
Identification of Cellular Factors Required for SARS-CoV-2 Replication

 , , and
, , and
Abstract
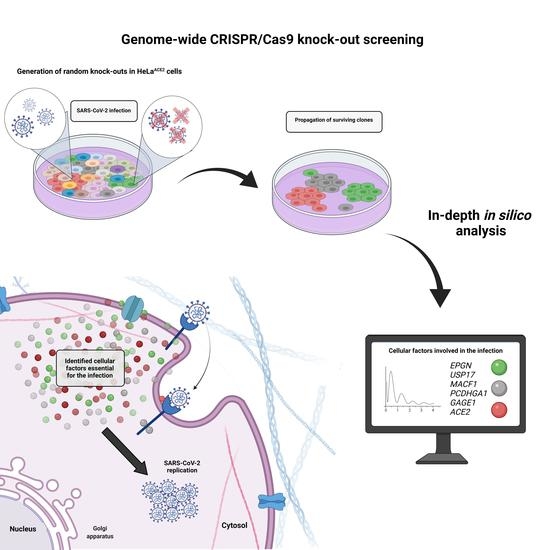
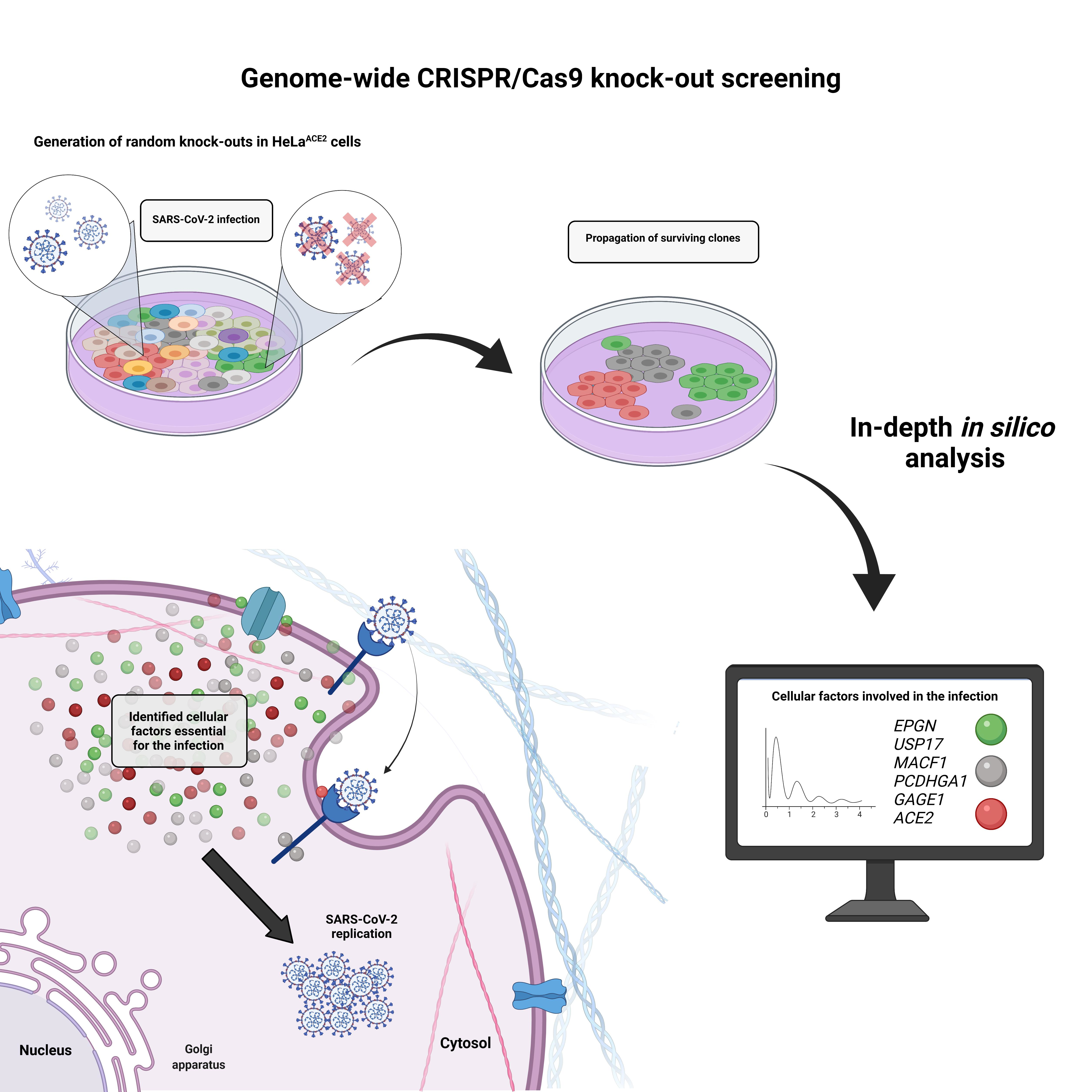
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus
2.3. GeCKO Plasmids Library Preparation
2.4. Lentiviral Particle Production
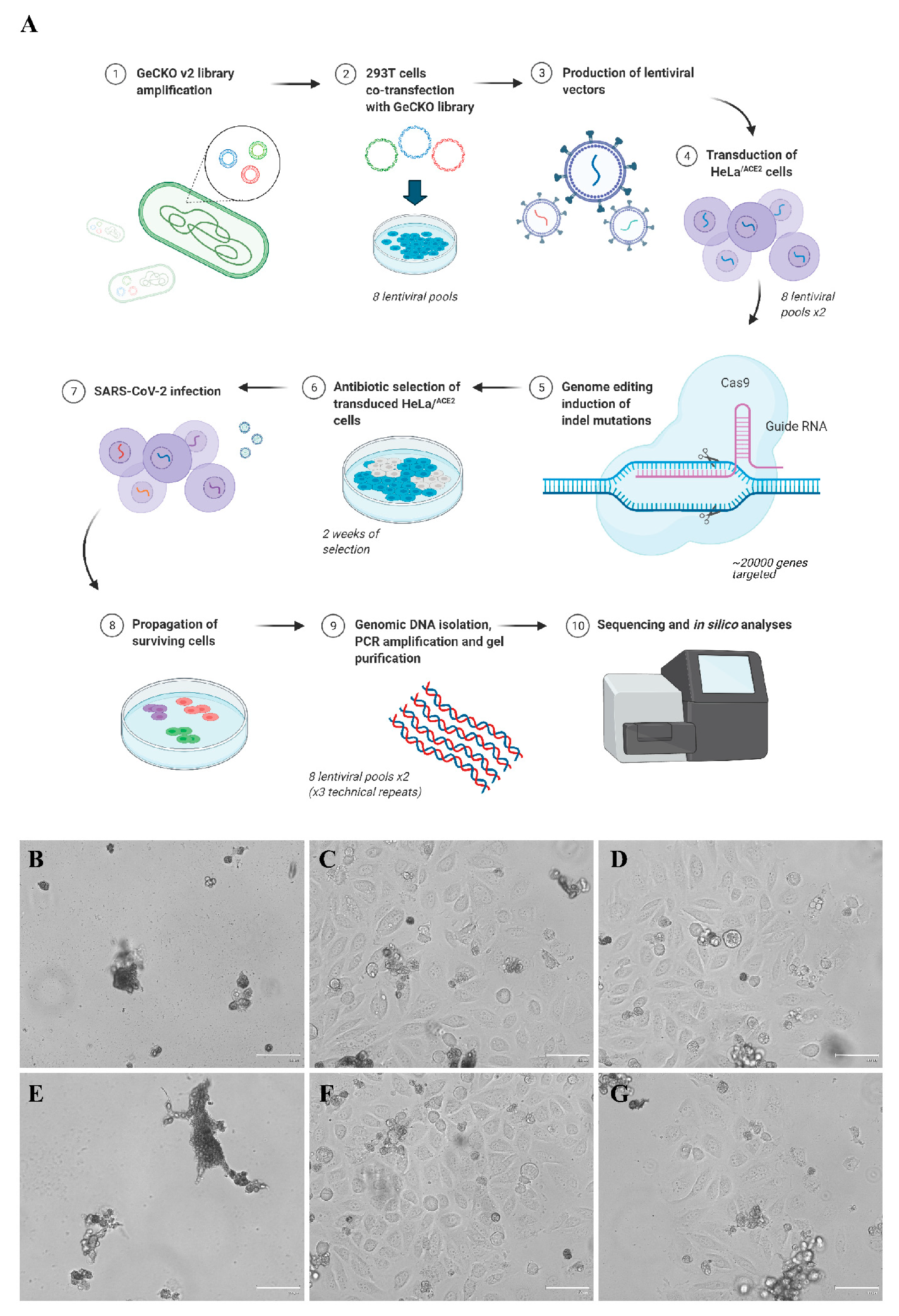
2.5. Transduction, Selection, and Genome-Wide Screening
2.6. Generation of HeLaACE2 Cells
2.7. Generation of A549ACE2/TMPRSS2 Cells
2.8. Immunofluorescence Assay
2.9. Genomic DNA Isolation
2.10. Preparation of NGS Library
2.11. Sequencing and Data Processing
