, 210, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix, E.; Godlee, C.; Cerny, O.; Blundell, S.; Tocci, R.; Matthews, S.; Liu, M.; Pruneda, J.N.; Swatek, K.N.; Komander, D.; et al. The Tumour Suppressor TMEM127 Is a Nedd4-Family E3 Ligase Adaptor Required by Salmonella SteD to Ubiquitinate and Degrade MHC Class II Molecules. Cell Host Microbe 2020, 28, 54–68.e57. [Google Scholar] [CrossRef] [PubMed]
- Vilches, C.; Parham, P. KIR: Diverse, Rapidly Evolving Receptors of Innate and Adaptive Immunity. Annu. Rev. Immunol. 2002, 20, 217–251. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.S.; Purdy, A.K. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011, 132, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; Zhou, C. Killer immunoglobulin-like receptors/human leukocyte antigen class-I, a crucial immune pathway in cancer. Ann. Transl. Med. 2020, 8, 244. [Google Scholar] [CrossRef]
- Thielens, A.; Vivier, E.; Romagné, F. NK cell MHC class I specific receptors (KIR): From biology to clinical intervention. Curr. Opin. Immunol. 2012, 24, 239–245. [Google Scholar] [CrossRef]
- Duncan, L.M.; Piper, S.; Dodd, R.; Saville, M.K.; Sanderson, C.M.; Luzio, J.P.; Lehner, P.J. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J. 2006, 25, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Van den Boomen, D.; Lehner, P. Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Mol. Immunol. 2015, 68, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Kuchroo, V.K. Tim-3, Lag-3, and TIGIT. Curr. Top. Microbiol. Immunol. 2017, 410, 127–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, J. Functions of Immune Checkpoint Molecules beyond Immune Evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.-L.E.; Lee, H.-H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T Cell/Transmembrane, Ig, and Mucin-3 Allelic Variants Differentially Recognize Phosphatidylserine and Mediate Phagocytosis of Apoptotic Cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, M.M.; Choi, J.-Y.; Voelker, D.R. Phosphatidylserine Transport to the Mitochondria Is Regulated by Ubiquitination. J. Biol. Chem. 2002, 277, 51033–51042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.-F.; Joller, N.; Tan, D.J.; Teng, M.; Smyth, M.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molfetta, R.; Milito, N.D.; Zitti, B.; Lecce, M.; Fionda, C.; Cippitelli, M.; Santoni, A.; Paolini, R. The Ubiquitin-proteasome pathway regulates Nectin2/CD112 expression and impairs NK cell recognition and killing. Eur. J. Immunol. 2019, 49, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Q.; Xin, N.; Wang, W.; Zhao, C. CD 155, an onco-immunologic molecule in human tumors. Cancer Sci. 2017, 108, 1934–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, Y.; Fan, J.; Chen, W.; Luan, J.; Mei, X.; Wang, S.; Li, Y.; Ye, L.; Li, S.; et al. Blocking CD47 efficiently potentiated therapeutic effects of anti-angiogenic therapy in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 346. [Google Scholar] [CrossRef] [Green Version]
- Logtenberg, M.E.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, J.; Ju, D. Insights into CD47/SIRPα axis-targeting tumor immunotherapy. Antib. Ther. 2018, 1, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; Van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Liu, H.; Liang, Z.; Wang, F.; Zhou, C.; Zheng, X.; Zhang, Y.; Song, Y.; Hu, J.; He, X.; et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 2020, 10, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Sugimura-Nagata, A.; Koshino, A.; Inoue, S.; Matsuo-Nagano, A.; Komura, M.; Riku, M.; Ito, H.; Inoko, A.; Murakami, H.; Ebi, M.; et al. Expression and Prognostic Significance of CD47–SIRPA Macrophage Checkpoint Molecules in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 2690. [Google Scholar] [CrossRef] [PubMed]
- Piccione, E.C.; Juarez, S.; Tseng, S.; Liu, J.; Stafford, M.; Narayanan, C.; Wang, L.; Weiskopf, K.; Majeti, R. SIRPα-Antibody Fusion Proteins Selectively Bind and Eliminate Dual Antigen-Expressing Tumor Cells. Clin. Cancer Res. 2016, 22, 5109–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Saito, Y.; Kotani, T.; Matozaki, T. Blockade of CD47 or SIRPα: A new cancer immunotherapy. Expert Opin. Ther. Targets 2020, 24, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Song, X.; Wavelet, C.M.; Wan, Y. Cullin 4-DCAF Proteins in Tumorigenesis. Adv. Exp. Med. Biol. 2020, 1217, 241–259. [Google Scholar] [CrossRef]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, J.; Zhu, T.; Zhuo, W.; Yi, Y.; Hu, S.; Yin, J.; Zhang, W.; Zhou, H.; Liu, Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 2020, 19, 133. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Greten, F. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Evaristo, C.; Spranger, S.; Barnes, S.E.; Miller, M.L.; Molinero, L.; Locke, F.L.; Gajewski, T.F.; Alegre, M.-L. Cutting Edge: Engineering Active IKKβ in T Cells Drives Tumor Rejection. J. Immunol. 2016, 196, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I. TNFR1-activated NF-κB signal transduction: Regulation by the ubiquitin/proteasome system. Curr. Opin. Chem. Biol. 2014, 23, 71–77. [Google Scholar] [CrossRef]
