
Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Obesity and Associated Metabolic Diseases
1.2. Adipose Tissue Dysfunction in the Pathogenesis of NAFLD
1.3. Differential Functions of Adipose Tissue Depots in Pathogenesis of NAFLD
1.4. Brown Adipose Tissue and NAFLD
2. Pathophysiology of NAFLD during Obesity
2.1. The Prevalence of NAFLD
2.2. Two Hit or Multiple Hit Mechanisms of NAFLD
2.3. Dysregulation of Fat Metabolism in the Liver
2.4. Pathogenesis of Liver Inflammation
2.5. Hepatocyte Fat Deposition and Regulation of Fat Metabolism
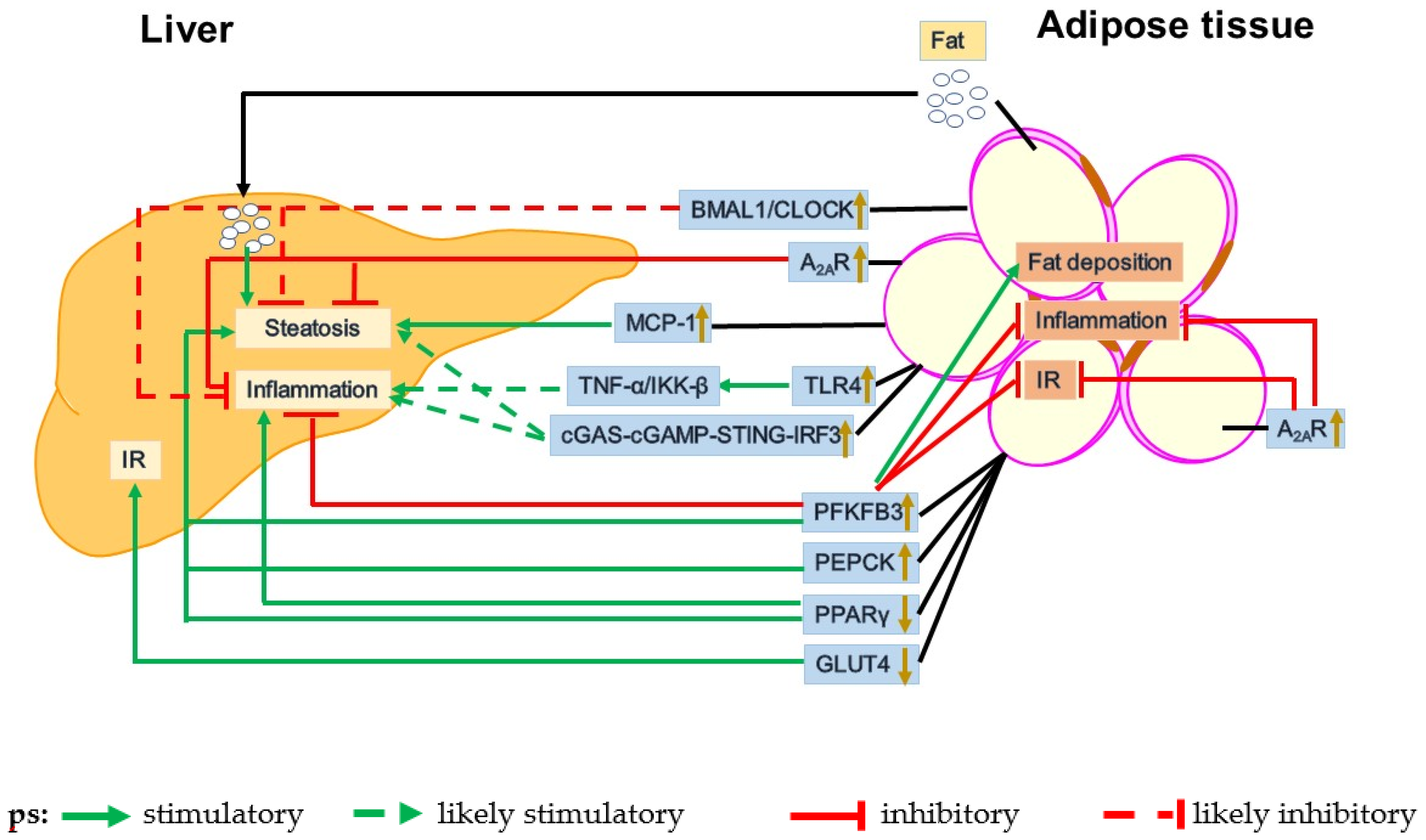
3. Dysfunctional Adipose-Liver Crosstalk in the Pathogenesis of NAFLD
3.1. GLUT4
3.2. PEPCK
3.3. PFKFB3
3.4. PPARγ
4. Obesity-Associated Adipose Tissue Dysfunction
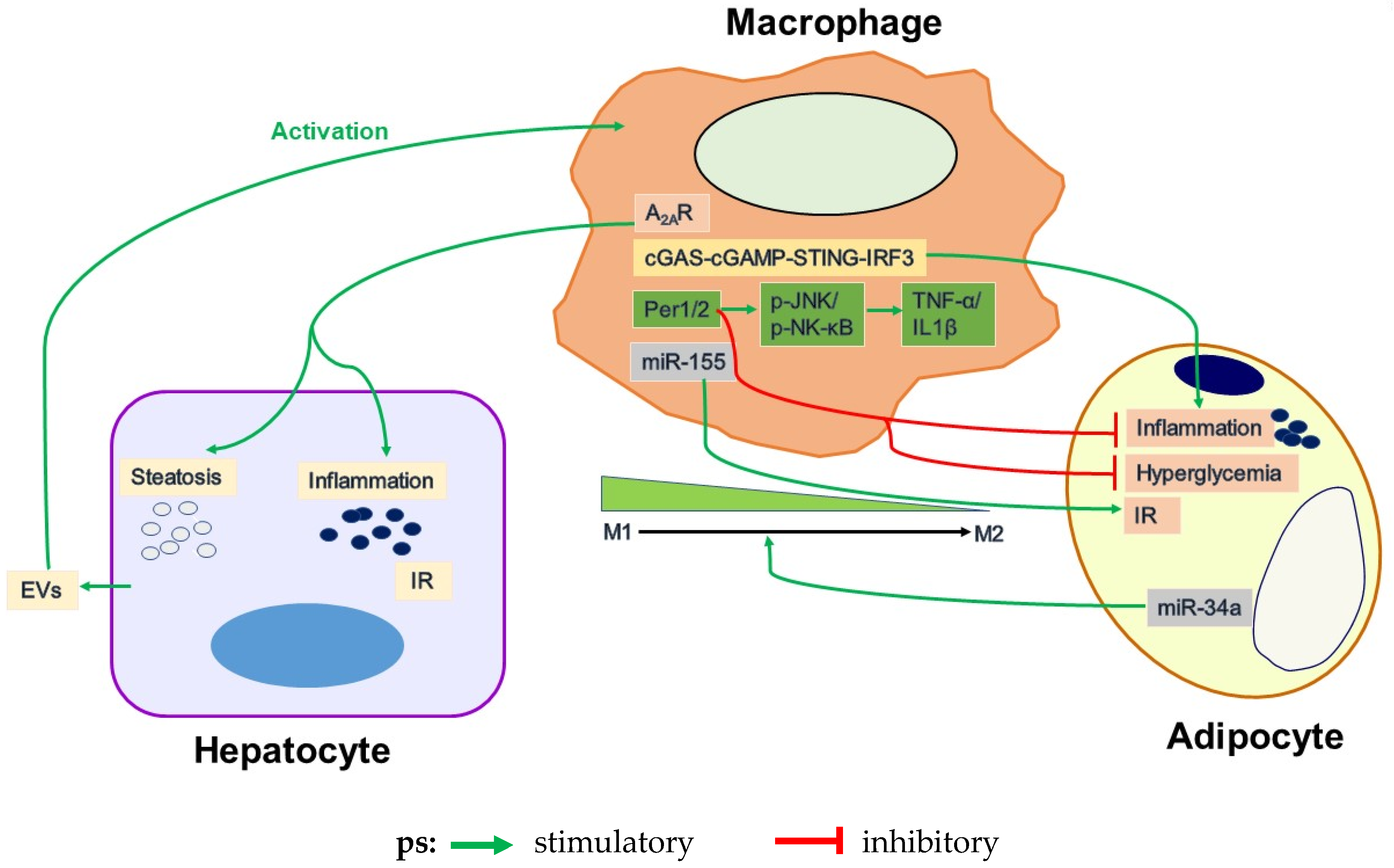
4.1. Adipose Tissue Inflammation and Its Regulation by Macrophages
4.2. Uncoupling Fat Deposition and Inflammation
4.3. Circadian Dysregulation
4.4. Adipocyte and Macrophage Inflammatory Signaling
4.4.1. TLR4
4.4.2. MCP1
4.4.3. JNK
4.4.4. A2AR
4.4.5. cGAS-cGAMP-STING-IRF3 Signaling Pathway
4.4.6. Circadian Regulation
4.5. Dysregulation of Adipocyte-Macrophage Crosstalk in Obesity-Related WAT Inflammation
5. Perspective and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, M.; Mayhew, L.; Richardson, J.; Rickayzen, B. Waist-to-height ratio is more predictive of years of life lost than body mass index. PLoS ONE 2014, 9, e103483. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- WHO. Obesity and Overweight; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Holtcamp, W. Obesogens: An environmental link to obesity. Environ. Health Perspect. 2012, 120, a62–a68. [Google Scholar] [CrossRef]
- Tan, P.Y.; Mitra, S.R.; Amini, F. Lifestyle Interventions for Weight Control Modified by Genetic Variation: A Review of the Evidence. Public Health Genom. 2018, 21, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Śledzińska, E.; Baturo, A.; Jończyk, I.; Maleszko, A.; Samborski, P.; Begier-Krasińska, B.; Dobrowolska, A. Obesity and inflammation. Eur. Cytokine Netw. 2018, 29, 83–94. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Kahn, C.R. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Que, S.; Xu, J.; Peng, T. Alanine aminotransferase-old biomarker and new concept: A review. Int. J. Med. Sci. 2014, 11, 925–935. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Gholam, P.M.; Kotler, D.P.; Flancbaum, L.J. Liver pathology in morbidly obese patients undergoing Roux-en-Y gastric bypass surgery. Obes. Surg. 2002, 12, 49–51. [Google Scholar] [CrossRef]
- Marcos, A.; Fisher, R.A.; Ham, J.M.; Olzinski, A.T.; Shiffman, M.L.; Sanyal, A.J.; Luketic, V.A.C.; Sterling, R.K.; Olbrisch, M.E.; Posner, M.P. Selection and outcome of living donors for adult to adult right lobe transplantation. Transplantation 2000, 69, 2410–2415. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G. Nonalcoholic steatohepatitis: A study of 49 patients. Hum. Pathol. 1989, 20, 594–598. [Google Scholar] [CrossRef]
