
Gene Expression Profiling of Mycosis Fungoides in Early and Tumor Stage—A Proof-of-Concept Study Using Laser Capture/Single Cell Microdissection and NanoString Analysis




Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Tissue Staining and Laser Capture Single-Cell Microdissection
2.3. RNA Extraction and Purification
2.4. Reverse Transcriptase Polymerase Chain Reaction (RT–PCR) for the Optimization Step
2.5. cDNA Synthesis and Amplification for NanoString
2.6. NanoString Analysis
2.7. Immunohistochemistry
3. Results
3.1. Optimization of a Protocol to Generate Sufficient cDNA for NanoString
3.2. Generation of cDNA Samples from Singly Dissected MF Cells
3.3. NanoString Analysis
3.4. Target Validation Using Immunohistochemistry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mycosis Fungoides. In WHO Classification of Hematological Malignancy, 4th ed.; Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) World Health Organization Classification of Tumours: Lyon, France, 2017; Volume 2, pp. 385–389. [Google Scholar]
- Vergier, B.; de Muret, A.; Beylot-Barry, M.; Vaillant, L.; Ekouevi, D.; Chene, G.; Carlotti, A.; Franck, N.; Dechelotte, P.; Souteyrand, P.; et al. Transformation of mycosis fungoides: Clinicopathological and prognostic features of 45 cases. French study group of cutaneous lymphomas. Blood 2000, 95, 2212–2218. [Google Scholar] [PubMed]
- Dmitrovsky, E.; Matthews, M.J.; Bunn, P.A.; Schechter, G.P.; Makuch, R.W.; Winkler, C.F.; Eddy, J.; Sausville, E.A.; Ihde, D.C. Cytologic transformation in cutaneous T cell lymphoma: A clinicopathologic entity associated with poor prognosis. J. Clin. Oncol. 1987, 5, 208–215. [Google Scholar] [CrossRef]
- van Santen, S.; Roach, R.E.; van Doorn, R.; Horváth, B.; Bruijn, M.S.; Sanders, C.J.; de Pooter, J.C.; van Rossum, M.M.; de Haas, E.R.; Veraart, J.C.; et al. Clinical Staging and Prognostic Factors in Folliculotropic Mycosis Fungoides. JAMA Dermatol. 