
Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Chronic Kidney Disease (CKD)
2.3. Assessment of Kidney Function
2.4. Renal Histology
2.5. Mitochondrial Isolation Procedure
2.6. Measurement of Mitochondrial Oxphos Conductance
2.7. Mitochondrial Hydrogen Peroxide (H2O2) Production and Electron Leak
2.8. NAD(P)H/NAD(P)+ Redox Potential
2.9. Mitochondrial Membrane Potential (Δψ)
2.10. Assessment of Mitochondrial Dehydrogenase Activity
2.11. Hydroxyacyl-CoA Dehydrogenase Activity
2.12. ATP Synthase Activity (Complex V)
2.13. Mitochondrial OXPHOS Complex Activities
2.14. Tmt-Assisted Proteomics
2.15. Data Analysis for Mitochondrial Proteomics
2.16. Statistical Analysis
3. Results
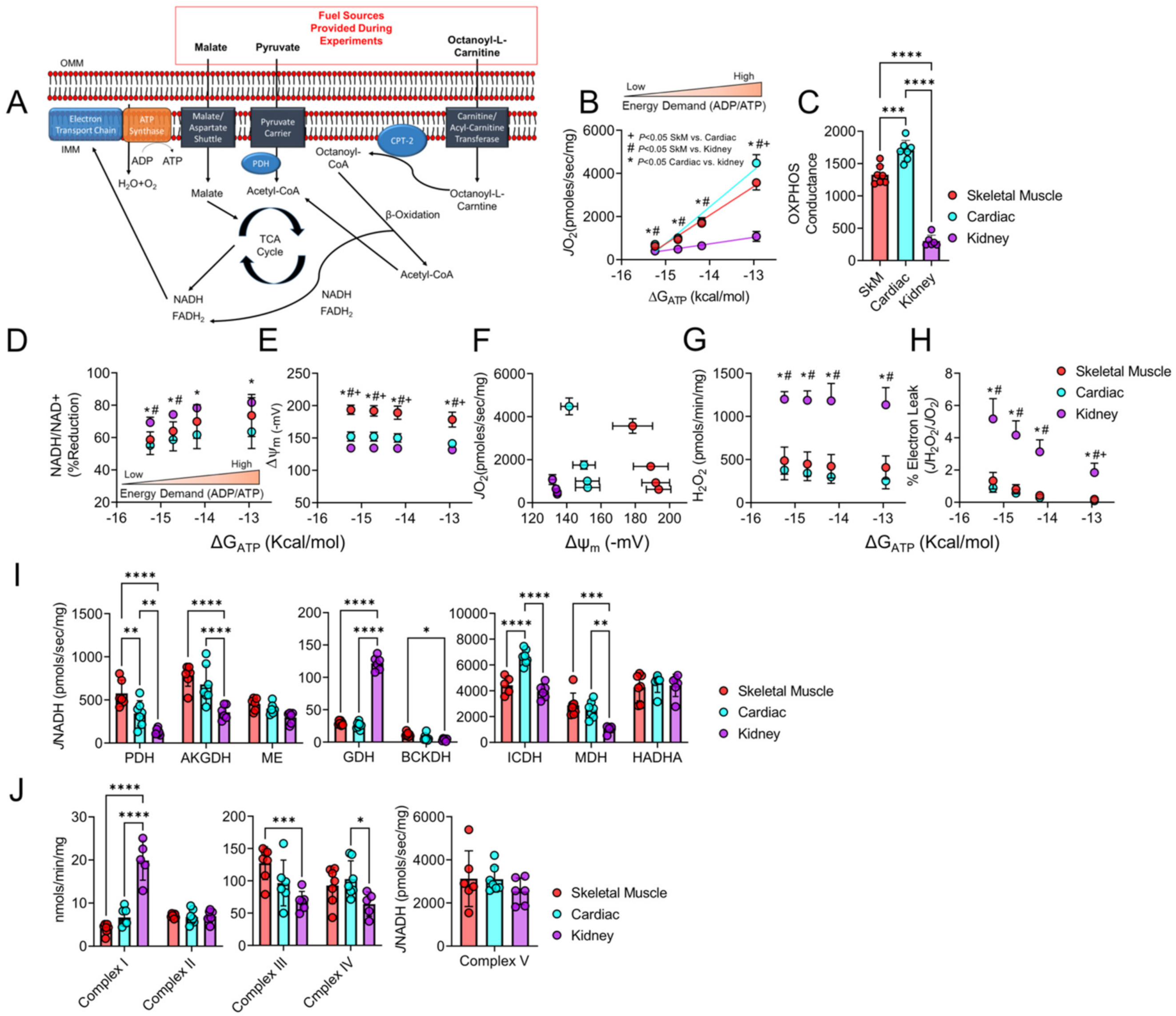
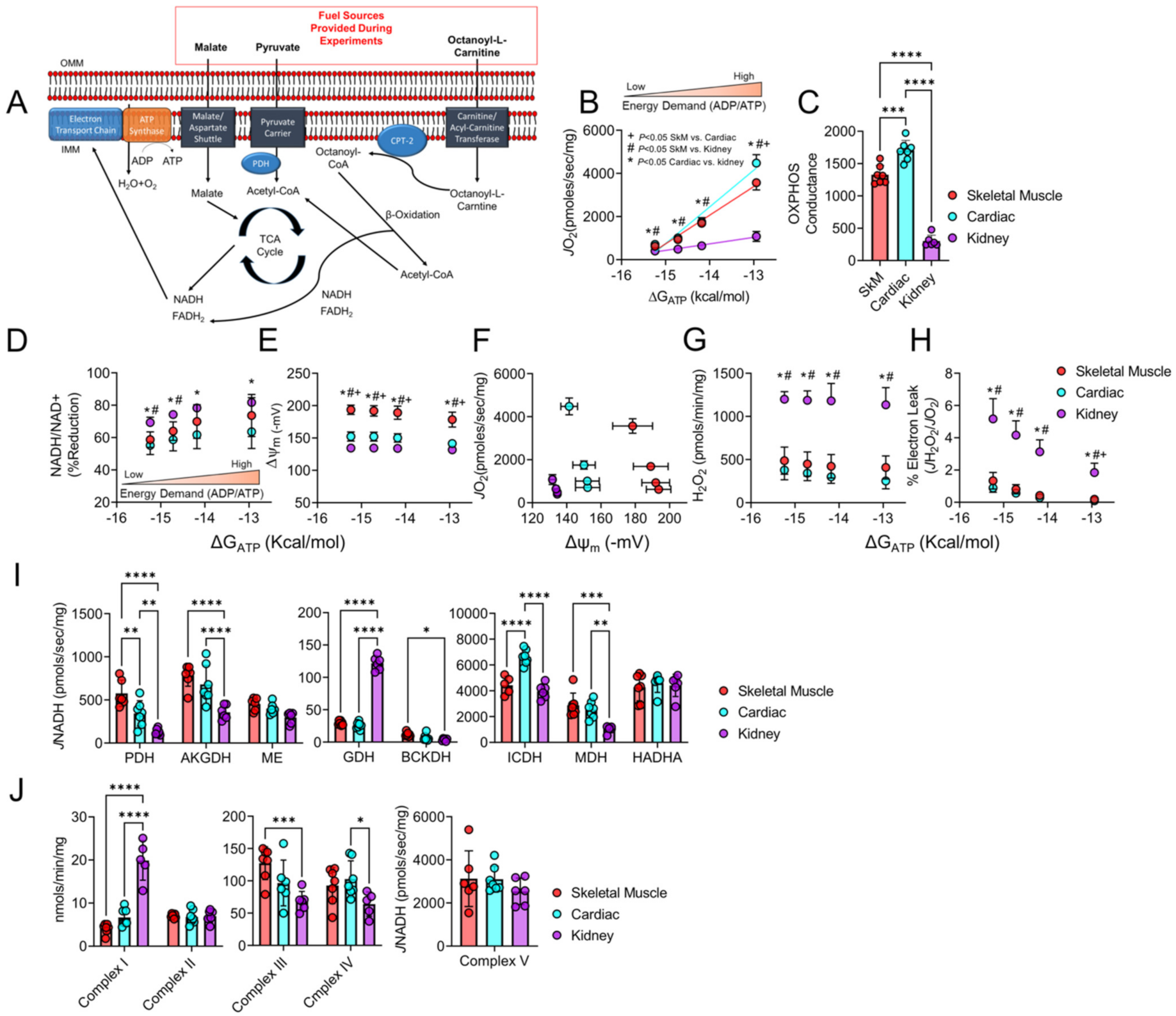
3.1. Mitochondrial Phenotyping Identifies Organ-Specific Bioenergetic Profiles
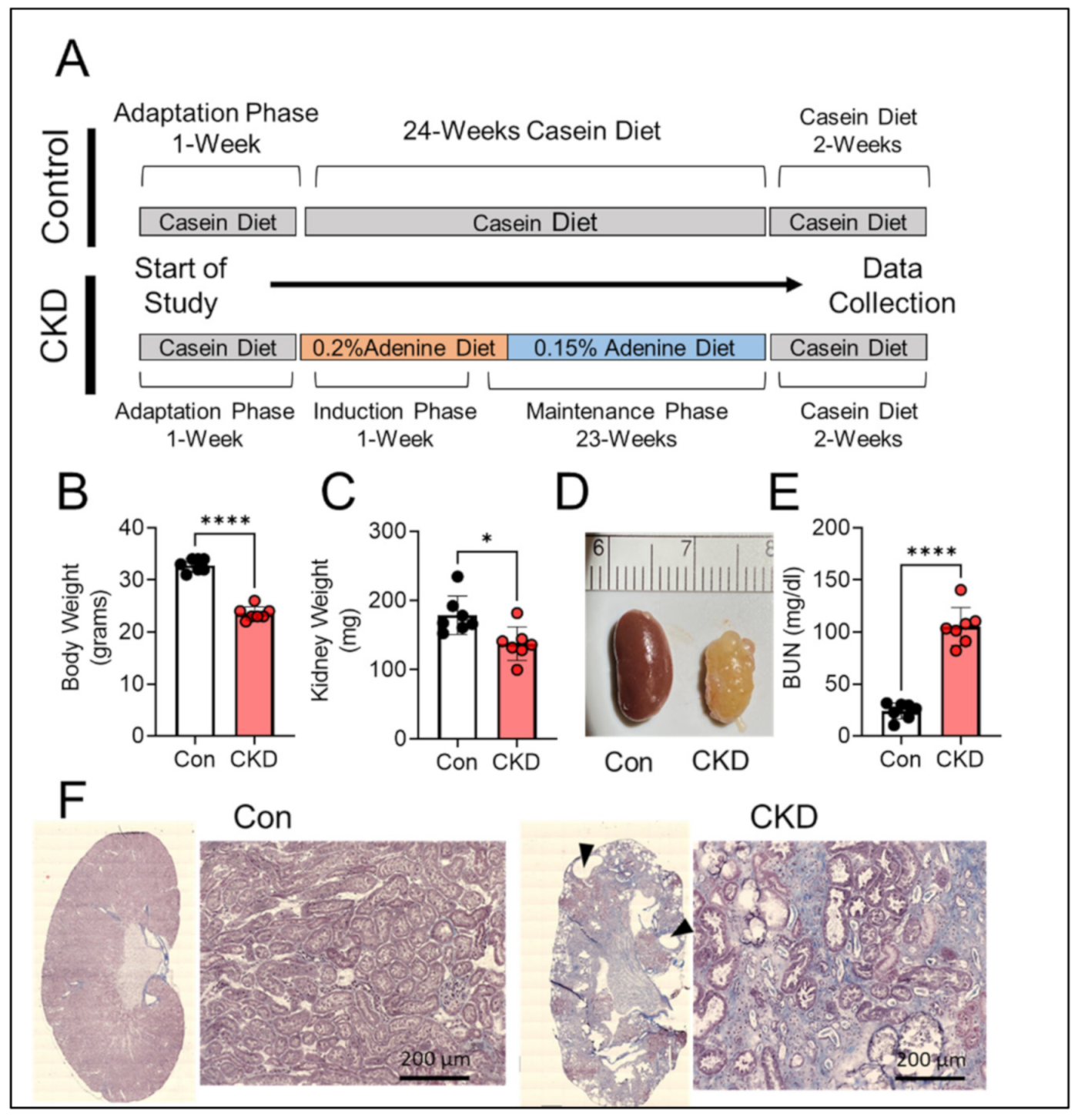
3.2. Long-Term Adenine Feeding Causes Severe CKD
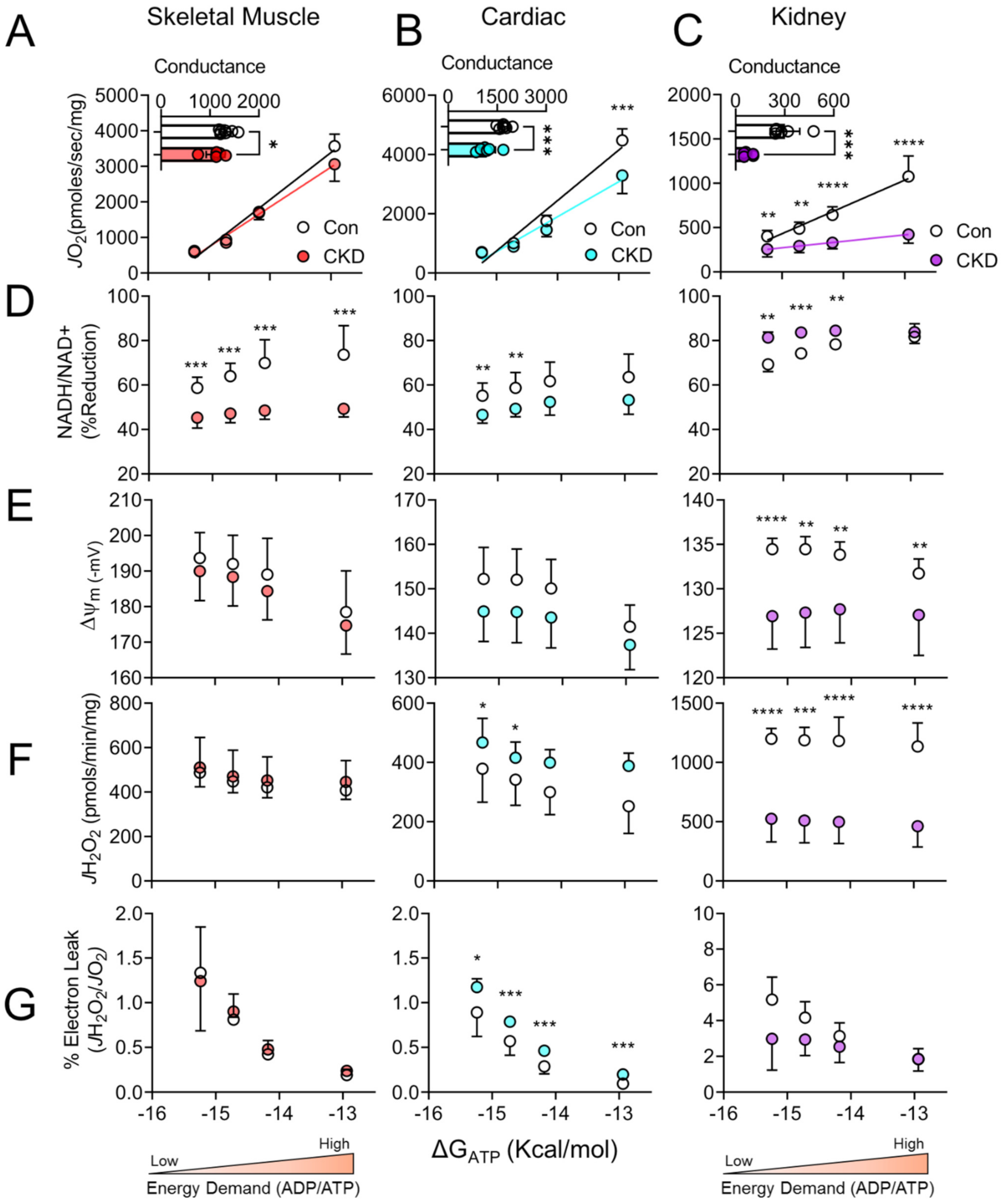
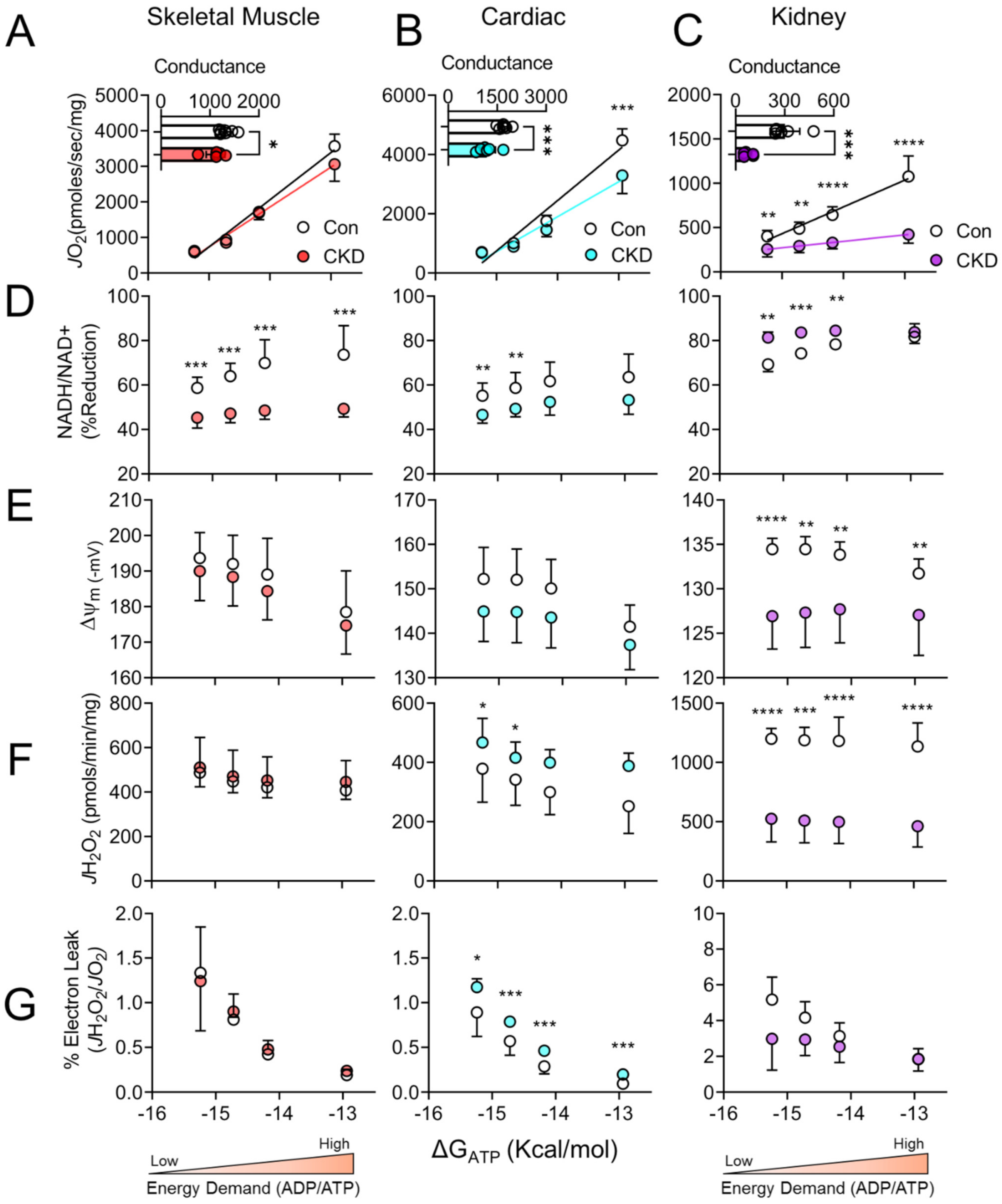
3.3. Chronic Kidney Disease Impairs Mitochondrial Energy Transduction in Skeletal Muscle, Cardiac Muscle, and Renal Tissue
3.4. Tissue-Specific Changes in Mitochondrial Redox and Membrane Potential in CKD
3.5. H2O2 Production and Electron Leak Are Elevated in Cardiac but Not Skeletal Muscle or Kidney Mitochondria in CKD
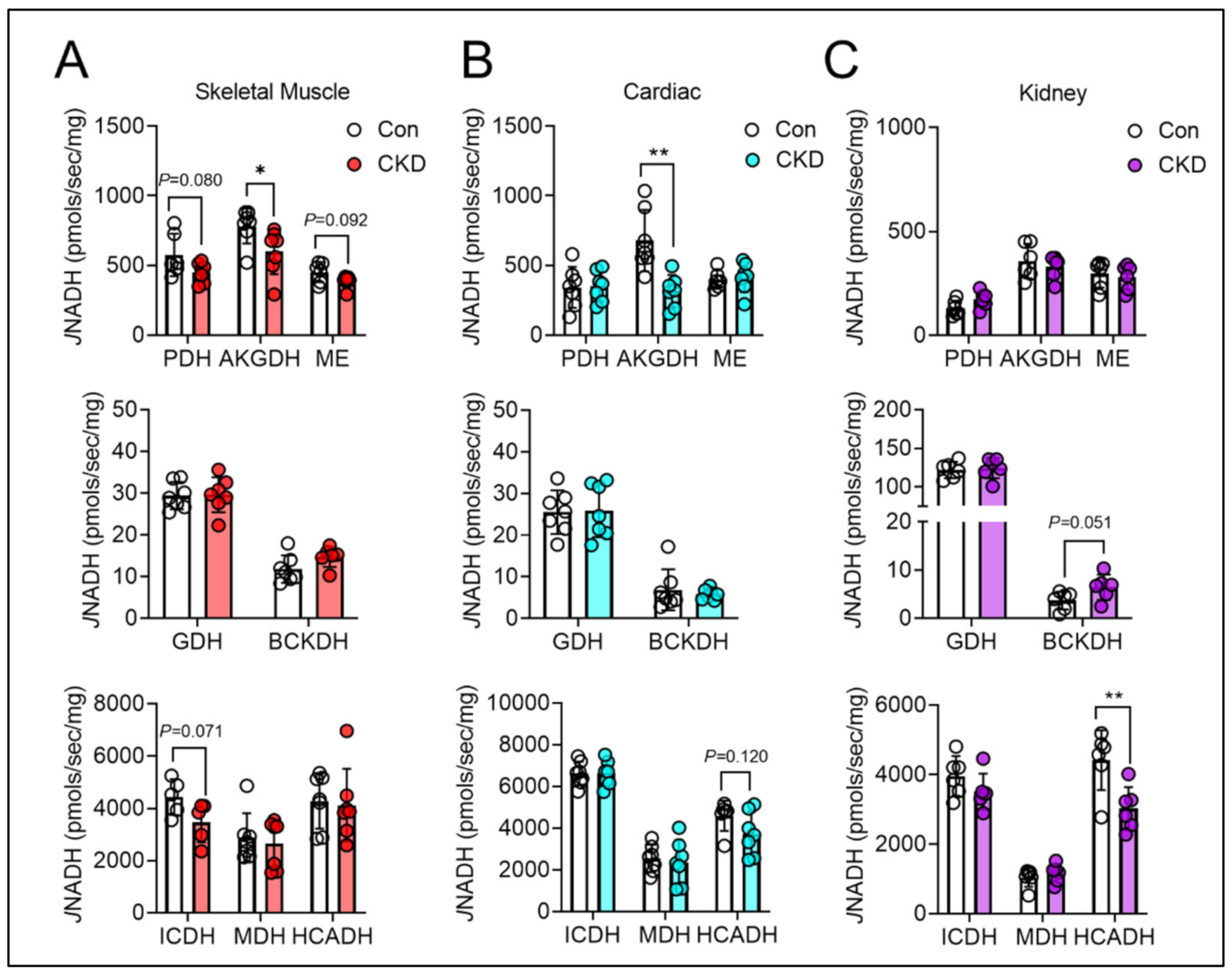
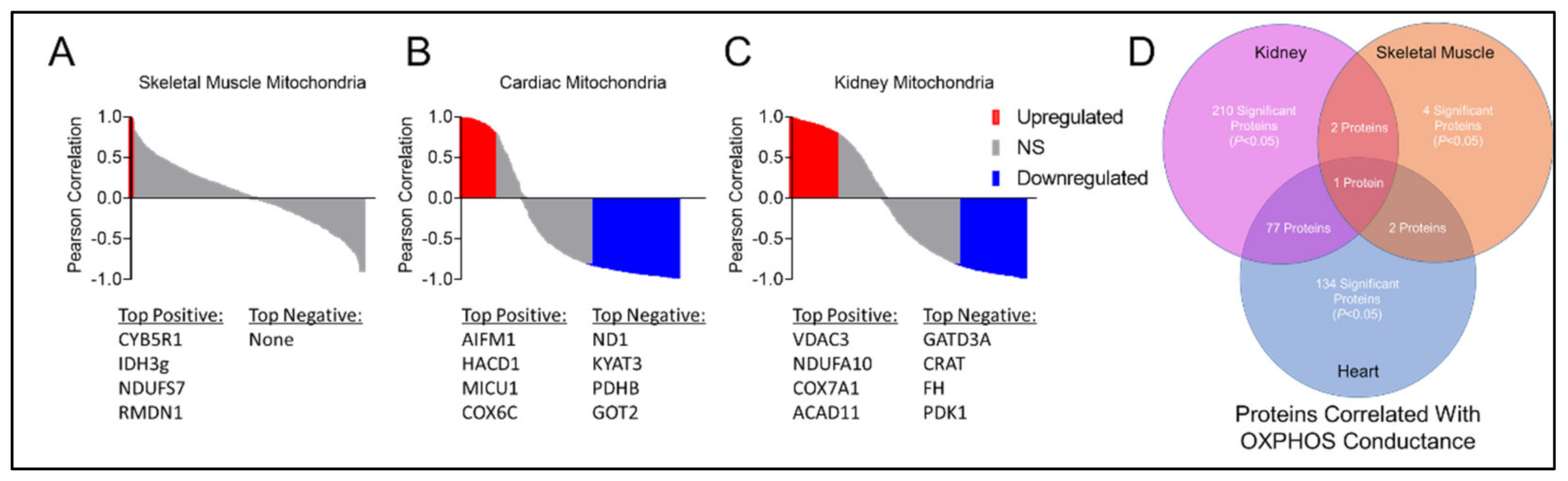
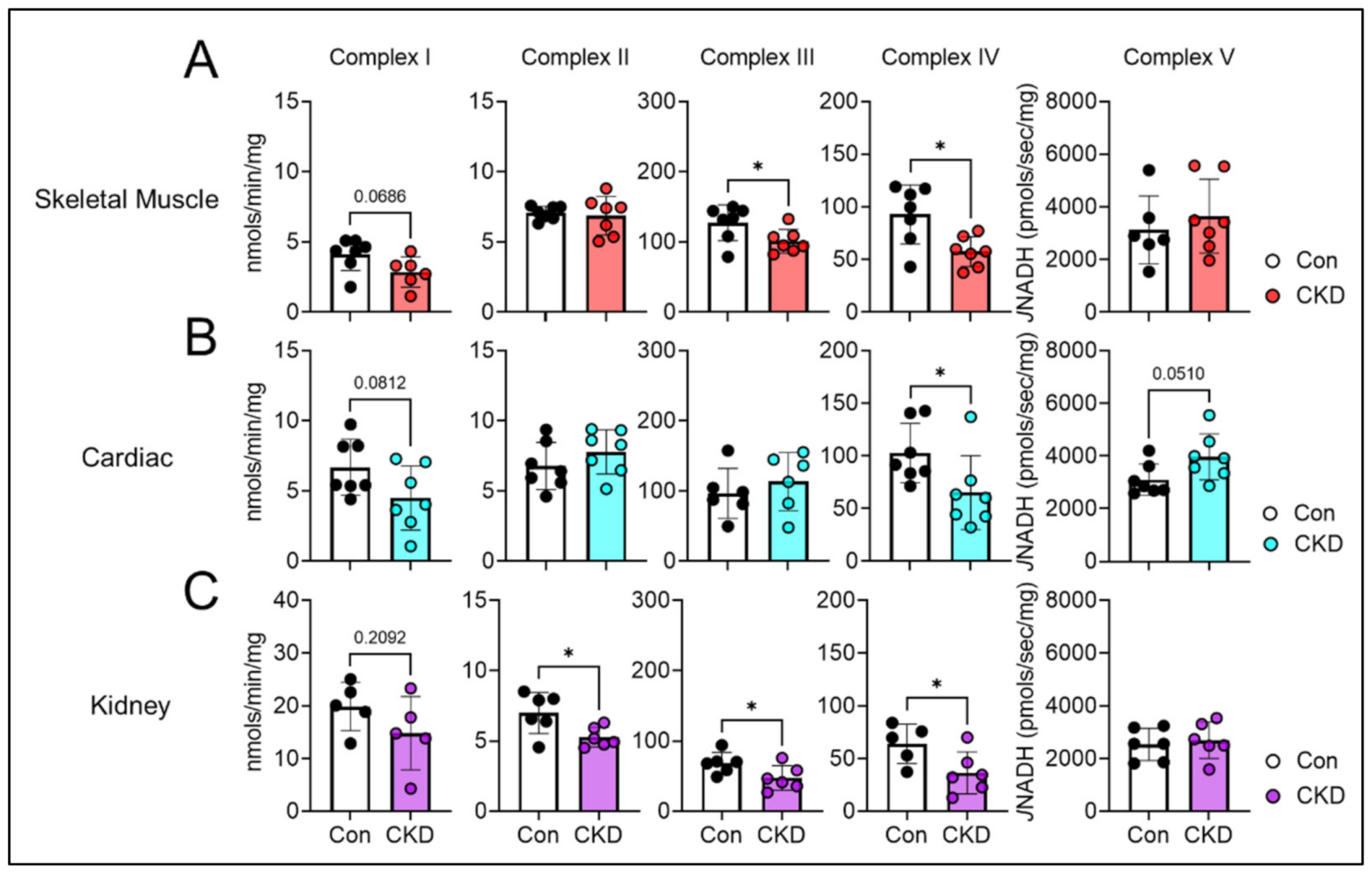
3.6. Mitochondrial Matrix Enzyme Activities and Production of NADH Are Differentially Altered across Tissues
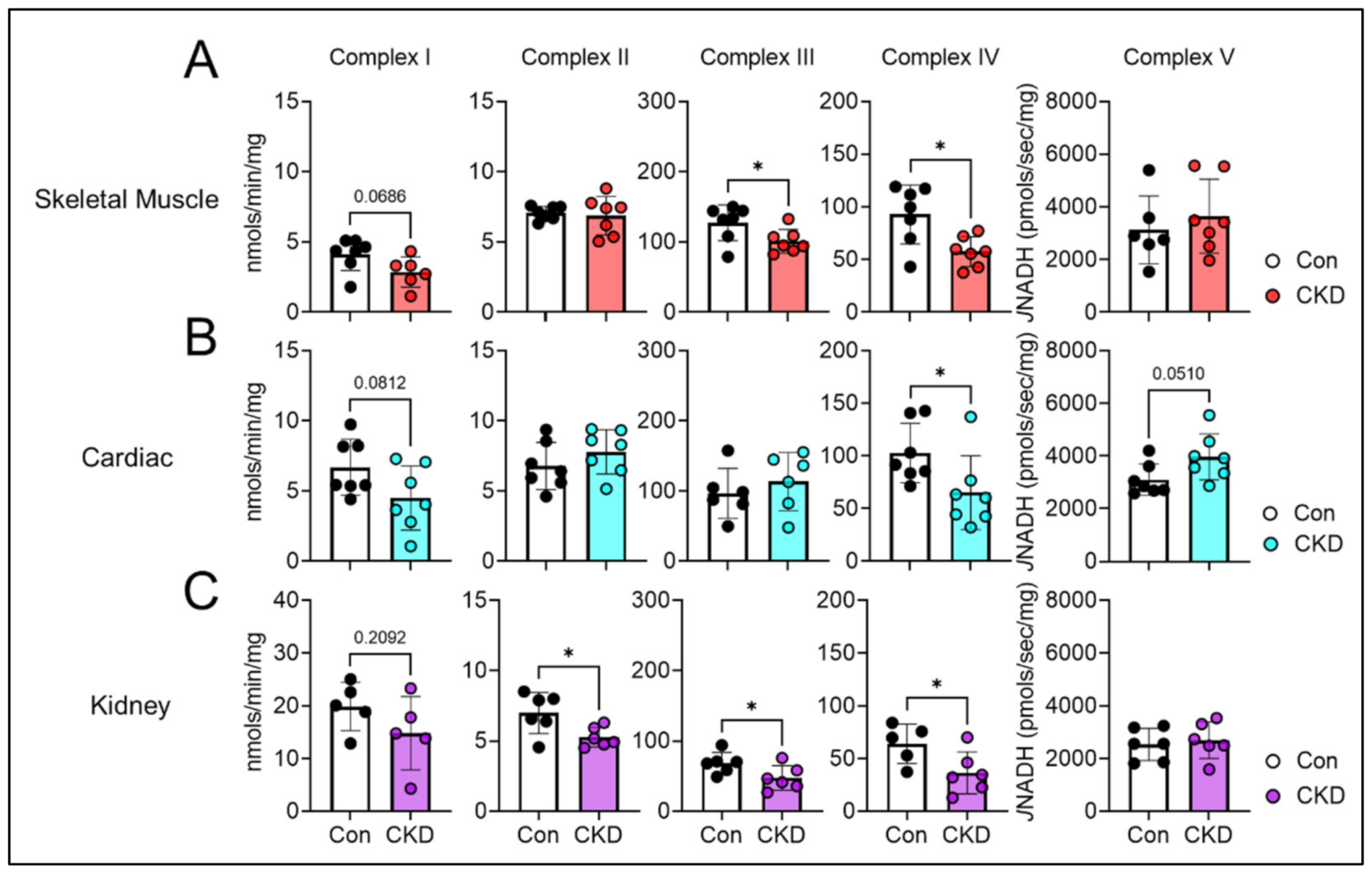