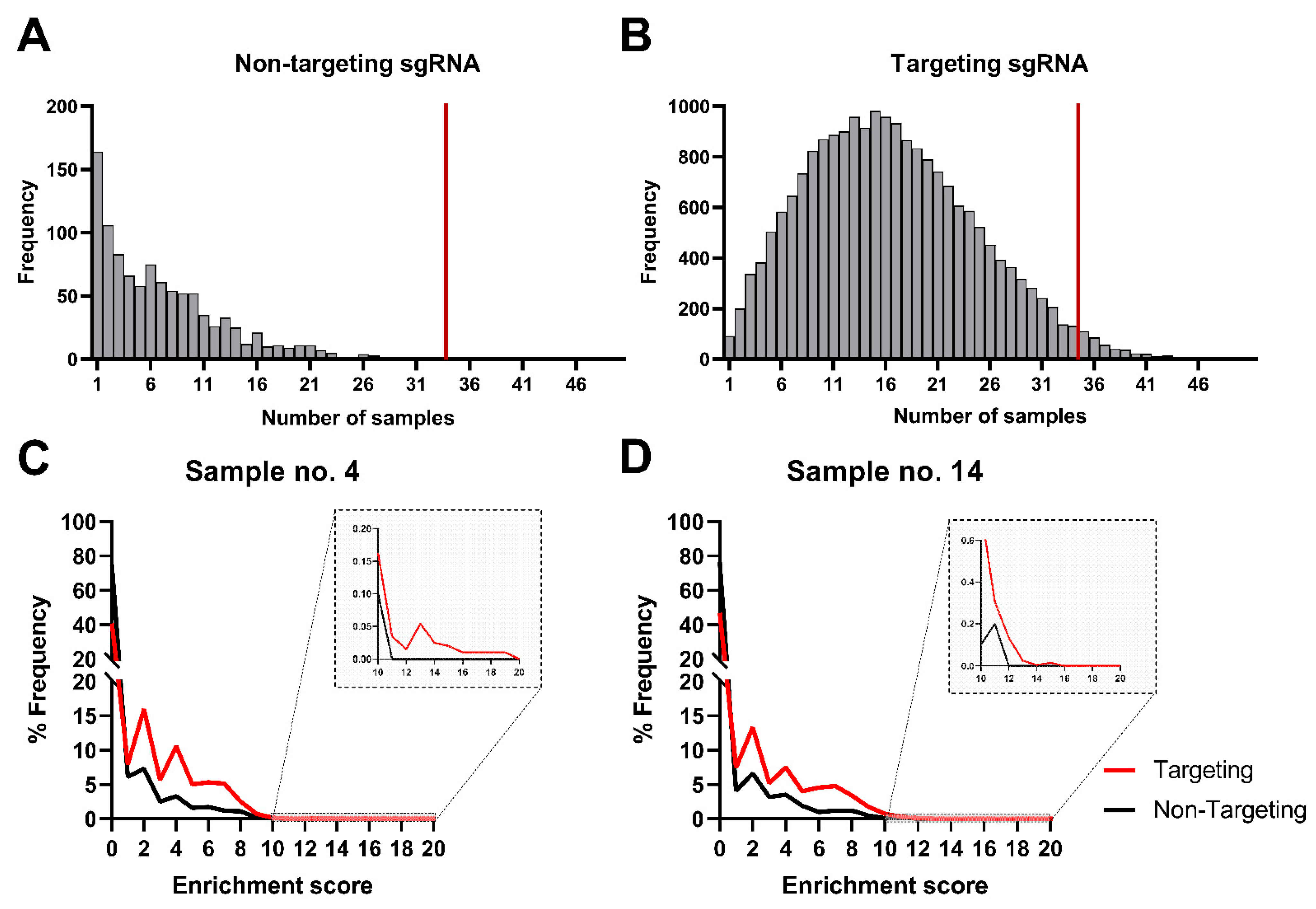
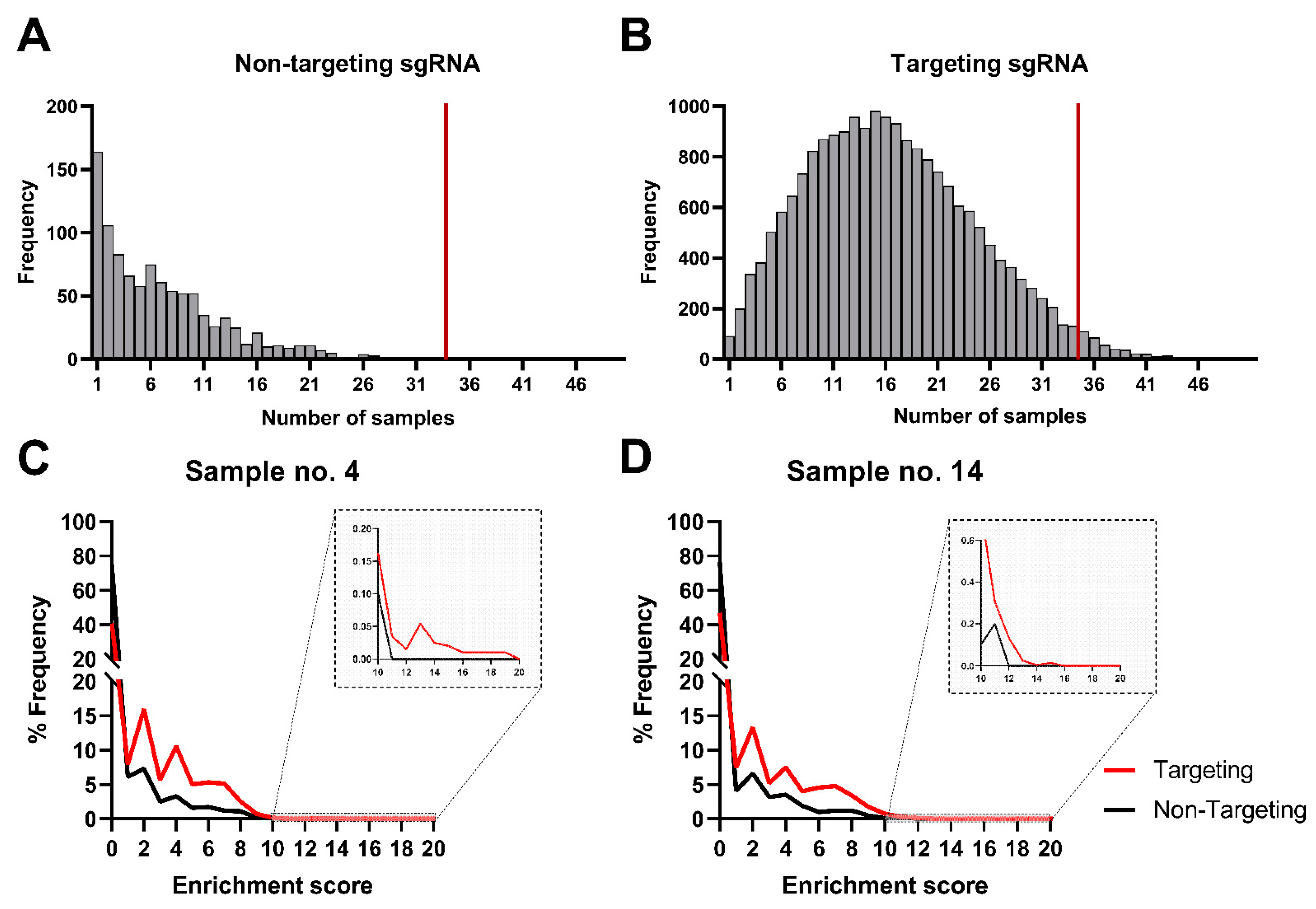
2.12. Between- and Within-Sample Normalization and Enrichment Score Calculations
2.13. Selection of Candidate Genes and Gene Set Enrichment Analysis
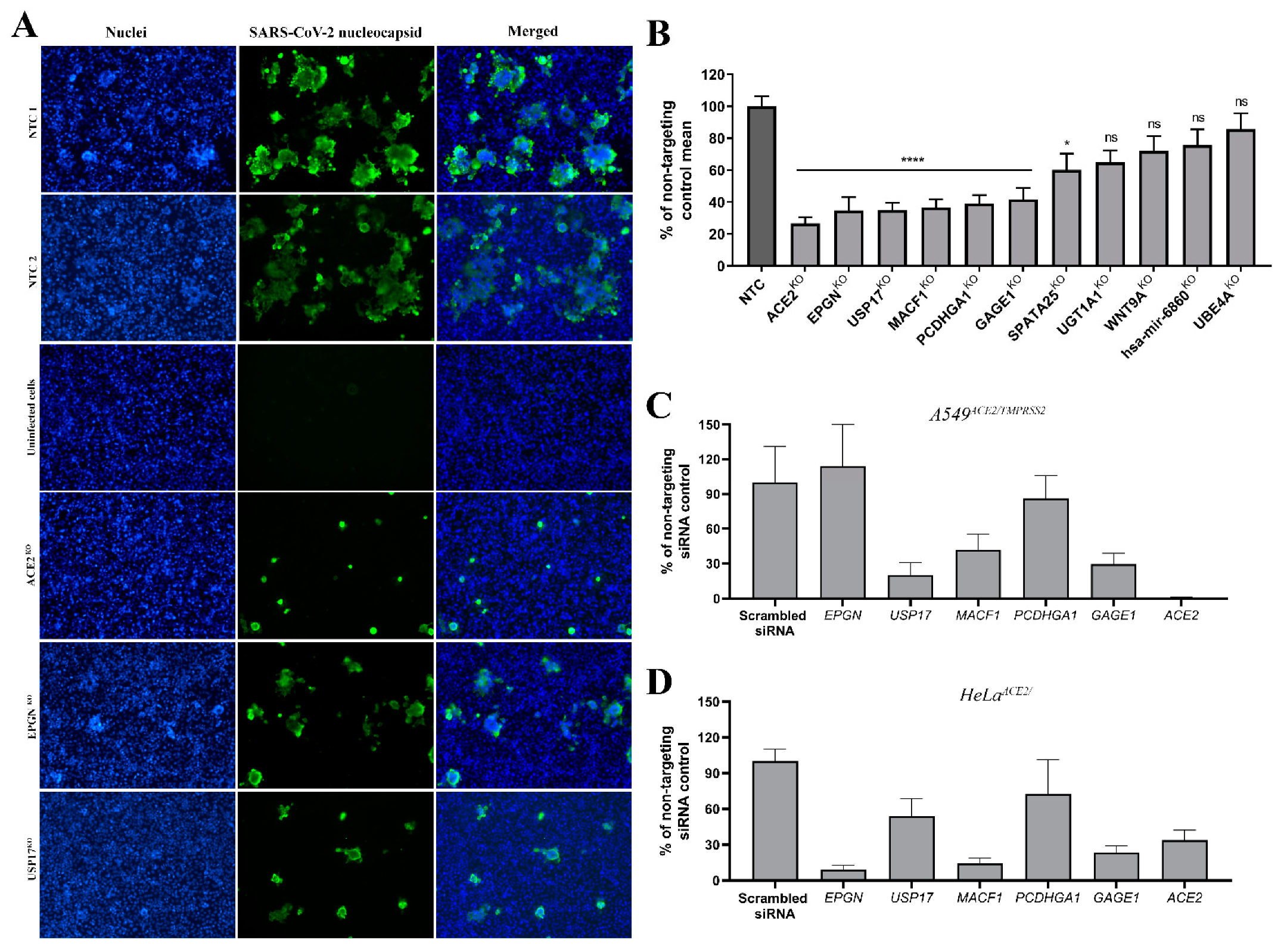
2.14. Gene Knock-Out by CRISPR/Cas9
2.15. siRNA Silencing of Target Genes
2.16. Virus Replication Assay
2.17. RNA Isolation
2.18. Reverse Transcription
2.19. SYBR qPCR
2.20. RT-qPCR (Viral RNA Yield Analysis)
2.21. Statistical Analysis of in Vitro Experiments
3. Results and Discussion
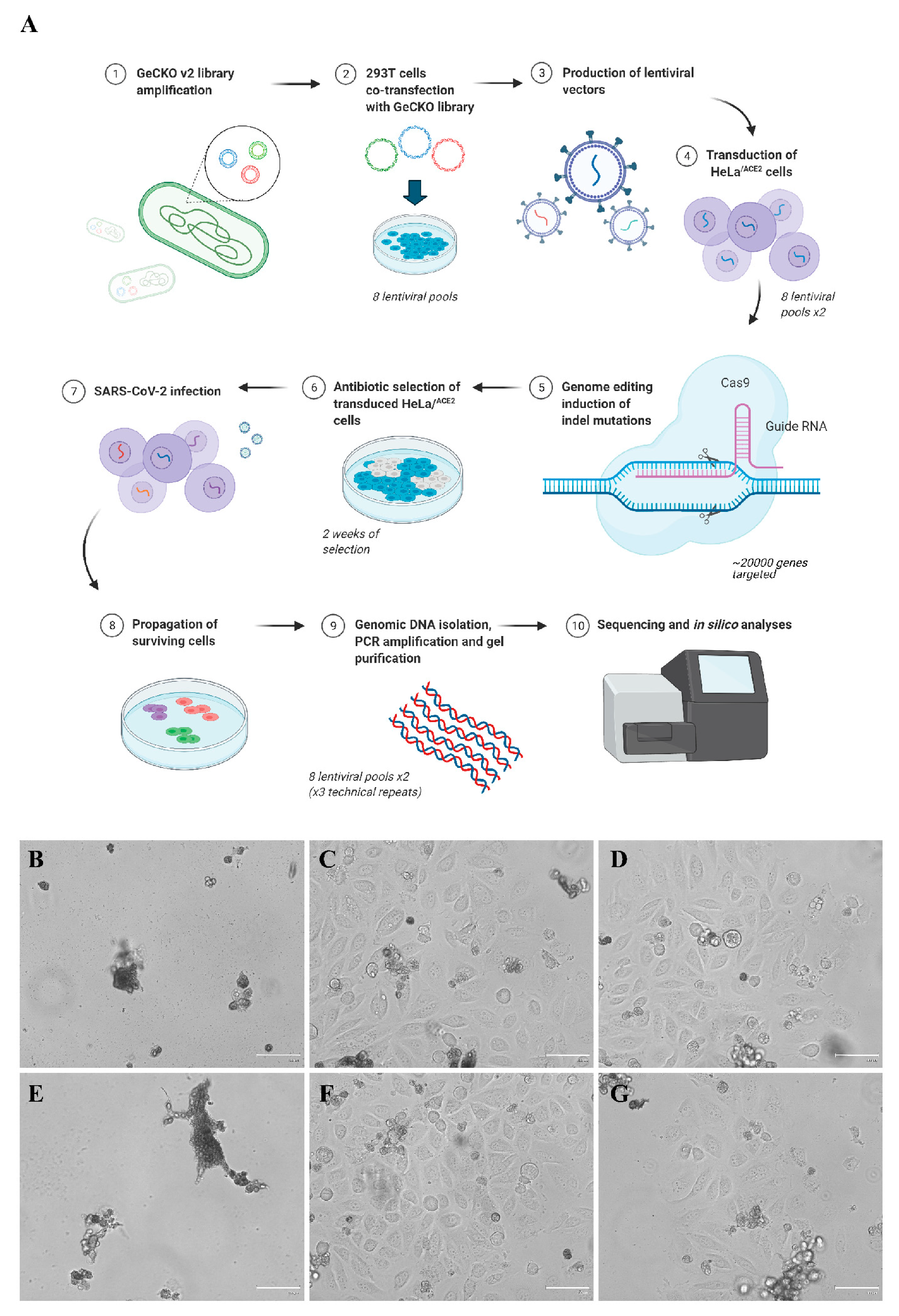