2016, 152, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Lei, Q.; Zhang, S.; Kong, L.; Qin, B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol. Rep. 2017, 38, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Morais-Rodrigues, F.; Silverio-Machado, R.; Kato, R.B.; Rodrigues, D.L.N.; Valdez-Baez, J.; Fonseca, V.; San, E.J.; Gomes, L.G.R.; dos Santos, R.G.; Vinicius Canário Viana, M.; et al. Analysis of the microarray gene expression for breast cancer progression after the application modified logistic regression. Gene 2020, 726, 144168. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Hashikawa, K.; Yasumoto, S.; Nakashima, K.; Arakawa, F.; Kiyasu, J.; Kimura, Y.; Saruta, H.; Nakama, T.; Yasuda, K.; Tashiro, K.; et al. Microarray analysis of gene expression by microdissected epidermis and dermis in mycosis fungoides and adult T-cell leukemia/lymphoma. Int. J. Oncol. 2014, 45, 1200–1208. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Y.; Yu, R.; Huang, Y.; Su, M.; Xiao, C.; Martinka, M.; Dutz, J.P.; Zhang, X.; Zheng, Z.; et al. Molecular Markers of Early-Stage Mycosis Fungoides. J. Investig. Dermatol. 2012, 132, 1698–1706. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.; Li, J.; Huang, Y.; Almowaled, M.; Barger, C.J.; Karpf, A.R.; Wang, P.; Chen, W.; Turner, S.D.; Lai, R. NPM-ALK Is a Key Regulator of the Oncoprotein FOXM1 in ALK-Positive Anaplastic Large Cell Lymphoma. Cancers 2019, 11, 1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliafierro, L.; Bonawitz, K.; Glenn, O.C.; Chiba-Falek, O. Gene Expression Analysis of Neurons and Astrocytes Isolated by Laser Capture Microdissection from Frozen Human Brain Tissues. Front. Mol. Neurosci. 2016, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Katz, I.K.; Lamprecht, R. Fear conditioning leads to alteration in specific genes expression in cortical and thalamic neurons that project to the lateral amygdala. J. Neurochem. 2015, 132, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Huso, T.H.; Resar, L.M. The high mobility group A1 molecular switch: Turning on cancer-can we turn it off? Expert Opin. Ther. Targets 2014, 18, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuldenfrei, A.; Belton, A.; Kowalski, J.; Talbot, J.; Conover, C.; Di Cello, F.; Poh, W.; Tsai, H.; Shah, S.N.; Huso, T.H.; et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genom. 2011, 12, 549. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hu, L.; Zheng, Y.; Guo, L. HMGA1 in cancer: Cancer classification by location. J. Cell. Mol. Med. 2019, 23, 2293–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard-Chapeau, E.; Li, S.; Ding, J.; Zhang, S.; Zhu, H.; Princen, F.; Fang, D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 Acts as a Tumor Suppressor in Hepatocellular Carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Hu, F.; Tai, Y.; Du, J.; Mao, B.; Yuan, Z.; Wang, Y.; Wei, L. The tumor suppressor role of Src homology phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 637–646. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef] [Green Version]
- Rampias, T.; Giagini, A.; Siolos, S.; Matsuzaki, H.; Sasaki, C.; Scorilas, A.; Psyrri, A. Crosstalk and Cetuximab Resistance in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 2933–2946. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Fan, H.; Zhang, I.Y.; Liu, B.; Feng, B.; Meng, L.; Zhang, R.; Sadeghi, S.; Guo, H.; Pang, Q. HMGA1 expression in human gliomas and its correlation with tumor proliferation, invasion and angiogenesis. J. Neurooncol. 2012, 106, 543–549. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.; Mondéjar, R.; García-díaz, N.; Cereceda, L.; León, A.; Montes, S.; Durán Vian, C.; Pérez Paredes, M.G.; González-morán, A.; Miguel, V.; et al. Advanced-stage mycosis fungoides: Role of the signal transducer and activator of transcription 3, nuclear factor-κB and nuclear factor of activated T cells pathways. Br. J. Derm. 2020, 182, 16. [Google Scholar] [CrossRef] [PubMed]


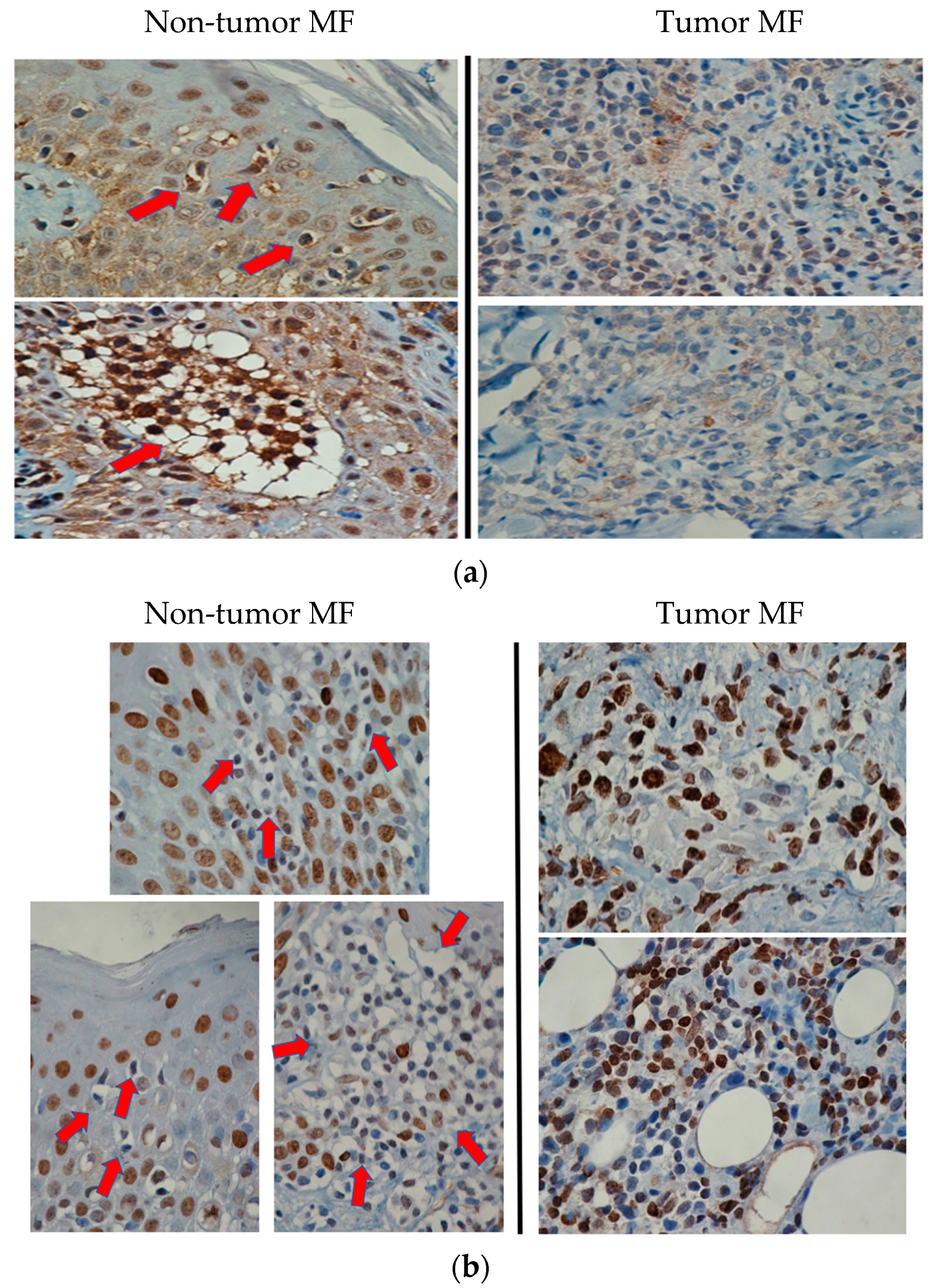

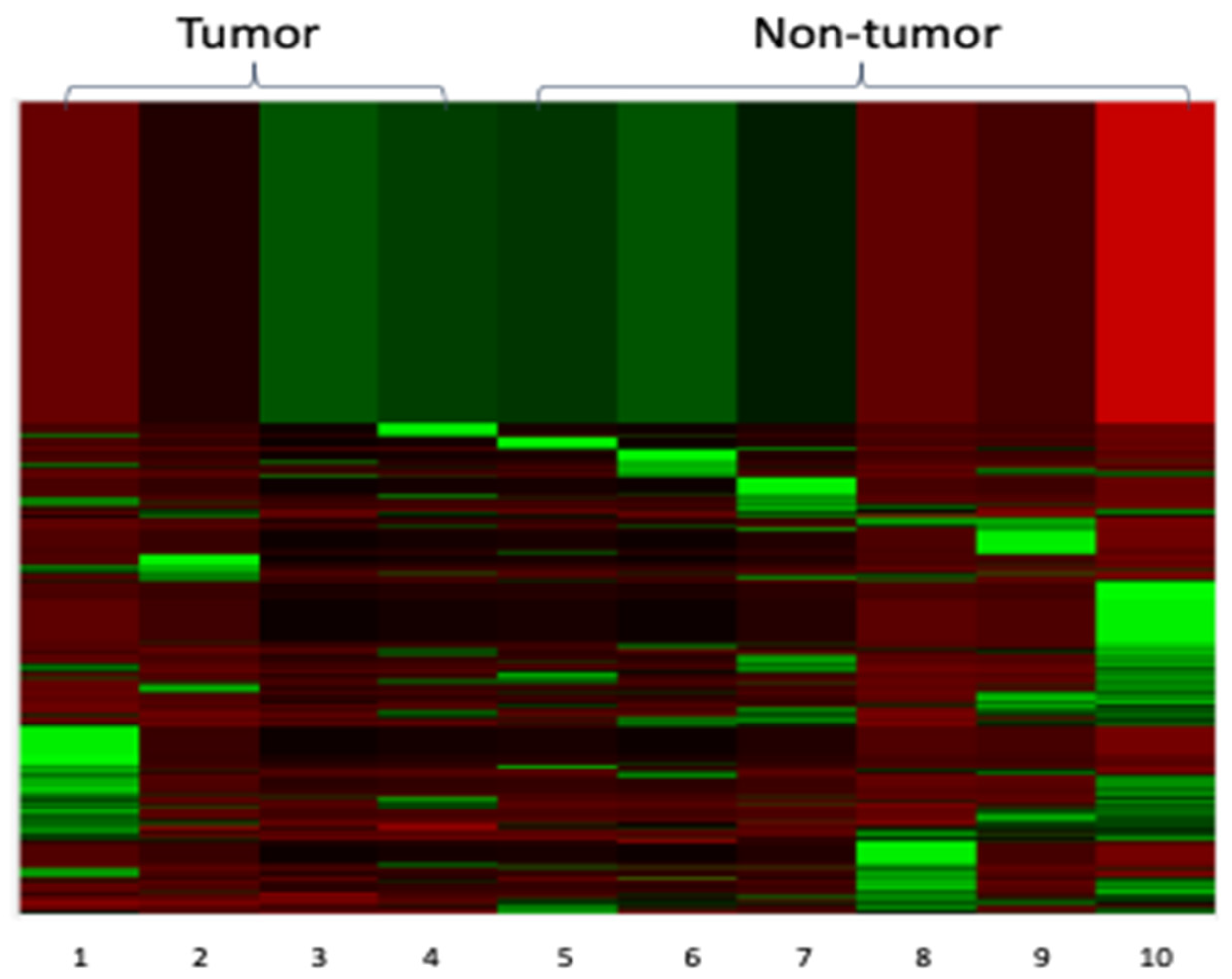{kind=link}
{kind=link}
{kind=link}
| Gene Name | Mean of Non-Tumor MF | Mean of Tumor MF | SD of Non-Tumor MF | SD of Tumor MF | p-Value | Tumor/Non-Tumor |
|---|---|---|---|---|---|---|
| FGF9 | 1.60 | 3.12 | 0.63 | 1.97 | 0.22 | 1.95 |
| B2M | 1.83 | 3.54 | 0.77 | 1.79 | 0.15 | 1.94 |
| COL5A1 | 1.36 | 2.55 | 0.13 | 1.52 | 0.21 | 1.88 |
| PRKACG | 1.55 | 2.89 | 0.38 | 2.16 | 0.31 | 1.86 |
| SSX1 | 1.36 | 2.45 | 0.13 | 1.24 | 0.18 | 1.80 |
| WEE1 | 1.36 | 2.42 | 0.13 | 1.93 | 0.35 | 1.78 |
| HMGA1 | 2.01 | 3.44 | 0.68 | 2.47 | 0.33 | 1.71 |