- Elton, L.; Carpentier, I.; Verhelst, K.; Staal, J.; Beyaert, R. The multifaceted role of the E3 ubiquitin ligase HOIL-1: Beyond linear ubiquitination. Immunol. Rev. 2015, 266, 208–221. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, B.; Aleithan, F.; Abdul-Sater, Z.; Abdul-Sater, A.A. The Evolving Role of TRAFs in Mediating Inflammatory Responses. Front. Immunol. 2019, 10, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, W.; Zhang, S. TRIM proteins in lung cancer: Mechanisms, biomarkers and therapeutic targets. Life Sci. 2021, 268, 118985. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.J.; Snowden, J.; Zeidler, M.; Danson, S. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Coria, L.; Rosemblit, C.; Rivas, M.A.; Proietti, C.; Flaqué, M.C.D.; Beguelin, W.; Frahm, I.; Charreau, E.H.; Cassataro, J.; et al. Targeting Stat3 Induces Senescence in Tumor Cells and Elicits Prophylactic and Therapeutic Immune Responses against Breast Cancer Growth Mediated by NK Cells and CD4+ T Cells. J. Immunol. 2012, 189, 1162–1172. [Google Scholar] [CrossRef] [Green Version]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interf. Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Trifinopoulos, J.; Sexl, V.; Putz, E.M. JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front. Immunol. 2019, 10, 2590. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Evel-Kabler, K.; Strube, R.; Chen, S.-Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat. Biotechnol. 2004, 22, 1546–1553. [Google Scholar] [CrossRef]
- Rakesh, K.; Agrawal, D.K. Controlling cytokine signaling by constitutive inhibitors. Biochem. Pharmacol. 2005, 70, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Heusinkveld, M.; Van Der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Barros, M.H.M.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage Polarisation: An Immunohistochemical Approach for Identifying M1 and M2 Macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [Green Version]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Qi, J.; Zhao, X.; Gao, C. Smurf1 Protein Negatively Regulates Interferon-γ Signaling through Promoting STAT1 Protein Ubiquitination and Degradation. J. Biol. Chem. 2012, 287, 17006–17015. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.W.; Kornbluth, J. E3 ubiquitin ligase NKLAM ubiquitinates STAT1 and positively regulates STAT1-mediated transcriptional activity. Cell. Signal. 2016, 28, 1833–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. SLIM Is a Nuclear Ubiquitin E3 Ligase that Negatively Regulates STAT Signaling. Immunity 2005, 22, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Jiang, M.; Wang, W.; Liu, W.; Song, X.; Ma, Z.; Zhang, S.; Liu, L.; Liu, Y.; Cao, X. Nuclear RNF2 inhibits interferon function by promoting K33-linked STAT1 disassociation from DNA. Nat. Immunol. 2018, 19, 41–52. [Google Scholar] [CrossRef]
- Guo, X.; Ma, P.; Li, Y.; Yang, Y.; Wang, C.; Xu, T.; Wang, H.; Li, C.; Mao, B.; Qi, X. RNF220 mediates K63-linked polyubiquitination of STAT1 and promotes host defense. Cell Death Differ. 2021, 28, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Zhou, M.-T.; Hu, M.-M.; Hu, Y.-H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.-B. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLOS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Li, Q.; Lin, L.; Tong, Y.; Liu, Y.; Mou, J.; Wang, X.; Wang, X.; Gong, Y.; Zhao, Y.; Liu, Y.; et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov. 2018, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lian, Q.; Yang, B.; Yan, S.; Zhou, H.; He, L.; Lin, G.; Lian, Z.; Jiang, Z.; Sun, B. TRIM30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLoS Pathog. 2015, 11, e1005012. [Google Scholar] [CrossRef]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016, 22, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I Dose-Escalation Study of LCL161, an Oral Inhibitor of Apoptosis Proteins Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef]