3.7. CKD Impairs Electron Transport System Enzyme Activity in a Tissue-Specific Manner
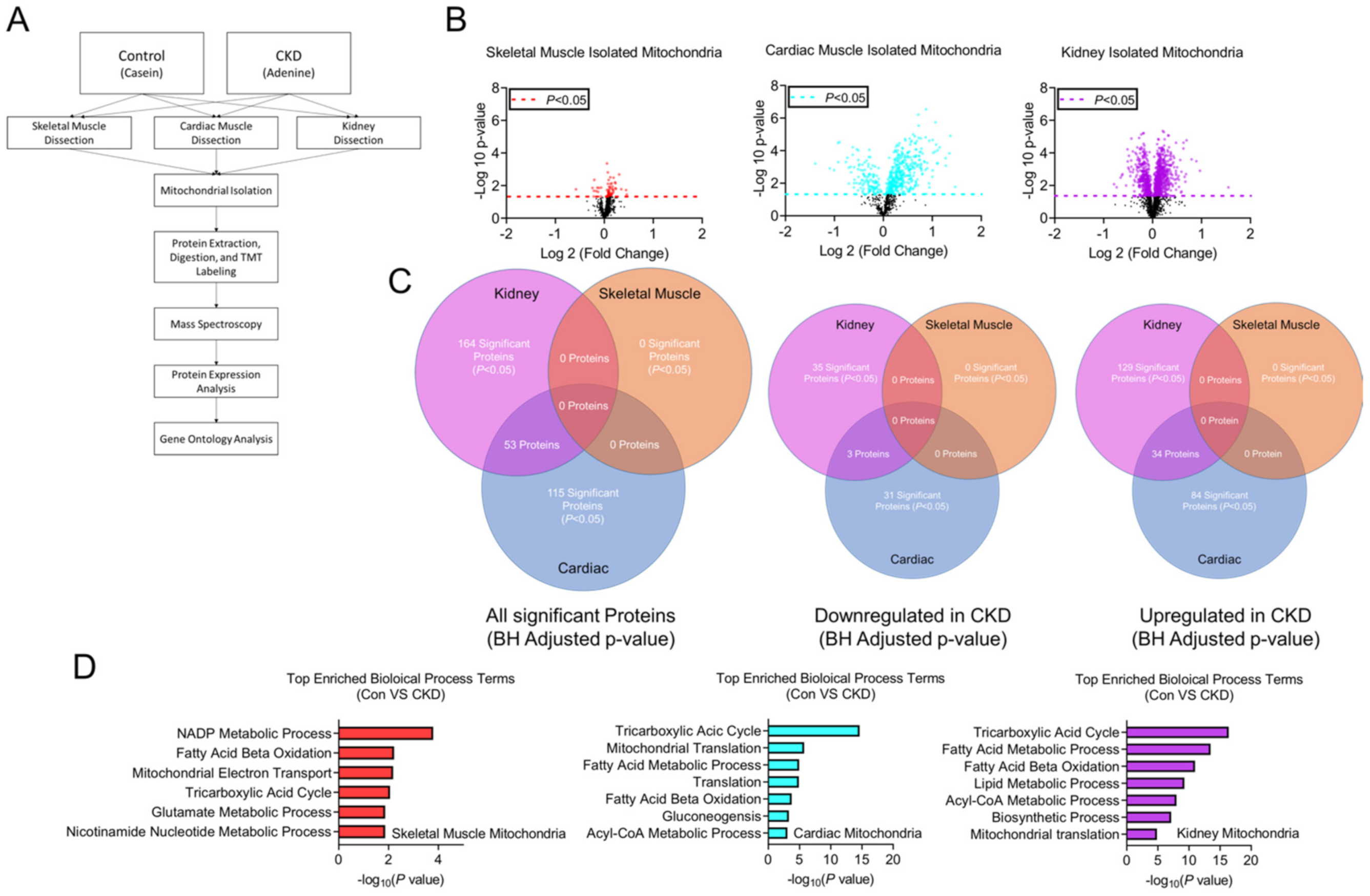
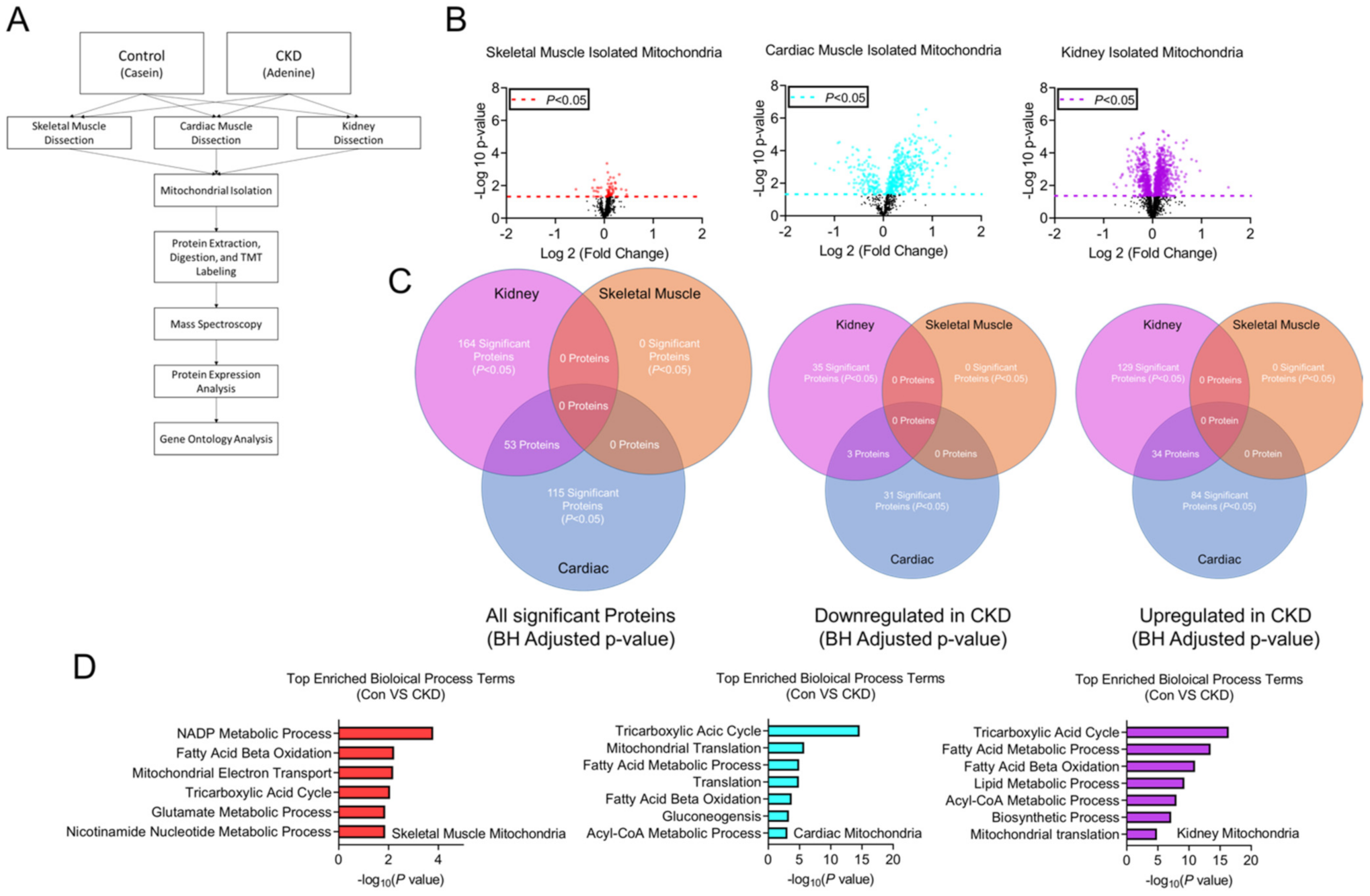
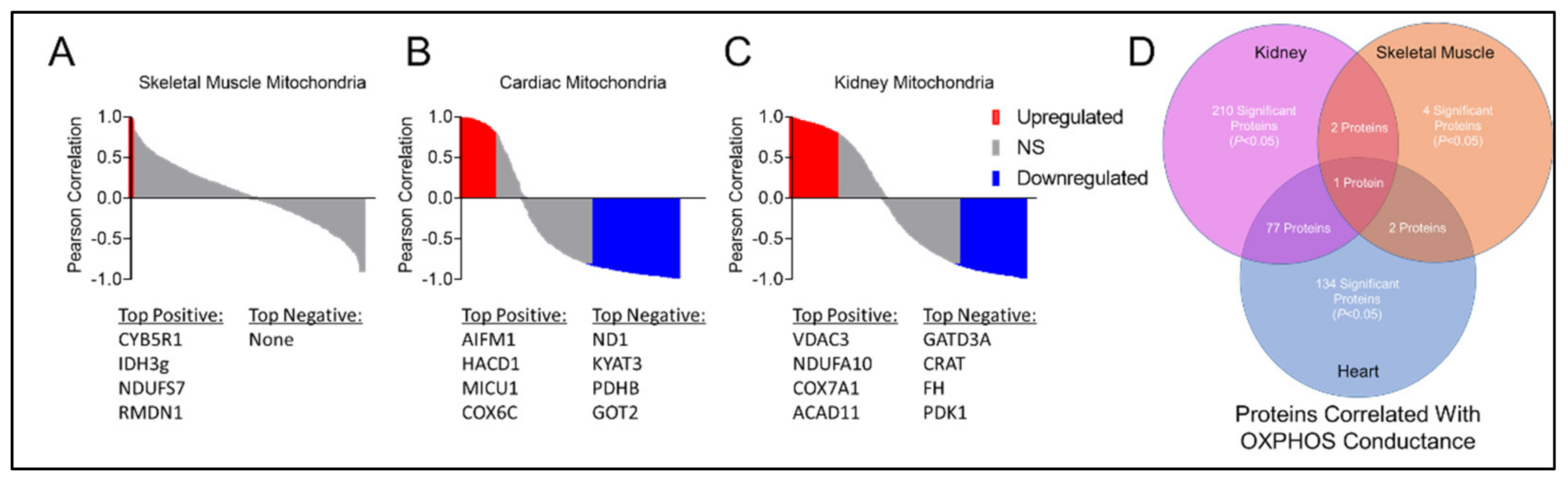
3.8. Tissue-Specific Mitochondrial Proteomes in CKD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Kraut, J.A.; Kurtz, I. Metabolic Acidosis of CKD: Diagnosis, Clinical Characteristics, and Treatment. Am. J. Kidney Dis. 2005, 45, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Slee, A.D. Exploring metabolic dysfunction in chronic kidney disease. Nutr. Metab. 2012, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.-Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.H.; Mitch, W.E. Muscle wasting from kidney failure—A model for catabolic conditions. Int. J. Biochem. Cell Biol. 2013, 45, 2230–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.S.; Mitch, W.E. Mechanisms stimulating muscle wasting in chronic kidney disease: The roles of the ubiquitin-proteasome system and myostatin. Clin. Exp. Nephrol. 2013, 17, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Roshanravan, B.; Kestenbaum, B.; Gamboa, J.; Jubrias, S.A.; Ayers, E.; Curtin, L.; Himmelfarb, J.; de Boer, I.H.; Conley, K.E. CKD and Muscle Mitochondrial Energetics. Am. J. Kidney Dis. 2016, 68, 658–659. [Google Scholar] [CrossRef] [Green Version]
- Roshanravan, B.; Gamboa, J.; Wilund, K. Exercise and CKD: Skeletal Muscle Dysfunction and Practical Application of Exercise to Prevent and Treat Physical Impairments in CKD. Am. J. Kidney Dis. 2017, 69, 837–852. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Billings, F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef]