| ERCC2 | 1.61 | 2.75 | 0.69 | 2.59 | 0.45 | 1.71 |
| BMP5 | 2.03 | 3.24 | 1.16 | 1.42 | 0.21 | 1.60 |
| ITGA3 | 1.55 | 2.48 | 0.38 | 0.67 | 0.06 | 1.60 |
| FGF16 | 2.22 | 3.51 | 0.94 | 1.72 | 0.24 | 1.59 |
| PAK3 | 1.55 | 2.45 | 0.38 | 1.24 | 0.25 | 1.58 |
| TGFB3 | 1.36 | 2.13 | 0.13 | 0.82 | 0.15 | 1.57 |
| DUSP6 | 1.36 | 2.11 | 0.13 | 1.34 | 0.34 | 1.56 |
| BAIAP3 | 1.81 | 2.81 | 0.72 | 1.02 | 0.15 | 1.55 |
| BCL2 | 1.36 | 2.10 | 0.13 | 0.69 | 0.12 | 1.55 |
| RASA4 | 1.36 | 2.10 | 0.13 | 0.69 | 0.12 | 1.55 |
| DDB2 | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| FGF2 | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| GNG4 | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| LEFTY2 | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| MAP2K6 | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| PPP3CC | 1.36 | 2.09 | 0.13 | 1.27 | 0.33 | 1.54 |
| NPM1 | 2.13 | 3.17 | 1.78 | 2.03 | 0.44 | 1.49 |
| GRIN2A | 1.77 | 2.47 | 0.56 | 1.26 | 0.36 | 1.39 |
| MLLT3 | 1.74 | 2.42 | 0.84 | 1.93 | 0.55 | 1.39 |
| SPRY4 | 1.79 | 2.47 | 0.64 | 1.26 | 0.38 | 1.38 |
| STAT3 | 1.79 | 2.47 | 0.64 | 1.26 | 0.38 | 1.38 |
| FGF23 | 1.55 | 2.13 | 0.38 | 0.82 | 0.26 | 1.38 |
| MCM7 | 2.05 | 2.76 | 0.79 | 1.80 | 0.50 | 1.35 |
| Gene Name | Mean of Non-Tumor MF | Mean of Tumor MF | SD of Non-Tumor MF | SD of Tumor MF | p-Value | Tumor/Non-Tumor |
|---|---|---|---|---|---|---|
| BID | 3.54 | 1.43 | 1.67 | 0.09 | 0.03 | 0.40 |
| SIN3A | 5.77 | 2.48 | 1.89 | 0.67 | 0.01 | 0.43 |
| PTPN11 | 3.04 | 1.43 | 2.25 | 0.09 | 0.14 | 0.47 |
| NKD1 | 2.94 | 1.43 | 0.57 | 0.09 | 0.00 | 0.48 |
| EFNA3 | 5.32 | 2.55 | 4.29 | 2.31 | 0.22 | 0.48 |
| EFNA2 | 2.89 | 1.43 | 0.86 | 0.09 | 0.01 | 0.49 |
| EPOR | 3.60 | 1.77 | 1.60 | 0.65 | 0.04 | 0.49 |
| HNF1A | 2.93 | 1.43 | 1.03 | 0.09 | 0.02 | 0.49 |
| HGF | 3.70 | 1.80 | 1.61 | 0.80 | 0.04 | 0.49 |
| CAMK2B | 3.51 | 1.77 | 1.91 | 0.65 | 0.08 | 0.50 |
| DDIT3 | 2.79 | 1.43 | 1.39 | 0.09 | 0.06 | 0.51 |
| SFRP1 | 2.80 | 1.43 | 1.77 | 0.09 | 0.12 | 0.51 |
| C19orf40 | 2.76 | 1.43 | 1.40 | 0.09 | 0.07 | 0.52 |
| PRKAR1B | 2.67 | 1.43 | 1.42 | 0.09 | 0.08 | 0.53 |
| ITGA2 | 2.62 | 1.43 | 1.72 | 0.09 | 0.15 | 0.54 |
| PGF | 2.62 | 1.43 | 1.72 | 0.09 | 0.15 | 0.54 |