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.-Z.; et al. A novel SMAC mimetic APG-1387 exhibits dual antitumor effect on HBV-positive hepatocellular carcinoma with high expression of cIAP2 by inducing apoptosis and enhancing innate anti-tumor immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Flygare, J.; Fairbrother, W.J. Small-molecule pan-IAP antagonists: A patent review. Expert Opin. Ther. Pat. 2010, 20, 251–267. [Google Scholar] [CrossRef]
- Michie, J.; Kearney, C.J.; Hawkins, E.D.; Silke, J.; Oliaro, J. The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells 2020, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Flygare, J.A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I.T.; Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S.G.; et al. Discovery of a Potent Small-Molecule Antagonist of Inhibitor of Apoptosis (IAP) Proteins and Clinical Candidate for the Treatment of Cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2–p53 Protein–Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, I.; Zhang, S.; Navaraj, A.; Zhou, L.; Dizon, D.; Safran, H.; El-Deiry, W.S. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef]
- De Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Macé, S.; Tuffal, G.; et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Song, J.; Wu, H.; Yang, M.; Lu, L.; Weng, X.; Liu, L.; Nie, G. MDM2 inhibitor RG7388 potently inhibits tumors by activating p53 pathway in nasopharyngeal carcinoma. Cancer Biol. Ther. 2019, 20, 1328–1336. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Maki, C.G. Pharmacologic Activation of p53 by Small-Molecule MDM2 Antagonists. Curr. Pharm. Des. 2011, 17, 560–568. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 Ligase Inhibitors Using a Cell-Based Ubiquitination Assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Cui, D.; Xiong, X.; Zhao, Y. Targeting Cullin-RING Ubiquitin Ligases and the Applications in PROTACs. Adv. Exp. Med. Biol. 2020, 1217, 317–347. [Google Scholar] [CrossRef]
- Frost, J.; Galdeano, C.; Soares, P.; Gadd, M.; Grzes, K.M.; Ellis, L.; Epemolu, O.; Shimamura, S.; Bantscheff, M.; Grandi, P.; et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun. 2016, 7, 13312. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the p27 E3 ligase SCFSkp2 results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Grigoryan, A.V.; Li, Y.; Hao, B.; Pagano, M.; Cardozo, T.J. Specific Small Molecule Inhibitors of Skp2-Mediated p27 Degradation. Chem. Biol. 2012, 19, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-H.; Morrow, J.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Malek, E.; Abdel-Malek, M.A.Y.; Jagannathan, S.; Vad, N.; Karns, R.; Jegga, A.G.; Broyl, A.; Van Duin, M.; Sonneveld, P.; Cottini, F.; et al. Pharmacogenomics and chemical library screens reveal a novel SCFSKP2 inhibitor that overcomes Bortezomib resistance in multiple myeloma. Leukemia 2017, 31, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Chen, Y.; Zhou, Y.; Zhang, S.; Hong, M.; Yu, X.; Wei, S.; Fan, X.; Li, S.; Wang, Q.; et al. Phytochemical library screening reveals betulinic acid as a novel Skp2-SCF E3 ligase inhibitor in non–small cell lung cancer. Cancer Sci. 2021, 112, 3218–3232. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, X.; Li, M.; Gong, G.; Liu, W.; Li, T.; Zuo, H.; Li, W.; Gao, F.; Liu, H. Cdh1-mediated Skp2 degradation by dioscin reprogrammes aerobic glycolysis and inhibits colorectal cancer cells growth. EBioMedicine 2020, 51, 102570. [Google Scholar] [CrossRef]
- Jiang, W.; Lin, M.; Wang, Z. Dioscin: A new potential inhibitor of Skp2 for cancer therapy. EBioMedicine 2020, 51, 102593. [Google Scholar] [CrossRef] [Green Version]
- Blees, J.S.; Bokesch, H.R.; Rübsamen, D.; Schulz, K.; Milke, L.; Bajer, M.M.; Gustafson, K.R.; Henrich, C.J.; McMahon, J.B.; Colburn, N.H.; et al. Erioflorin Stabilizes the Tumor Suppressor Pdcd4 by Inhibiting Its Interaction with the E3-ligase β-TrCP1. PLoS ONE 2012, 7, e46567. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallampalli, R.K.; Coon, T.A.; Glasser, J.R.; Wang, C.; Dunn, S.R.; Weathington, N.M.; Zhao, J.; Zou, C.; Zhao, Y.; Chen, B.B. Targeting F Box Protein Fbxo3 To Control Cytokine-Driven Inflammation. J. Immunol. 2013, 191, 5247–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; King, R.W. An APC/C inhibitor stabilizes cyclin B1 by prematurely terminating ubiquitination. Nat. Chem. Biol. 2012, 8, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharm. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Thyagarajan-Sahu, A.; Krchnak, V.; Jedinak, A.; Sandusky, G.E.; Sliva, D. NAHA, a Novel Hydroxamic Acid-Derivative, Inhibits Growth and Angiogenesis of Breast Cancer In Vitro and In Vivo. PLoS ONE 2012, 7, e34283. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Jedinak, A.; Sliva, D. Ganodermanontriol (GDNT) exerts its effect on growth and invasiveness of breast cancer cells through the down-regulation of CDC20 and uPA. Biochem. Biophys. Res. Commun. 2011, 415, 325–329. [Google Scholar] [CrossRef]