- Hilden, M.; Christoffersen, P.; Juhl, E.; Dalgaard, J.B. Liver histology in a ‘normal’ population—Examinations of 503 consecutive fatal traffic casualties. Scand. J. Gastroenterol. 1977, 12, 593–597. [Google Scholar] [CrossRef]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus white adipose tissue: A mini-review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef]
- Leitner, B.; Huang, S.; Brychta, R.J.; Duckworth, C.J.; Baskin, A.S.; McGehee, S.; Tal, I.; Dieckmann, W.; Gupta, G.; Kolodny, G.M.; et al. Mapping of human brown adipose tissue in lean and obese young men. Proc. Natl. Acad. Sci. USA 2017, 114, 8649–8654. [Google Scholar] [CrossRef] [PubMed]
- Hajer, G.R.; van Haeften, T.W.; Visseren, F.L. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur. Heart J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Guerre-Millo, M. Adipose tissue and adipokines: For better or worse. Diabetes Metab. 2004, 30, 13–19. [Google Scholar] [CrossRef]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.-S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Shabalala, S.C.; Dludla, P.V.; Mabasa, L.; Kappo, A.P.; Basson, A.K.; Pheiffer, C.; Johnson, R. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed. Pharmacother. 2020, 131, 110785. [Google Scholar] [CrossRef]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef]
- Xie, X.; Yan, D.; Li, H.; Zhu, Q.; Li, J.; Fang, Y.-P.; Cheung, C.W.; Irwin, M.G.; Xia, Z.; Lian, Q. Enhancement of Adiponectin Ameliorates Nonalcoholic Fatty Liver Disease via Inhibition of FoxO1 in Type I Diabetic Rats. J. Diabetes Res. 2018, 2018, 6254340. [Google Scholar] [CrossRef] [PubMed]
- Nobarani, S.; Alaei-Shahmiri, F.; Aghili, R.; Malek, M.; Poustchi, H.; Lahouti, M.; Khamseh, M.E. Visceral Adipose Tissue and Non-alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes. Dig. Dis. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Franczyk, M.P.; He, M.; Yoshino, J. Removal of Epididymal Visceral Adipose Tissue Prevents Obesity-Induced Multi-organ Insulin Resistance in Male Mice. J. Endocr. Soc. 2021, 5, bvab024. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, J.; Tan, J.; Song, Y.; Liu, L.; Zhang, F.; Zhang, Y.; Li, X.; Chi, Y.; Liu, Y. Mesenteric adipose tissue B lymphocytes promote local and hepatic inflammation in non-alcoholic fatty liver disease mice. J. Cell. Mol. Med. 2019, 23, 3375–3385. [Google Scholar] [CrossRef] [PubMed]
- Drolet, R.; Richard, C.; Sniderman, A.D.; Mailloux, J.; Fortier, M.; Huot, C.; Rhéaume, C.; Tchernof, A. Hypertrophy and hyperplasia of abdominal adipose tissues in women. Int. J. Obes. 2008, 32, 283–291. [Google Scholar] [CrossRef]
- Ahmed, B.A.; Ong, F.J.; Barra, N.G.; Blondin, D.P.; Gunn, E.; Oreskovich, S.M.; Szamosi, J.C.; Syed, S.A.; Hutchings, E.K.; Konyer, N.B.; et al. Lower brown adipose tissue activity is associated with non-alcoholic fatty liver disease but not changes in the gut microbiota. Cell Rep. Med. 2021, 2, 100397. [Google Scholar] [CrossRef]