| TNFRSF10A | 2.64 | 1.43 | 1.52 | 0.09 | 0.11 | 0.54 |
| SMC3 | 3.77 | 2.09 | 1.65 | 1.27 | 0.11 | 0.55 |
| SOS1 | 3.14 | 1.76 | 1.25 | 0.60 | 0.05 | 0.56 |
| KIT | 2.49 | 1.43 | 1.41 | 0.09 | 0.12 | 0.57 |
| DTX1 | 2.50 | 1.43 | 1.09 | 0.09 | 0.06 | 0.57 |
| PKMYT1 | 3.16 | 1.80 | 0.65 | 0.80 | 0.03 | 0.57 |
| IL22RA2 | 2.50 | 1.43 | 1.13 | 0.09 | 0.07 | 0.57 |
| DKK4 | 3.09 | 1.76 | 1.43 | 0.60 | 0.08 | 0.57 |
| U2AF1 | 2.47 | 1.43 | 1.32 | 0.09 | 0.11 | 0.58 |
| WNT2 | 3.05 | 1.76 | 2.02 | 0.60 | 0.19 | 0.58 |
| MMP7 | 2.48 | 1.43 | 0.99 | 0.09 | 0.05 | 0.58 |
| LAMA1 | 3.53 | 2.09 | 2.31 | 1.27 | 0.24 | 0.59 |
| DAXX | 2.41 | 1.43 | 1.64 | 0.09 | 0.20 | 0.59 |
| CREB5 | 4.79 | 2.81 | 2.51 | 1.02 | 0.13 | 0.59 |
| Downregulated in Tumor MF | Upregulated in Tumor MF | ||||
|---|---|---|---|---|---|
| Gene | Log2 Fold Change | p-Value | Gene | Log2 Fold Change | p-Value |
| SIN3A | −1.31 | 0.0728 | B2M | 0.908 | 0.232 |
| BID | −1.41 | 0.127 | FGF9 | 0.945 | 0.241 |
| HGF | −1.18 | 0.156 | HMGA1 | 0.727 | 0.318 |
| RHOA * | −0.601 | 0.159 | |||
| EPOR | −1.1 | 0.187 | |||
| CAMK2B | −1.09 | 0.189 | |||
| CREB5 | −0.866 | 0.196 | |||
| PTPN11 | −1.18 | 0.201 | |||
| NKD1 | −1.12 | 0.221 | |||
| HNF1A | −1.12 | 0.222 | |||
| EFNA2 | −1.12 | 0.222 | |||
| SMC3 | −0.925 | 0.226 | |||
| SFRP1 | −1.1 | 0.231 | |||
| PKMYT1 | −0.905 | 0.273 | |||
| C19orf40 | −1 | 0.273 | |||
| DDIT3 | −1 | 0.273 | |||
| SOS1 | −0.902 | 0.275 | |||
| DKK4 | −0.901 | 0.275 | |||
| PRKAR1B | −0.997 | 0.276 | |||
| TNFRSF10A | −0.995 | 0.277 | |||
| WNT2 * | −0.892 | 0.281 | |||
| LAMA1 | −0.819 | 0.286 | |||
| ITGA2 | −0.978 | 0.288 | |||
| PGF | −0.978 | 0.288 | |||
| MEN1 * | −0.795 | 0.335 | |||
| GPC4 * | −0.792 | 0.337 | |||
| GLI3 * | −0.791 | 0.338 | |||
| Pathway | Upregulated Genes | Downregulated Genes |
|---|---|---|
| PI3K | FGF9, COL5A1, ITGA3, FGF16, BCL2, FGF2, GNG4, FGF23 | EFNA3, EFNA2, EPOR, HGF, ITGA2, PGF, SOS1, KIT, LAMA1, CREB5 |
| RAS | FGF9, PRKACG, FGF16, PAK3, RASA4, FGF2, GNG4, GRIN2A, FGF23 | PTPN11, EFNA3, EFNA2, HGF, PGF, SOS1, KIT |
| Cell Cycle and Apoptosis | PRKACG, WEE1, TGFB3, BCL2, DDB2, PPP3CC, MCM7 | BID, PRKAR1B, TNFRSF10A, SMC3, PKMTY1 |