- Ábrigo, J.; Elorza, A.A.; Riedel, C.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxidative Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ishimori, N.; Takada, S.; Saito, A.; Kadoguchi, T.; Furihata, T.; Fukushima, A.; Matsushima, S.; Yokota, T.; Kinugawa, S.; et al. AST-120 ameliorates lowered exercise capacity and mitochondrial biogenesis in the skeletal muscle from mice with chronic kidney disease via reducing oxidative stress. Nephrol. Dial. Transplant. 2015, 30, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Bellido, B.; Pedraza-Chaverri, J. Mitochondrial dysfunction and endoplasmic reticulum stress in the promotion of fibrosis in obstructive nephropathy induced by unilateral ureteral obstruction. BioFactors 2020, 46, 716–733. [Google Scholar] [CrossRef]
- Cantarin, M.P.M.; Whitaker-Menezes, D.; Lin, Z.; Falkner, B. Uremia induces adipose tissue inflammation and muscle mitochondrial dysfunction. Nephrol. Dial. Transplant 2017, 32, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Chronic impairment of mitochondrial bioenergetics and β-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Bigelman, E.; Cohen, L.; Aharon-Hananel, G.; Levy, R.; Rozenbaum, Z.; Saada, A.; Keren, G.; Entin-Meer, M. Pathological presentation of cardiac mitochondria in a rat model for chronic kidney disease. PLoS ONE 2018, 13, e0198196. [Google Scholar] [CrossRef]
- Doi, K.; Noiri, E. Mitochondrial Dysfunction in Cardiorenal Syndrome. Antioxid. Redox Signal. 2016, 25, 200–207. [Google Scholar] [CrossRef]