- Wibmer, A.G.; Becher, T.; Eljalby, M.; Crane, A.; Andrieu, P.C.; Jiang, C.S.; Vaughan, R.; Schöder, H.; Cohen, P. Brown adipose tissue is associated with healthier body fat distribution and metabolic benefits independent of regional adiposity. Cell Rep. Med. 2021, 2, 100332. [Google Scholar] [CrossRef]
- Blondin, D.P.; Tingelstad, H.C.; Noll, C.; Frisch, F.; Phoenix, S.; Guérin, B.; Turcotte, É.E.; Richard, D.; Haman, F.; Carpentier, A.C. Dietary fatty acid metabolism of brown adipose tissue in cold-acclimated men. Nat. Commun. 2017, 8, 14146. [Google Scholar] [CrossRef]
- Wang, G.-X.; Zhao, X.-Y.; Meng, Z.-X.; Kern, M.; Dietrich, A.; Chen, Z.; Cozacov, Z.; Zhou, D.; Okunade, A.L.; Su, X.; et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat. Med. 2014, 20, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, C.; Cai, Y.; Fang, S.; Zeng, Y.; Zhang, Y.; Lin, X.; Zhang, H.; Xue, Y.; Guan, M. Transplantation of brown adipose tissue up-regulates miR-99a to ameliorate liver metabolic disorders in diabetic mice by targeting NOX4. Adipocyte 2020, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Trueba, J.L.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e1447. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales., A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2021. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Liao, C.-Y.; Song, M.J.; Gao, Y.; Mauer, A.S.; Revzin, A.; Malhi, H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front. Immunol. 2018, 9, 2980. [Google Scholar] [CrossRef] [PubMed]
- Saiman, Y.; Friedman, S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [PubMed]
- Stanton, M.C.; Chen, S.-C.; Jackson, J.V.; Rojas-Triana, A.; Kinsley, D.; Cui, L.; Fine, J.S.; Greenfeder, S.; Bober, L.A.; Jenh, C.-H. Inflammatory Signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J. Inflamm. 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, W.; Mao, M.; Gao, R.; Shi, W.; Li, D.; Calderone, R.; Sui, B.; Tian, X.; Meng, X. Similarities and Differences: A Comparative Review of the Molecular Mechanisms and Effectors of NAFLD and AFLD. Front. Physiol. 2021, 12, 710285. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takamura, T.; Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef]
- Cai, Y.; Li, H.; Liu, M.; Pei, Y.; Zheng, J.; Zhou, J.; Luo, X.; Huang, W.; Ma, L.; Yang, Q.; et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 2018, 68, 48–61. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef]
- Wang, X.; Rao, H.; Zhao, J.; Wee, A.; Li, X.; Fei, R.; Huang, R.; Wu, C.; Liu, F.; Wei, L. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab. Investig. 2020, 100, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.-L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients with NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e1974. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.-H.; Luo, W.-J.; Xu, Q.-Y.; Hua, J. Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J. Gastroenterol. 2016, 22, 2533–2544. [Google Scholar] [CrossRef]