| MAPK | FGF9, PRKACG, FGF16, TGFB3, DUSP6, FGF2, MAP2K6, PPP3CC, FGF23 | DDIT3, SOS1, DAXX |
| Wnt | PRKACG, PPP3CC | NKD1, CAMK2B, SFRP1, DKK4, WNT2, MMP7 |
| Driver Gene | B2M, BCL2, NPM1 | PTPN11, HNF1A, KIT, U2AF1, DAXX |
| JAK-STAT | SPRY4, STAT3 | PTPN11, EPOR, SOS1, IL22RA2 |
| Transcriptional Regulation | SSX1, DUSP6, BAIAP3, MLLT3 | SIN3A, DDIT3 |
| TGF-Beta | BMP5, TGFB3, LEFTY2 | |
| DNA Repair | ERCC2 | C19orf40 |
| Hedge Hog | PRKACG | WNT2 |
| Chromatin Modification | HMGA1 | |
| Notch | DTX1 |
| Patient No. | Non-Tumor MF or Tumor MF | NanoString Data | SHP2 | HMGA1 |
|---|---|---|---|---|
| 1 | non-tumor | Yes | >50% | ≤50% |
| 1 | tumor | Yes | ≤50% | >50% |
| 1 | tumor | Yes | ≤50% | >50% |
| 2 | non-tumor | Yes | >50% | ≤50% |
| 2 | tumor | Yes | ≤50% | >50% |
| 3 | tumor | Yes | ≤50% | >50% |
| 4 | non-tumor | No | >50% | ≤50% |
| 4 | tumor | No | ≤50% | >50% |
| 5 | non-tumor | No | >50% | ≤50% |
| 5 | non-tumor | No | >50% | ≤50% |
| 6 | non-tumor | No | >50% | ≤50% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, J.; Li, J.; Gniadecki, R.; Lai, R. Gene Expression Profiling of Mycosis Fungoides in Early and Tumor Stage—A Proof-of-Concept Study Using Laser Capture/Single Cell Microdissection and NanoString Analysis. Cells 2021, 10, 3190. https://doi.org/10.3390/cells10113190
Lai J, Li J, Gniadecki R, Lai R. Gene Expression Profiling of Mycosis Fungoides in Early and Tumor Stage—A Proof-of-Concept Study Using Laser Capture/Single Cell Microdissection and NanoString Analysis. Cells. 2021; 10(11):3190. https://doi.org/10.3390/cells10113190
Chicago/Turabian StyleLai, Justine, Jing Li, Robert Gniadecki, and Raymond Lai. 2021. "Gene Expression Profiling of Mycosis Fungoides in Early and Tumor Stage—A Proof-of-Concept Study Using Laser Capture/Single Cell Microdissection and NanoString Analysis" Cells 10, no. 11: 3190. https://doi.org/10.3390/cells10113190
APA StyleLai, J., Li, J., Gniadecki, R., & Lai, R. (2021). Gene Expression Profiling of Mycosis Fungoides in Early and Tumor Stage—A Proof-of-Concept Study Using Laser Capture/Single Cell Microdissection and NanoString Analysis. Cells, 10(11), 3190. https://doi.org/10.3390/cells10113190