- Burchill, L.; Velkoska, E.; Dean, R.G.; Lew, R.A.; Smith, A.I.; Levidiotis, V.; Burrell, L.M. Acute kidney injury in the rat causes cardiac remodelling and increases angiotensin-converting enzyme 2 expression. Exp. Physiol. 2008, 93, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bhandari, S.; Seymour, A.-M.L. Mitochondrial dysfunction in uremic cardiomyopathy. Am. J. Physiol. Renal Physiol. 2015, 308, F579–F587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, T.; Salyers, Z.R.; Kumar, R.A.; Hahn, D.; Berru, F.N.; Ferreira, L.F.; Scali, S.T.; Ryan, T.E. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am. J. Physiol. Cell Physiol. 2019, 317, C701–C713. [Google Scholar] [CrossRef] [PubMed]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2021, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachex Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, S.; Zhou, J.; Liu, Y.; Du, C.; Yang, K.; Bi, X.; Liu, M.; Han, W.; Wang, K.; et al. IRF1-mediated downregulation of PGC1α contributes to cardiorenal syndrome type 4. Nat. Commun. 2020, 11, 4664. [Google Scholar] [CrossRef]
- Tamura, M.; Aizawa, R.; Hori, M.; Ozaki, H. Progressive renal dysfunction and macrophage infiltration in interstitial fibrosis in an adenine-induced tubulointerstitial nephritis mouse model. Histochem. Cell Biol. 2009, 131, 483–490. [Google Scholar] [CrossRef]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal myopathy in CKD: A comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am. J. Physiol. Renal Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef] [PubMed]