- Brenke, J.K.; Popowicz, G.M.; Schorpp, K.; Rothenaigner, I.; Roesner, M.; Meininger, I.; Kalinski, C.; Ringelstetter, L.; R’Kyek, O.; Jürjens, G.; et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J. Biol. Chem. 2018, 293, 13191–13203. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Rotblat, B.; Ansell, K.; Amelio, I.; Caraglia, M.; Misso, G.; Bernassola, F.; Cavasotto, C.; Knight, R.; Ciechanover, A.; et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014, 5, e1203. [Google Scholar] [CrossRef] [Green Version]
- Aronchik, I.; Kundu, A.; Quirit, J.G.; Firestone, G.L. The Antiproliferative Response of Indole-3-Carbinol in Human Melanoma Cells Is Triggered by an Interaction with NEDD4-1 and Disruption of Wild-Type PTEN Degradation. Mol. Cancer Res. 2014, 12, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirit, J.G.; Lavrenov, S.N.; Poindexter, K.; Xu, J.; Kyauk, C.; Durkin, K.A.; Aronchik, I.; Tomasiak, T.; Solomatin, Y.A.; Preobrazhenskaya, M.N.; et al. Indole-3-carbinol (I3C) analogues are potent small molecule inhibitors of NEDD4-1 ubiquitin ligase activity that disrupt proliferation of human melanoma cells. Biochem. Pharmacol. 2017, 127, 13–27. [Google Scholar] [CrossRef]
- Watt, J.E.; Hughes, G.R.; Walpole, S.; Monaco, S.; Stephenson, G.R.; Page, P.C.B.; Hemmings, A.M.; Angulo, J.; Chantry, A. Discovery of Small Molecule WWP2 Ubiquitin Ligase Inhibitors. Chem. A Eur. J. 2018, 24, 17677–17680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Li, X.; Xing, L.; Ding, Y.; Shi, P.; Zhang, Y.; Guo, S.; Shu, X.; Shan, B. Bortezomib prevents oncogenesis and bone metastasis of prostate cancer by inhibiting WWP1, Smurf1 and Smurf2. Int. J. Oncol. 2014, 45, 1469–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, M.; Zeng, T.; Liu, M.; Han, S.; Lin, H.; Lin, Q.; Li, L.; Jiang, T.; Li, G.; Lin, H.; et al. A cell-based high-throughput screening method based on a ubiquitin-reference technique for identifying modulators of E3 ligases. J. Biol. Chem. 2019, 294, 2880–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cao, R.; Lian, C.; Cao, T.; Shi, Y.; Ma, J.; Wang, P.; Xia, J. Nitidine chloride suppresses NEDD4 expression in lung cancer cells. Aging 2020, 13, 782–793. [Google Scholar] [CrossRef]
- Mund, T.; Lewis, M.J.; Maslen, S.; Pelham, H.R. Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2014, 111, 16736–16741. [Google Scholar] [CrossRef] [Green Version]
- Ricci-López, J.; Vidal-Limon, A.; Zunñiga, M.; Jimenez, V.; Alderete, J.B.; Brizuela, C.A.; Aguila, S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PLoS ONE 2019, 14, e0213028. [Google Scholar] [CrossRef]
- Chen, L.; Ruan, Y.; Wang, X.; Min, L.; Shen, Z.; Sun, Y.; Qin, X. BAY 11-7082, a nuclear factor-κB inhibitor, induces apoptosis and S phase arrest in gastric cancer cells. J. Gastroenterol. 2013, 49, 864–874. [Google Scholar] [CrossRef]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin Suppresses NF-κB Activation by Selectively Inhibiting Linear Ubiquitin Chain Assembly Complex (LUBAC). ACS Chem. Biol. 2015, 10, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Alonso, F.; Whiting, A.L.; Kim, Y.J.; Bernal, F. Biophysical and biological evaluation of optimized stapled peptide inhibitors of the linear ubiquitin chain assembly complex (LUBAC). Bioorg. Med. Chem. 2018, 26, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as Attractive Anti-cancer Targets. Curr. Pharm. Des. 2013, 19, 3215–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. Engl. 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. BioMed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, F.; Milani, M.; Mastrangelo, E.; Lecis, D. Targeting the BIR Domains of Inhibitor of Apoptosis (IAP) Proteins in Cancer Treatment. Comput. Struct. Biotechnol. J. 2019, 17, 142–150. [Google Scholar] [CrossRef]