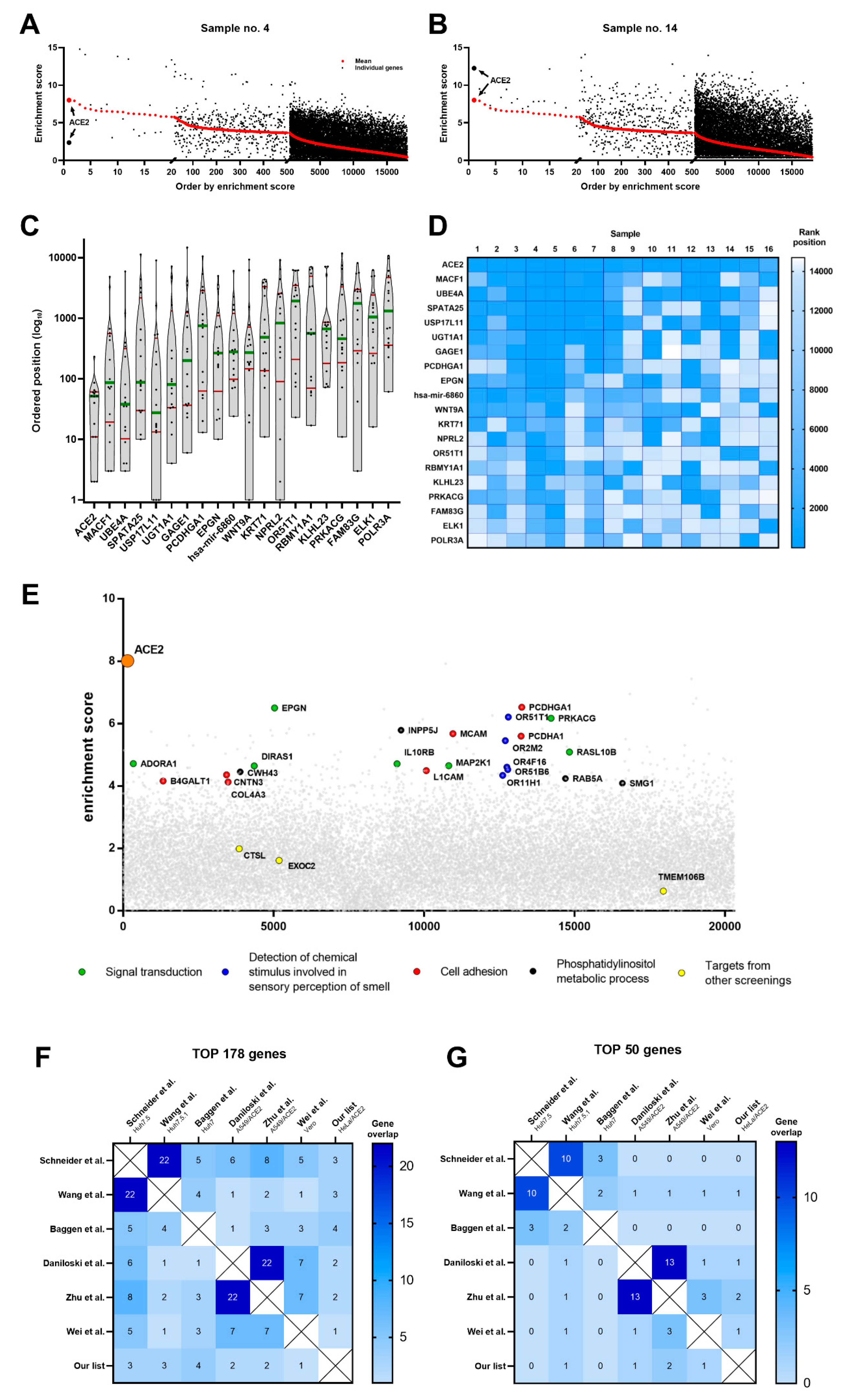
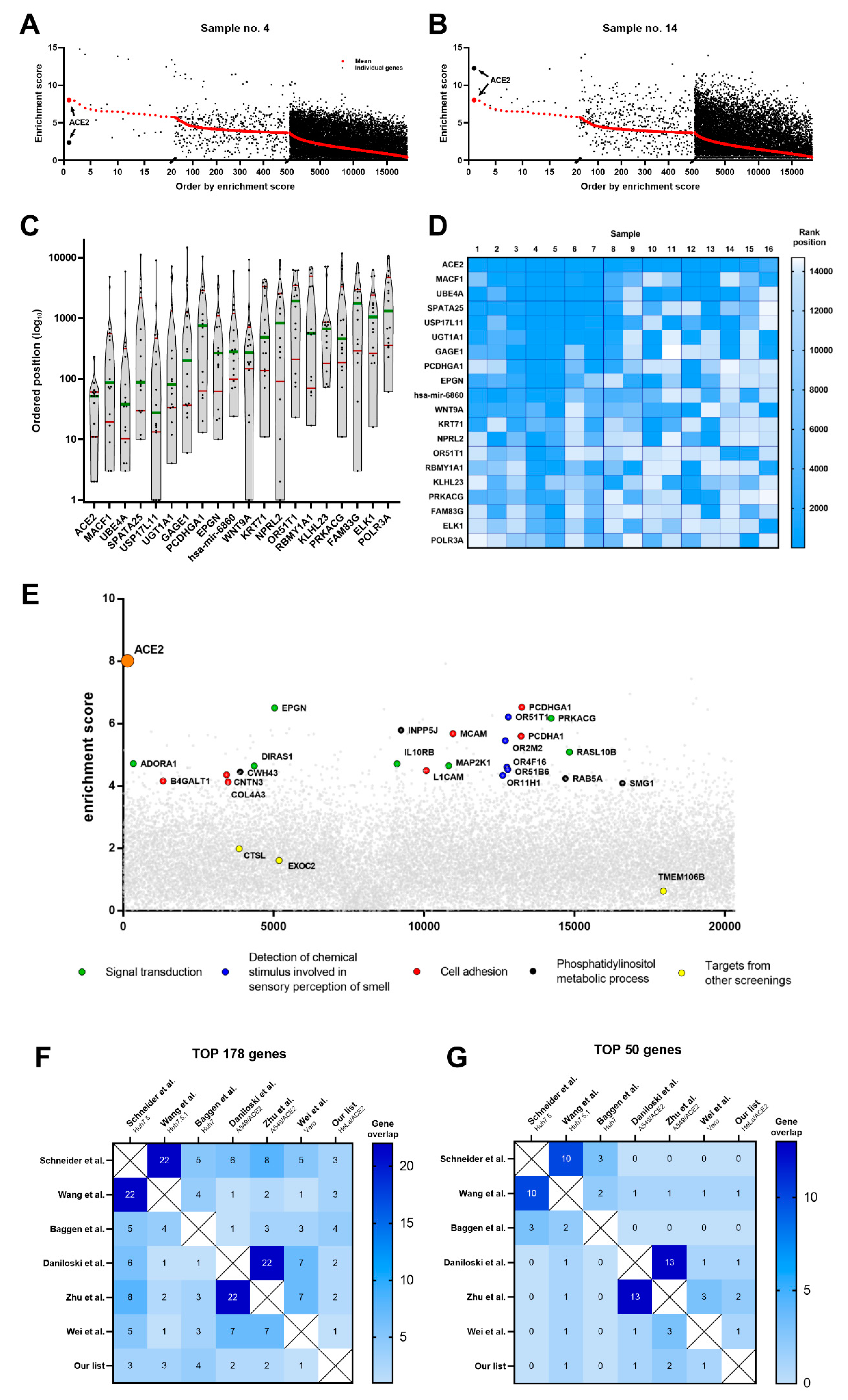
3.1. CRISPR-Cas9 Genome-Wide Screening
3.2. In Silico Analysis of Cellular Targets
3.3. Pathway Analysis
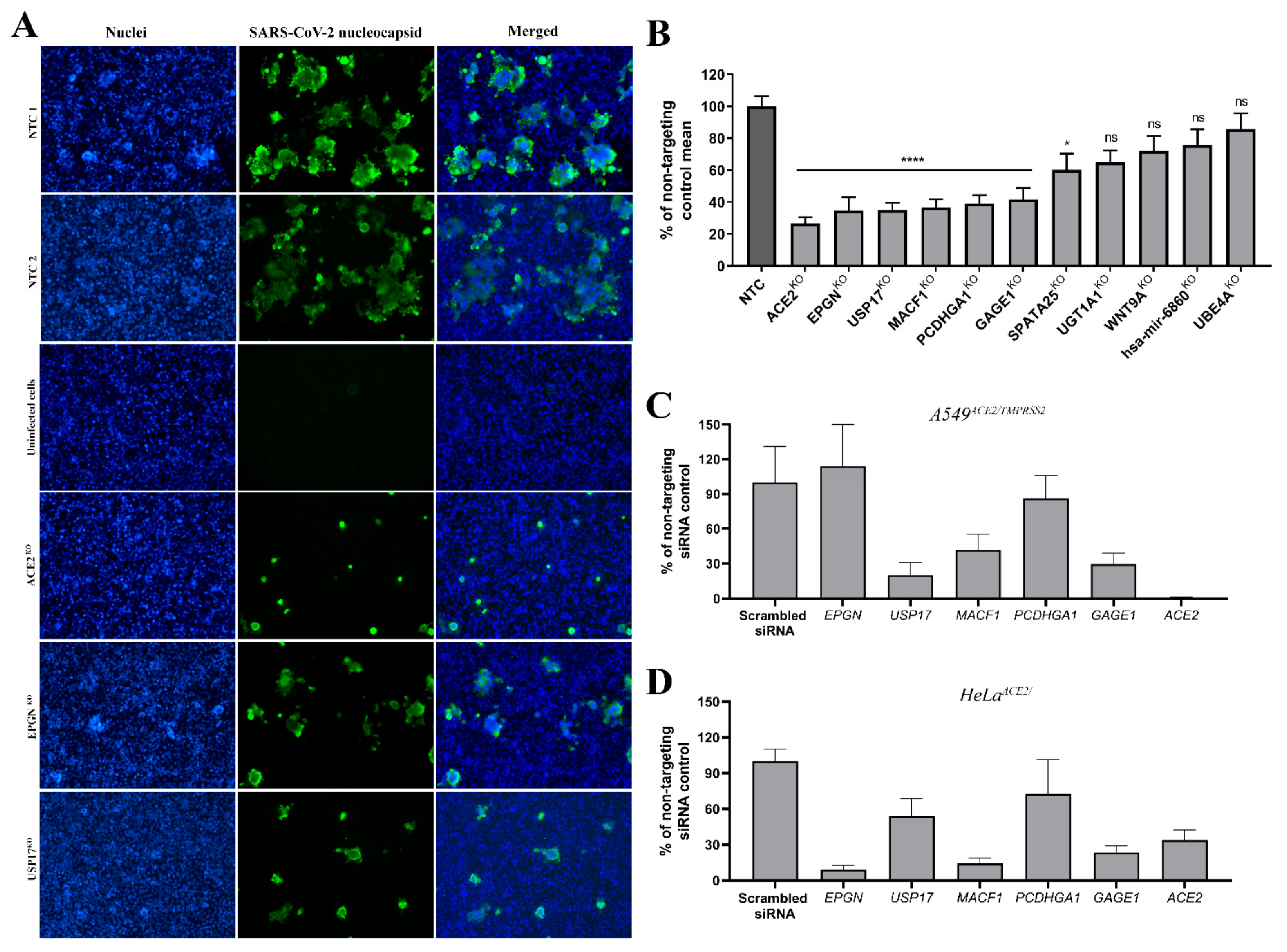
3.4. Validation of Top Hits
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Al-Khannaq, M.N.; Ng, K.T.; Oong, X.Y.; Pang, Y.K.; Takebe, Y.; Chook, J.B.; Hanafi, N.S.; Kamarulzaman, A.; Tee, K.K. Molecular epidemiology and evolutionary histories of human coronavirus OC43 and HKU1 among patients with upper respiratory tract infections in Kuala Lumpur, Malaysia. Virol. J. 