- Berru, F.N.; Gray, S.E.; Thome, T.; Kumar, R.A.; Salyers, Z.R.; Coleman, M.; Le, D.; O’Malley, K.; Ferreira, L.F.; Berceli, S.A.; et al. Chronic kidney disease exacerbates ischemic limb myopathy in mice via altered mitochondrial energetics. Sci. Rep. 2019, 9, 15547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Spangenburg, E.E.; Neufer, P.D.; McClung, J.M. Targeted Expression of Catalase to Mitochondria Protects Against Ischemic Myopathy in High-Fat Diet–Fed Mice. Diabetes 2016, 65, 2553–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher-Wellman, K.H.; Davidson, M.; Narowski, T.M.; Lin, C.-T.; Koves, T.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018, 24, 3593–3606.e3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher-Wellman, K.H.; Draper, J.A.; Davidson, M.; Williams, A.S.; Narowski, T.M.; Slentz, D.H.; Ilkayeva, O.R.; Stevens, R.D.; Wagner, G.R.; Najjar, R.; et al. Respiratory Phenomics across Multiple Models of Protein Hyperacylation in Cardiac Mitochondria Reveals a Marginal Impact on Bioenergetics. Cell Rep. 2019, 26, 1557–1572.e1558. [Google Scholar] [CrossRef] [Green Version]
- Scaduto, R.C.; Grotyohann, L.W. Measurement of Mitochondrial Membrane Potential Using Fluorescent Rhodamine Derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Krumschnabel, G.; Eigentler, A.; Fasching, M.; Gnaiger, E. Use of Safranin for the Assessment of Mitochondrial Membrane Potential by High-Resolution Respirometry and Fluorometry. Methods Enzymol. 2014, 542, 163–181. [Google Scholar] [CrossRef]
- Barrientos, A.; Fontanesi, F.; Díaz, F. Evaluation of the Mitochondrial Respiratory Chain and Oxidative Phosphorylation System Using Polarography and Spectrophotometric Enzyme Assays. Curr. Protoc. Hum. Genet. 2009, 63, 19.3.1–19.3.14. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Anders, H.