- Fulda, S. Smac mimetics as IAP antagonists. Semin. Cell Dev. Biol. 2015, 39, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; McCall, N.S.; Wiedemann, N.; Vuagniaux, G.; Yuan, Z.-Y.; Lu, B. SMAC Mimetic Debio 1143 and Ablative Radiation Therapy Synergize to Enhance Antitumor Immunity against Lung Cancer. Clin. Cancer Res. 2019, 25, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Landré, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-Ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Aminu, B.; Roscow, O.; Zhang, W. Targeting the Ubiquitin Signaling Cascade in Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 791. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. The VHL Tumor Suppressor: Master Regulator of HIF. Curr. Pharm. Des. 2009, 15, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert Opin. Ther. Pat. 2018, 28, 919–937. [Google Scholar] [CrossRef]
- Tao, X.; Yin, L.; Xu, L.; Peng, J. Dioscin: A diverse acting natural compound with therapeutic potential in metabolic diseases, cancer, inflammation and infections. Pharmacol. Res. 2018, 137, 259–269. [Google Scholar] [CrossRef]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Li, Y.; Yu, D.; Thomas-Tikhonenko, A.; Spiegelman, V.S.; Fuchs, S.Y. Targeting -Transducin Repeat-Containing Protein E3 Ubiquitin Ligase Augments the Effects of Antitumor Drugs on Breast Cancer Cells. Cancer Res. 2005, 65, 1904–1908. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Cui, D.; Xiong, X.; Zhao, Y. The characteristics and roles of β-TrCP1/2 in carcinogenesis. FEBS J. 2021, 288, 3351–3374. [Google Scholar] [CrossRef]
- Ye, X.; Wang, L.; Shang, B.; Wang, Z.; Wei, W. NEDD4: A Promising Target for Cancer Therapy. Curr. Cancer Drug Targets 2014, 14, 549–556. [Google Scholar] [CrossRef]
- Yang, B.; Gay, D.L.; MacLeod, M.K.L.; Cao, X.; Hala, T.; Sweezer, E.M.; Kappler, J.; Marrack, P.; Oliver, P.M. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat. Immunol. 2008, 9, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Aki, D.; Li, H.; Zhang, W.; Zheng, M.; Elly, C.; Lee, J.H.; Zou, W.; Liu, Y.-C. The E3 ligases Itch and WWP2 cooperate to limit TH2 differentiation by enhancing signaling through the TCR. Nat. Immunol. 2018, 19, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liao, B.; Wang, S.; Yan, B.; Jin, Y.; Shu, H.-B.; Wang, Y.-Y. E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 5115–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venuprasad, K.; Zeng, M.; Baughan, S.; Massoumi, R. Multifaceted role of the ubiquitin ligase Itch in immune regulation. Immunol. Cell Biol. 2015, 93, 452–460. [Google Scholar] [CrossRef]
- Aki, D.; Zhang, W.; Liu, Y.-C. The E3 ligase Itch in immune regulation and beyond. Immunol. Rev. 2015, 266, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wyatt, C.J.; Han, T.; Smalley, K.S.; Wan, L. ITCH as a potential therapeutic target in human cancers. Semin. Cancer Biol. 2020, 67, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Eto, D.; Elly, C.; Peng, G.; Crotty, S.; Liu, Y.-C. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat. Immunol. 2014, 15, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.; Jin, Y. Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell Res. 2010, 20, 332–344. [Google Scholar] [CrossRef]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jiang, X.; Luo, Z. WWP2: A Multifunctional Ubiquitin Ligase Gene. Pathol. Oncol. Res. 2014, 20, 799–803. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.; Luo, W.; Luo, Z.; Shi, S. WWP2 Is One Promising Novel Oncogene. Pathol. Oncol. Res. 2019, 25, 443–446. [Google Scholar] [CrossRef]
- Johansson, H.; Tsai, Y.-C.I.; Fantom, K.; Chung, C.-W.; Kümper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.-L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, V.; Johnson, C.; Barlow, V.; Hastie, J.; Knebel, A.; Trost, M. The MALDI-TOF E2/E3 Ligase Assay as Universal Tool for Drug Discovery in the Ubiquitin Pathway. Cell Chem. Biol. 2018, 25, 1117–1127.e1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.I.; Johansson, H.; Dixon, D.; Martin, S.; Chung, C.-W.; Clarkson, J.; House, D.; Rittinger, K. Single-Domain Antibodies as Crystallization Chaperones to Enable Structure-Based Inhibitor Development for RBR E3 Ubiquitin Ligases. Cell Chem. Biol. 2020, 27, 83–93.e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors I karos and A iolos via modulation of the E 3 ubiquitin ligase complex CRL 4(CRBN). Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Handa, H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int. J. Hematol. 2016, 104, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Handa, H. Molecular mechanisms of thalidomide and its derivatives. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2020, 96, 189–203. [Google Scholar] [CrossRef]
- Grant, S. Targeting cereblon in AML. Blood 2021, 137, 584–586. [Google Scholar] [CrossRef]