- Ma, M.; Duan, R.; Zhong, H.; Liang, T.; Guo, L. The Crosstalk between Fat Homeostasis and Liver Regional Immunity in NAFLD. J. Immunol. Res. 2019, 2019, 3954890. [Google Scholar] [CrossRef]
- Kim, S.; Feng, D.; Guillot, A.; Dai, S.; Liu, F.; Hwang, S.; Parker, R.; Seo, W.; He, Y.; Godlewski, G.; et al. Adipocyte Death Preferentially Induces Liver Injury and Inflammation through the Activation of Chemokine (C-C Motif) Receptor 2-Positive Macrophages and Lipolysis. Hepatology 2019, 69, 1965–1982. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kang, J.E.; Peng, L.-J.; Li, H.; Khan, S.A.; Hillard, C.J.; Okar, D.A.; Lange, A.J. Enhancing hepatic glycolysis reduces obesity: Differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metab. 2005, 2, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; Titos, E.; Rius, B.; González-Périz, A.; García-Alonso, V.; López-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Clària, J. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. Metabolic interplay between white, beige, brown adipocytes and the liver. J. Hepatol. 2016, 64, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Jacome-Sosa, M.M.; Parks, E.J. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr. Opin. Lipidol. 2014, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Kahn, B.B. Glucose transporters and insulin action--implications for insulin resistance and diabetes mellitus. N. Engl. J. Med. 1999, 341, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Peroni, O.D.; Kim, J.; Kim, Y.-B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef]
- Jaubert, A.-M.; Penot, G.; Niang, F.; Durant, S.; Forest, C. Rapid nitration of adipocyte phosphoenolpyruvate carboxykinase by leptin reduces glyceroneogenesis and induces fatty acid release. PLoS ONE 2012, 7, e40650. [Google Scholar] [CrossRef]
- Franckhauser, S.; Antras-Ferry, J.; Robin, P.; Robin, D.; Granner, D.K.; Forest, C. Expression of the phosphoenolpyruvate carboxykinase gene in 3T3-F442A adipose cells: Opposite effects of dexamethasone and isoprenaline on transcription. Biochem. J. 1995, 305 Pt 1, 65–71. [Google Scholar] [CrossRef]
- Franckhauser, S.; Muñoz, S.; Pujol, A.; Casellas, A.; Riu, E.; Otaegui, P.; Su, B.; Bosch, F. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes 2002, 51, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Franckhauser, S.; Muñoz, S.; Elias, I.; Ferre, T.; Bosch, F. Adipose overexpression of phosphoenolpyruvate carboxykinase leads to high susceptibility to diet-induced insulin resistance and obesity. Diabetes 2006, 55, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Järvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Nishio, T.; Niwa, H.; Takeuchi, J.; Bando, H.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes 2005, 54, 3349–3357. [Google Scholar] [CrossRef][Green Version]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.; Rousseau, G.G.; Hue, L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Guo, X.; Li, H.; Xu, H.; Halim, V.; Zhang, W.; Wang, H.; Fan, Y.-Y.; Ong, K.T.; Woo, S.-L.; et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J. Biol. Chem. 2012, 287, 21492–21500. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Guo, X.; Li, H.; Wang, H.; Zhang, W.; Wang, Y.; Zhou, H.; Gao, Z.; Telang, S.; Chesney, J.; et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J. Biol. Chem. 2010, 285, 3713–3721. [Google Scholar] [CrossRef]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Sabatini, S.; Carli, F.; Gaggini, M.; Bril, F.; Belfort-DeAguiar, R.; Positano, V.; Barb, D.; Kadiyala, S.; Harrison, S.; et al. PPAR-gamma-induced changes in visceral fat and adiponectin levels are associated with improvement of steatohepatitis in patients with NASH. Liver Int. 2021, 41, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Adipose Tissue-Endothelial Cell Interactions in Obesity-Induced Endothelial Dysfunction. Front. Cardiovasc. Med. 2021, 8, 681581. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef]
- Tong, X.; Wei, L.; Wang, T.; Han, R. Remodeling of Macrophages in White Adipose Tissue under the Conditions of Obesity as well as Lipolysis. Oxid. Med. Cell. Longev. 2021, 2021, 9980877. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef]
- Zhao, H.; Shang, Q.; Pan, Z.; Bai, Y.; Li, Z.; Zhang, H.; Zhang, Q.; Guo, C.; Zhang, L.; Wang, Q. Exosomes From Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity Through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes 2018, 67, 235–247. [Google Scholar] [CrossRef]
- Guo, X.; Xu, K.; Zhang, J.; Li, H.; Zhang, W.; Wang, H.; Lange, A.J.; Chen, Y.E.; Huo, Y.; Wu, C. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of peroxisome proliferator-activated receptor gamma activation in mice. J. Biol. Chem. 2010, 285, 23711–23720. [Google Scholar] [CrossRef]
- Stienstra, R.; Duval, C.; Keshtkar, S.; van der Laak, J.; Kersten, S.; Muller, M. Peroxisome proliferator-activated receptor gamma activation promotes infiltration of alternatively activated macrophages into adipose tissue. J. Biol. Chem. 2008, 283, 22620–22627. [Google Scholar] [CrossRef]
- Clément, S.; Juge-Aubry, C.; Sgroi, A.; Conzelmann, S.; Pazienza, V.; Pittet-Cuenod, B.; Meier, C.A.; Negro, F. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology 2008, 48, 799–807. [Google Scholar] [CrossRef]
- De Rocha, A.R.F.; Morais, N.D.S.; Priore, S.E.; Franceschini, S.D.C.C. Inflammatory Biomarkers and Components of Metabolic Syndrome in Adolescents: A Systematic Review. Inflammation 2021. [Google Scholar] [CrossRef]
- Ruf, W.; Samad, F. Tissue factor pathways linking obesity and inflammation. Hamostaseologie 2015, 35, 279–283. [Google Scholar] [CrossRef]
- Gao, H.; Mejhert, N.; Fretz, J.; Arner, E.; Lorente-Cebrián, S.; Ehrlund, A.; Dahlman-Wright, K.; Gong, X.; Strömblad, S.; Douagi, I.; et al. Early B cell factor 1 regulates adipocyte morphology and lipolysis in white adipose tissue. Cell Metab. 2014, 19, 981–992. [Google Scholar] [CrossRef]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Weng, S.; Shen, F.; Chang, Y.; Lian, W.; Hsieh, C.; Chuang, J.; Lin, T.; Liou, C.; Chang, C.; et al. Abrogation of Toll-Like Receptor 4 Mitigates Obesity-Induced Oxidative Stress, Proinflammation, and Insulin Resistance Through Metabolic Reprogramming of Mitochondria in Adipose Tissue. Antioxid Redox Signal. 2020, 33, 66–86. [Google Scholar] [CrossRef]