-J.; Schlöndorff, D. Murine Models of Renal Disease: Possibilities and Problems in Studies Using Mutant Mice. Nephron 2000, 8, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Yazdi, P.G.; Moradi, H.; Yang, J.-Y.; Wang, P.H.; Vaziri, N.D. Skeletal muscle mitochondrial depletion and dysfunction in chronic kidney disease. Int. J. Clin. Exp. Med. 2013, 6, 532–539. [Google Scholar]
- Wang, D.; Wei, L.; Yang, Y.; Liu, H. Dietary supplementation with ketoacids protects against CKD-induced oxidative damage and mitochondrial dysfunction in skeletal muscle of 5/6 nephrectomised rats. Skelet. Muscle 2018, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Kasimumali, A.; Guo, X.; Lu, R.; Xie, K.; Zhu, M.; Qian, Y.; Chen, X.; Pang, H.; Wang, Q.; et al. Reduction of mitochondria and up regulation of pyruvate dehydrogenase kinase 4 of skeletal muscle in patients with chronic kidney disease. Nephrology 2019, 25, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Luptak, I.; Balschi, J.A.; Xing, Y.; Leone, T.C.; Kelly, D.P.; Tian, R. Decreased Contractile and Metabolic Reserve in Peroxisome proliferator-activated receptor-alpha-null Hearts Can Be Rescued by Increasing Glucose Transport and Utilization. Circulation 2005, 112, 2339–2346. [Google Scholar] [CrossRef] [Green Version]
- Luptak, I.; Sverdlov, A.; Panagia, M.; Qin, F.; Pimentel, D.R.; Croteau, D.; Siwik, D.A.; Ingwall, J.S.; Bachschmid, M.M.; Balschi, J.A.; et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J. Mol. Cell. Cardiol. 2018, 116, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tocchi, A.; Quarles, E.