- Surka, C.; Jin, L.; Mbong, N.; Lu, C.-C.; Jang, I.S.; Rychak, E.; Mendy, D.; Clayton, T.; Tindall, E.A.; Hsu, C.; et al. CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells. Blood 2021, 137, 661–677. [Google Scholar] [CrossRef]
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; Von Soly, S.K.; Kayser, F.; et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019, 10, 140. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.H.G.; Spoel, S.H. The ubiquitin–proteasome system as a transcriptional regulator of plant immunity. J. Exp. Bot. 2018, 69, 4529–4537. [Google Scholar] [CrossRef] [Green Version]
- Furniss, J.; Spoel, S.H. Cullin-RING ubiquitin ligases in salicylic acid-mediated plant immune signaling. Front. Plant Sci. 2015, 6, 154. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Lee, B.-H. Chemically Induced Cellular Proteolysis: An Emerging Therapeutic Strategy for Undruggable Targets. Mol. Cells 2018, 41, 933–942. [Google Scholar] [CrossRef]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Crews, C.M. Waste disposal—An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Pagano, M. Ubiquitin ligases in cancer: Functions and clinical potentials. Cell Chem. Biol. 2021, 28, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Huang, W.; Zheng, X.; Cheng, L.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Belkina, A.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Gadd, M.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Tanaka, M.; Roberts, J.; Seo, H.-S.; Souza, A.; Paulk, J.; Scott, T.; De Angelo, S.L.; Dhe-Paganon, S.; Bradner, J. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaide, S.; Riching, K.M.; Makukhin, N.; Vetma, V.; Whitworth, C.; Hughes, S.J.; Trainor, N.; Mahan, S.D.; Murphy, N.; Cowan, A.D.; et al. Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol. 2021, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Den Besten, W.; Lipford, J.R. Prospecting for molecular glues. Nat. Chem. Biol. 2020, 16, 1157–1158. [Google Scholar] [CrossRef]
- Schreiber, S.L. The Rise of Molecular Glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef]
- Isobe, Y.; Okumura, M.; McGregor, L.M.; Brittain, S.M.; Jones, M.D.; Liang, X.; White, R.; Forrester, W.; McKenna, J.M.; Tallarico, J.A.; et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol. 2020, 16, 1189–1198. [Google Scholar] [CrossRef]
- Yoon, K.W.; Byun, S.; Kwon, E.; Hwang, S.-Y.; Chu, K.; Hiraki, M.; Jo, S.-H.; Weins, A.; Hakroush, S.; Cebulla, A.; et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015, 349, 1261669. [Google Scholar] [CrossRef] [Green Version]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Chen, P.; Cao, L.; Li, Y.; Zeng, Z.; Cui, Y.; Wu, Q.; Li, J.; Wang, J.-H.; Dong, M.-Q.; et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 2020, 9, 59994. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.-C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Petzold, G.; Fischer, E.S.; Thomä, N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.-C.; Miller, K.; Fang, W.; Wang, N.-Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef]
- Hansen, J.D.; Correa, M.; Alexander, M.; Nagy, M.; Huang, D.; Sapienza, J.; Lu, G.; LeBrun, L.A.; Cathers, B.E.; Zhang, W.; et al. CC-90009: A Cereblon E3 Ligase Modulating Drug That Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 2021, 64, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.D.; Correa, M.; Nagy, M.A.; Alexander, M.; Plantevin, V.; Grant, V.; Whitefield, B.; Huang, D.; Kercher, T.; Harris, R.; et al. Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 2020, 63, 6648–6676. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Zhang, W.; Man, H.-W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; Lebrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. [Google Scholar] [CrossRef]
- Hagner, P.R.; Man, H.-W.; Fontanillo, C.; Wang, M.; Couto, S.S.; Breider, M.; Bjorklund, C.C.; Havens, C.G.; Lu, G.; Rychak, E.; et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 2015, 126, 779–789. [Google Scholar] [CrossRef]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, D. Fantastic Molecular Glues and Where to Find them. Chim. Int. J. Chem. 2021, 75, 439–441. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Agent | Mechanism | Tumor Types | Phase | Reference |
|---|---|---|---|---|---|
| RING-type E3 ligase inhibitor | |||||
| IAPs antagonists | LCL161 | Smac mimetic, induces degradation of cIAP-1 | Advanced solid tumors, hematologic neoplasms | 1, 2 | [3,156,157] |