- Guo, X.; Li, H.; Xu, H.; Halim, V.; Zhang, W.; Wang, H.; Ong, K.T.; Woo, S.-L.; Walzem, R.L.; Mashek, D.G.; et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS ONE 2012, 7, e39286. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhu, B.; Li, H.; Jiang, B.; Wang, Y.; Yin, Q.; Cai, J.; Glaser, S.; Francis, H.; Alpini, G.; et al. Adipocyte inducible 6-phosphofructo-2-kinase suppresses adipose tissue inflammation and promotes macrophage anti-inflammatory activation. J. Nutr. Biochem. 2021, 95, 108764. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, B.; Xu, H.; Li, H.; Jiang, B.; Wang, Y.; Zheng, B.; Glaser, S.; Alpini, G.; Wu, C. Adoptive transfer of Pfkfb3-disrupted hematopoietic cells to wild-type mice exacerbates diet-induced hepatic steatosis and inflammation. Liver Res. 2020, 4, 136–144. [Google Scholar] [CrossRef]
- Dyar, K.A.; Eckel-Mahan, K.L. Circadian Metabolomics in Time and Space. Front. Neurosci. 2017, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Morante, J.J.H.; Gomez-Santos, C.; Milagro, F.; Campion, J.; Martínez, J.A.; Zamora, S.; Garaulet, M. Expression of cortisol metabolism-related genes shows circadian rhythmic patterns in human adipose tissue. Int. J. Obes. 2009, 33, 473–480. [Google Scholar] [CrossRef]
- Xu, H.; Li, H.; Woo, S.-L.; Kim, S.-M.; Shende, V.R.; Neuendorff, N.; Guo, X.; Guo, T.; Qi, T.; Pei, Y.; et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J. Biol. Chem. 2014, 289, 16374–16388. [Google Scholar] [CrossRef]
- Shetty, A.; Hsu, J.W.; Manka, P.P.; Syn, W.-K. Role of the Circadian Clock in the Metabolic Syndrome and Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2018, 63, 3187–3206. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Costa, M.J.; Rivero-Gutierrez, B.; Ji, L.; Morgan, S.L.; Feldman, B.J. The Circadian Clock Regulates Adipogenesis by a Per3 Crosstalk Pathway to Klf15. Cell Rep. 2017, 21, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Esteban, J.P.; Dinani, A. Lifestyle Interventions Beyond Diet and Exercise for Patients with Nonalcoholic Fatty Liver Disease. Gastroenterol. Hepatol. 2020, 16, 119–130. [Google Scholar]
- Koopman, A.D.; Rauh, S.P.; Riet, E.V.T.; Groeneveld, L.; Van Der Heijden, A.A.; Elders, P.J.; Dekker, J.M.; Nijpels, G.; Beulens, J.W.; Rutters, F. The Association between Social Jetlag, the Metabolic Syndrome, and Type 2 Diabetes Mellitus in the General Population: The New Hoorn Study. J. Biol. Rhythms 2017, 32, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Stoner, L.; Castro, N.; Signal, L.; Skidmore, P.; Faulkner, J.; Lark, S.; Williams, M.A.; Muller, D.; Harrex, H. Sleep and Adiposity in Preadolescent Children: The Importance of Social Jetlag. Child. Obes. 2018, 14, 158–164. [Google Scholar] [CrossRef]