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, X.; Wang, X.; Cui, S.; Liu, R.; Liu, J.; Fu, B.; Gong, M.; Wang, C.; Shi, Y.; et al. Grb2 Induces Cardiorenal Syndrome Type 3: Roles of IL-6, Cardiomyocyte Bioenergetics, and Akt/mTOR Pathway. Front. Cell Dev. Biol. 2021, 9, 630412. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Dai, D.-F.; Rabinovitch, P. Mitochondrial oxidative stress and mammalian healthspan. Mech. Ageing Dev. 2010, 131, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayanna, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. 2016, 27, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-S.; Noh, M.R.; Ha, L.; Kim, J.; Padanilam, B.J. Proximal tubule cyclophilin D mediates kidney fibrogenesis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 2021, 321, F431–F442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021, 12, 847. [Google Scholar] [CrossRef]
- Su, H.; Wan, C.; Song, A.; Qiu, Y.; Xiong, W.; Zhang, C. Oxidative Stress and Renal Fibrosis: Mechanisms and Therapies. Adv. Exp. Med. Biol. 2019, 1165, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Booz, G.W.; Fan, F.; Wang, Y.; Roman, R.J. Oxidative Stress and Renal Fibrosis: Recent Insights for the Development of Novel Therapeutic Strategies. Front. Physiol. 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Sakemi, T.; Ohtsuka, N.; Tomiyosi, Y.; Morito, F. Attenuating Effect of Castration on Glomerular Injury Is Age-Dependent in Unilaterally Nephrectomized Male Sprague-Dawley Rats. Nephron 1997, 75, 342–349. [Google Scholar] [CrossRef]
- Neugarten, J.; Golestaneh, L.; Kolhe, N.V. Sex differences in acute kidney injury requiring dialysis. BMC Nephrol. 2018, 19, 131. [Google Scholar] [CrossRef]