| APG-1387 | Smac mimetic, induces proteasomal degradation of IAPs | Advanced solid tumors, hematologic neoplasms | 1, 2 | [158] | |
| Debio 1143 (AT-406) | Smac mimetic, inhibiting XIAP, cIAP-1 and cIAP-2 to promote apoptosis | Advanced solid tumors, lymphomas | 1, 2 | [159] | |
| Birinapant (TL32711) | Smac mimetic | Advanced solid tumors, hematologic neoplasms | 1, 2 | [160] | |
| AEG40826/ HGS1029 | Smac mimetic | Advanced solid tumors | 1 | [159,160] | |
| Compound 1 (GDC-0152) | Smac mimetic, binds to the BIR3 domains of cIAP1, cIAP2, and XIAP | Solid tumors | 1 | [161] | |
| Compound 13 (AEG40730) | Smac mimetic, binds to the BIR3 domains of cIAP1, cIAP2, and XIAP | N/A | Preclinical | [159] | |
| MDM2 antagonists | AMG 232 | Binds to MDM2 and inhibits the MDM2–p53 interaction | Advanced solid tumors, hematologic neoplasms | 1 | [3,162,163,164,165] |
| APG-115 | Targeting MDM2-p53 pathway | Advanced solid tumors, hematologic neoplasms | 1, 2 | [164,166] | |
| RG7112 | Binding to the p53 pocket on MDM2, effectively inhibits the MDM2-p53 interaction | Advanced solid tumors, hematologic neoplasms | 1 | [3,164] | |
| SAR405838 (MI-77301) | Binds selectively to HDM2, an oral spirooxindole derivative antagonist of HDM2 | Neoplasm malignant | 1 | [167] | |
| Idasanutlin (RG7388) | Blocking the MDM2–p53 interaction to reactivate the p53 pathway | Advanced solid tumors, hematologic neoplasms | 1, 2 | [168] | |
| Nutlin-3a | Inhibits the MDM2-p53 interaction, leading to p53 stabilization and activation of the p53 pathway | N/A | Preclinical | [169,170] | |
| HLI98 | Inhibits HDM2’s E3 activity | N/A | Preclinical | [171] | |
| MEL23, MEL24 | Inhibits the E3 ligase activity of the Mdm2-MdmX complex. | N/A | Preclinical | [172] | |
| pVHL antagonists | Compound 15, Compound 7, VH298 | The targeting of VHL disrupts the interaction of VHL with HIF-α | N/A | Preclinical | [173,174] |
| SKP2 antagonists | Compound A | Blocks the assembly of Skp2 into the SCF complex. | N/A | Preclinical | [175] |
| C1, C2, C16, C20 | Inhibits Skp2-Cks1-p27 interface and thereby inhibit p27 ubiquitination. | N/A | Preclinical | [176] | |
| Compound 25 | Prevents the formation of the Skp2-Skp1 complex and inhibits the activity of SCF-Skp2. | N/A | Preclinical | [177] | |
| DT204 | Reduces the binding of Skp2 to Cullin-1 and Commd1, a Cullin-1-binding protein, therefore decreasing SCFSkp2 ubiquitin ligase activity | N/A | Preclinical | [178] | |
| Betulinic acid (BA) | Binding to Skp2 decreases its stability by disrupting Skp1-Skp2 interactions, thereby inhibiting the Skp2-SCF E3 ligase and promoting the accumulation of its substrates | N/A | Preclinical | [179] | |
| Dioscin | A new Skp2 inhibitor | N/A | Preclinical | [180,181] | |
| Curcumin, Quercetin, Lycopene, Silibinin, Epigallocatechin-3-gallate, Vitamin D3 | Natural agents that inhibit the expression of Skp2 in human cancers | Variety tumors | 1,2,3,4 | [7] | |
| β-TrCP antagonists | Erioflorin | Inhibits the interaction of Pdcd4/β-TrCP1 | N/A | Preclinical | [182] |
| GS143 | Inhibits β-TrCP1 ubiquitination of IkB, suppresses NF-kB signaling | N/A | Preclinical | [3] | |
| UBP-036 | Competitive inhibition of substrate binding to β-TRCP | N/A | Preclinical | [183] | |
| Fbxo3 antagonist | BC-1215 | Disrupts the interaction of Fbxo3 with Fbxl2 | N/A | Preclinical | [184] |
| Met30 (yeast) antagonist | SMER3 | Inhibits SCF-Met30 effectively and selectively | N/A | Preclinical | [3] |
| Cdc20 antagonists | Tosyl-l-arginine methyl ester | Blocks the APC/C-Cdc20 interaction | N/A | Preclinical | [185,186] |
| Pro-TAME | Disrupted the APC-Cdc20/Cdh1 interaction to reduce APC activation | N/A | Preclinical | [187] | |
| Apcin | Binds to Cdc20 and inhibits APC/C-dependent ubiquitylation | N/A | Preclinical | [185,187] | |
| Withaferin A | Suppresses Cdc20 activity | N/A | Preclinical | [187] | |
| NAHA | Inhibits the expression of Cdc20 | N/A | Preclinical | [187,188] | |
| Ganodermanontriol (GDNT) | Inhibits cell proliferation via targeting Cdc20 | N/A | Preclinical | [187,189] | |
| TRAF6 antagonist | C25-140 | Reduces TRAF6 E3 ligase activity by interfering with the TRAF6–Ubc13 interaction | N/A | Preclinical | [190] |
| HECT-type E3 ligase inhibitor | |||||
| Itch antagonist | Clomipramine | Blocks p73 ubiquitylation by binding to ITCH and inhibiting its charging with Ub | N/A | Preclinical | [191] |
| NEDD4-1 antagonist | Indole-3-carbinol (I3C) analogues | The potent small molecule inhibitors of NEDD4-1 ubiquitin ligase activity | Adult solid tumor | 1 | [192,193] |
| WWP2 antagonist | Compound 20 | Binds to the WWP2 HECT domain | N/A | Preclinical | [194] |