- Mathew, G.M.; Hale, L.; Chang, A.M. Social jetlag, eating behaviours and BMI among adolescents in the USA. Br. J. Nutr. 2020, 124, 979–987. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araújo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.-Y.; Tang, J.; Hou, Y.-J.; Chang, Y.-X.; Tu, Q.-Q.; Feng, G.-S.; et al. Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011, 54, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Vianna, C.R.; Fukuda, M.; Berglund, E.D.; Liu, C.; Tao, C.; Sun, K.; Liu, T.; Harper, M.; Lee, C.E.; et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 3878. [Google Scholar] [CrossRef]
- Saberi, M.; Woods, N.-B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef]
- Tao, C.; Holland, W.L.; Wang, Q.; Shao, M.; Jia, L.; Sun, K.; Lin, X.; Kuo, Y.-C.; Johnson, J.A.; Gordillo, R.; et al. Short-Term versus Long-Term Effects of Adipocyte Toll-Like Receptor 4 Activation on Insulin Resistance in Male Mice. Endocrinology 2017, 158, 1260–1270. [Google Scholar] [CrossRef]
- Czemplik, M.; Kulma, A.; Wang, Y.F.; Szopa, J. Therapeutic Strategies of Plant-derived Compounds for Diabetes Via Regulation of Monocyte Chemoattractant Protein-1. Curr. Med. Chem. 2017, 24, 1453–1468. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.-L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, A.; Chung, S.K.; Cresser, J.H.; Sweeney, G.; Wong, R.L.; Lin, A.; Lam, K.S. Selective inactivation of c-Jun NH2-terminal kinase in adipose tissue protects against diet-induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes 2011, 60, 486–495. [Google Scholar] [CrossRef]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Li, H.; Cai, Y.; Zhou, J.; Luo, X.; Ma, L.; McDaniel, K.; Zeng, T.; Chen, Y.; Qian, X.; et al. Regulation of adipose tissue inflammation by adenosine 2A receptor in obese mice. J. Endocrinol. 2018, 239, 365–376. [Google Scholar] [CrossRef]
- DeOliveira, C.C.; Caria, C.R.E.P.; Gotardo, E.M.F.; Ribeiro, M.; Gambero, A. Role of A1 and A2A adenosine receptor agonists in adipose tissue inflammation induced by obesity in mice. Eur. J. Pharmacol. 2017, 799, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R.; Vance, C.O.; Morschl, E.; Wilson, C.N. Adenosine receptors and inflammation. Handb. Exp. Pharmacol. 2009, 215–269. [Google Scholar] [CrossRef]
- Alchera, E.; Rolla, S.; Imarisio, C.; Bardina, V.; Valente, G.; Novelli, F.; Carini, R. Adenosine A2a receptor stimulation blocks development of nonalcoholic steatohepatitis in mice by multilevel inhibition of signals that cause immunolipotoxicity. Transl. Res. 2017, 182, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Omura, H.; Ishitani, R.; Nureki, O. Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu. Rev. Biochem. 2017, 86, 541–566. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef]