- Hofer, A.; Wenz, T. Post-translational modification of mitochondria as a novel mode of regulation. Exp. Gerontol. 2014, 56, 202–220. [Google Scholar] [CrossRef]
- Hurd, T.R.; Prime, T.A.; Harbour, M.E.; Lilley, K.S.; Murphy, M. Detection of Reactive Oxygen Species-sensitive Thiol Proteins by Redox Difference Gel Electrophoresis: Implications for mitochondrial redox signaling. J. Biol. Chem. 2007, 282, 22040–22051. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.B.; Van Eyk, J.E.; Marbán, E.; O’Rourke, B. Redox signaling and protein phosphorylation in mitochondria: Progress and prospects. J. Bioenerg. Biomembr. 2009, 41, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.T.; Grimsrud, P.A.; Lai, L.; Draper, J.A.; Fisher-Wellman, K.H.; Narowski, T.M.; Abraham, D.M.; Koves, T.; Kelly, D.P.; Muoio, D.M. Extreme Acetylation of the Cardiac Mitochondrial Proteome Does Not Promote Heart Failure. Circ. Res. 2020, 127, 1094–1108. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thome, T.; Coleman, M.D.; Ryan, T.E. Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice. Cells 2021, 10, 3282. https://doi.org/10.3390/cells10123282
Thome T, Coleman MD, Ryan TE. Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice. Cells. 2021; 10(12):3282. https://doi.org/10.3390/cells10123282
Chicago/Turabian StyleThome, Trace, Madeline D. Coleman, and Terence E. Ryan. 2021. "Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice" Cells 10, no. 12: 3282. https://doi.org/10.3390/cells10123282
APA StyleThome, T., Coleman, M. D., & Ryan, T. E. (2021). Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice. Cells, 10(12), 3282. https://doi.org/10.3390/cells10123282