| SMURF1 antagonists | Bortezomib | Downregulated the protein level of SMURF1 by inhibiting SMURF1 mRNA levels | Neoplasm Malignant | 1, 2, 3, 4 | [195] |
| HS-152 | Blocked SMURF1-mediated RHOB ubiquitination | N/A | Preclinical | [196] | |
| NEDD4 antagonist | Nitidine chloride | A promising inhibitor of NEDD4 | N/A | Preclinical | [197] |
| Non-specific HECT antagonist | Heclin | Induces conformational change in HECT domain to inhibit activity | N/A | Preclinical | [198] |
| HUWE1 antagonists | BI8622, BI8626 | Inhibits HUWE1 to stabilize assembly of Myc-repressive MIZ1 complex on Myc-activated target genes | N/A | Preclinical | [3] |
| E6AP antagonists | Compound 12 | Inhibits E6AP–p53 interaction | N/A | Preclinical | [3] |
| Lutolein, CAF024 | Binds to viral E6 protein and prevents its association with E6AP | N/A | Preclinical | [3] | |
| Lig1, Lig2, Lig3 | Inhibits E6-E6AP interaction | N/A | Preclinical | [199] | |
| N-acetyl phenylalanine | Prevents the trimerization of E6AP and inhibits its E3 functionality | N/A | Preclinical | [3] | |
| CM11-1 | Prevents the poly-ubiquitination of Prx1 and p53 by E6AP | N/A | Preclinical | [3] | |
| RBR-type E3 ligase inhibitor | |||||
| LUBAC antagonists | HOIPIN-8 | Inhibits LUBAC activity and suppresses linear ubiquitination-mediated NF-κb activation. | Human lung carcinoma A549 cells and HEK293T cells | Preclinical | [9] |
| BAY11-7082 | Inactivates the E2-conjugating enzymes Ubc13 and UbcH7 and the E3 ligase LUBAC | pre-B ALL, natural killer/T-cell lymphomas, gastric cancer | Preclinical | [18,200] | |
| Gliotoxin | Inhibits LUBAC and suppresses NF-κB activation | N/A | Preclinical | [201] | |
| Stapled peptides | Inhibits LUBAC through the disruption of the HOIL-1L-HOIP interaction and loss of the functional complex | N/A | Preclinical | [202] | |
| HOIP antagonist | Bendamustine | Specifically inhibits HOIP | Solid tumors, hematologic neoplasms | FDA approved (Phase 4) | [3] |
| N/A: not applicable | |||||
| Drug Class | Agent | Mechanism | Tumor Types | Phase | Reference |
|---|---|---|---|---|---|
| E3 ligase agonists | |||||
| Cereblon (CRBN) agonists | Lenalidomide, Thalidomide, Pomalidomide | Modulation of the substrate specificity of the CRL4-CRBN E3 ubiquitin ligase, induces the ubiquitination of IKZF1 and IKZF3 | Multiple myeloma, diffuse large B-cell lymphoma | FDA approved (Phase 4) | [236,237,238,239] |
| CC-90009 | Promotes binding of cereblon to GSPT1, leading to enhanced ubiquitination and subsequent degradation | AML, leukemia, myelodysplastic syndromes | 1, 2 | [240,241] | |
| CC-122 (Avadomide), CC-220 (Iberdomide) | Cereblon E3 ligase modulators (CELMoDs) | AML, multiple myeloma, diffuse large B-cell lymphoma (DLBCL), advanced solid tumors, non-Hodgkin’s lymphoma (NHL), melanoma | 1, 2 | [239] | |
| β-TrCP agonists | NRX-252114, NRX-252262, NRX-1532, NRX-1933, NRX-2663, NRX-103094, RX-103095 | Promotes the interaction of β-TrCP with β-catenin | N/A | Preclinical | [242] |
| DCAF15 agonists | Indisulam(E7070), Tasisulam, CQS | Promotes the binding of Rbm39 to DCAF15 | Metastatic breast cancer, gastric cancer, leukemia, melanoma (skin), solid tumor, kidney neoplasms, adenocarcinoma, CRC | 1, 2 | [243] |
| TIR1 agonists | Hormone auxin | Binds to SCF F-box subunit TIR1 and promotes the interaction between TIR1 and its substrate | N/A | Preclinical | [244] |
| NPR agonists | Aalicylic acid (SA) | Regulates the effect of CRL3-NPR | N/A | Preclinical | [244,245] |
| COI1 agonists | Jasmonic acid (JA) | Facilitates the molecular association between SCF-COI1 ligase and its substrates | N/A | Preclinical | [244] |
| N/A: not applicable | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, P.; Chi, X.; Cha, J.-H.; Luo, S.; Yang, G.; Yan, X.; Yang, W.-H. Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges. Cells 2021, 10, 3309. https://doi.org/10.3390/cells10123309
Ye P, Chi X, Cha J-H, Luo S, Yang G, Yan X, Yang W-H. Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges. Cells. 2021; 10(12):3309. https://doi.org/10.3390/cells10123309
Chicago/Turabian StyleYe, Peng, Xiaoxia Chi, Jong-Ho Cha, Shahang Luo, Guanghui Yang, Xiuwen Yan, and Wen-Hao Yang. 2021. "Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges" Cells 10, no. 12: 3309. https://doi.org/10.3390/cells10123309
APA StyleYe, P., Chi, X., Cha, J.-H., Luo, S., Yang, G., Yan, X., & Yang, W.-H. (2021). Potential of E3 Ubiquitin Ligases in Cancer Immunity: Opportunities and Challenges. Cells, 10(12), 3309. https://doi.org/10.3390/cells10123309