- Guo, X.; Shu, C.; Li, H.; Pei, Y.; Woo, S.-L.; Zheng, J.; Liu, M.; Xu, H.; Botchlett, R.; Guo, T.; et al. Cyclic GMP-AMP Ameliorates Diet-induced Metabolic Dysregulation and Regulates Proinflammatory Responses Distinctly from STING Activation. Sci. Rep. 2017, 7, 6355. [Google Scholar] [CrossRef]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.-I.; Lemaire, S.A.; et al. STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Duez, H. Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front. Immunol. 2020, 11, 1630. [Google Scholar] [CrossRef]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Lazar, M.A. Interconnections between circadian clocks and metabolism. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Paschos, G.K.; Ibrahim, S.; Song, W.; Kunieda, T.; Grant, G.; Reyes, T.M.; A Bradfield, C.; Vaughan, C.H.; Eiden, M.; Masoodi, M.; et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat. Med. 2012, 18, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Bunger, M.K.; Wilsbacher, L.D.; Moran, S.M.; Clendenin, C.; Radcliffe, L.A.; Hogenesch, J.B.; Simon, M.C.; Takahashi, J.S.; Bradfield, C.A. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000, 103, 1009–1017. [Google Scholar] [CrossRef]
- Vitaterna, M.H.; King, D.P.; Chang, A.-M.; Kornhauser, J.M.; Lowrey, P.L.; McDonald, J.D.; Dove, W.F.; Pinto, L.H.; Turek, F.W.; Takahashi, J.S. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 1994, 264, 719–725. [Google Scholar] [CrossRef]
- Shostak, A.; Meyer-Kovac, J.; Oster, H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes 2013, 62, 2195–2203. [Google Scholar] [CrossRef]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef]
- Kennaway, D.J.; Varcoe, T.; Voultsios, A.; Boden, M.J. Global loss of bmal1 expression alters adipose tissue hormones, gene expression and glucose metabolism. PLoS ONE 2013, 8, e65255. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e312. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Sakane, S.; Hikita, H.; Shirai, K.; Myojin, Y.; Sasaki, Y.; Kudo, S.; Fukumoto, K.; Mizutani, N.; Tahata, Y.; Makino, Y.; et al. White Adipose Tissue Autophagy and Adipose-Liver Crosstalk Exacerbate Nonalcoholic Fatty Liver Disease in Mice. Cell Mol Gastroenterol. Hepatol. 2021, 12, 1683–1699. [Google Scholar] [CrossRef] [PubMed]
- Liao, N.; Pan, F.; Wang, Y.; Zheng, Y.; Xu, B.; Chen, W.; Gao, Y.; Cai, Z.; Liu, X.; Liu, J. Adipose tissue-derived stem cells promote the reversion of non-alcoholic fatty liver disease: An in vivo study. Int. J. Mol. Med. 2016, 37, 1389–1396. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Rao, H.; Liu, F.; Wei, L.; Li, H.; Wu, C. Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3300. https://doi.org/10.3390/cells10123300
Wang X, Rao H, Liu F, Wei L, Li H, Wu C. Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells. 2021; 10(12):3300. https://doi.org/10.3390/cells10123300
Chicago/Turabian StyleWang, Xiaoxiao, Huiying Rao, Feng Liu, Lai Wei, Honggui Li, and Chaodong Wu. 2021. "Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease" Cells 10, no. 12: 3300. https://doi.org/10.3390/cells10123300
APA StyleWang, X., Rao, H., Liu, F., Wei, L., Li, H., & Wu, C. (2021). Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells, 10(12), 3300. https://doi.org/10.3390/cells10123300