2016, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Barh, D.; Aljabali, A.A.; Tambuwala, M.M.; Tiwari, S.; Serrano-Aroca, A.; Alzahrani, K.J.; Silva Andrade, B.; Azevedo, V.; Ganguly, N.K.; Lundstrom, K. Predicting COVID-19-Comorbidity Pathway Crosstalk-Based Targets and Drugs: Towards Personalized COVID-19 Management. Biomedicines 2021, 9, 556. [Google Scholar] [CrossRef]
- Barh, D.; Tiwari, S.; Gabriel Rodrigues Gomes, L.; Weener, M.E.; Alzahrani, K.J.; Alsharif, K.F.; Aljabali, A.A.A.; Tambuwala, M.M.; Lundstrom, K.; Hassan, S.S.; et al. Potential Molecular Mechanisms of Rare Anti-Tumor Immune Response by SARS-CoV-2 in Isolated Cases of Lymphomas. Viruses 2021, 13, 1927. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nat. Microbiol. 2021, 6, 1219–1232. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e116. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sánchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132.e114. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e114. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Omer Javed, A.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Brandolini, M.; Taddei, F.; Marino, M.M.; Grumiro, L.; Scalcione, A.; Turba, M.E.; Gentilini, F.; Fantini, M.; Zannoli, S.; Dirani, G.; et al. Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management. Viruses 2021, 13, 1022. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- R-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology, R package version, 2(0); R Foundation for Statistical Computing: Vienna, Austria, 2010. [Google Scholar]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synowiec, A.; Gryniuk, I.; Pachota, M.; Strzelec, Ł.; Roman, O.; Kłysik-Trzciańska, K.; Zając, M.; Drebot, I.; Gula, K.; Andruchowicz, A.; et al. Cat flu: Broad spectrum polymeric antivirals. Antivir. Res. 2019, 170, 104563. [Google Scholar] [CrossRef]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Tsui, C.K.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017, 8, 15178. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Synowiec, A.; Szczepański, A.; Barreto-Duran, E.; Lie, L.K.; Pyrc, K. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): A Systemic Infection. Clin. Microbiol. Rev. 2021, 34, e00133-20. [Google Scholar] [CrossRef] [PubMed]
- Widera, M.; Wilhelm, A.; Toptan, T.; Raffel, J.M.; Kowarz, E.; Roesmann, F.; Grozinger, F.; Siemund, A.L.; Luciano, V.; Kulp, M.; et al. Generation of a Sleeping Beauty Transposon-Based Cellular System for Rapid and Sensitive Screening for Compounds and Cellular Factors Limiting SARS-CoV-2 Replication. Front. Microbiol. 2021, 12, 701198. [Google Scholar] [CrossRef] [PubMed]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695.e618. [Google Scholar] [CrossRef] [Green Version]
- Klann, K.; Bojkova, D.; Tascher, G.; Ciesek, S.; Münch, C.; Cinatl, J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell. 2020, 80, 164–174.e164. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, L.; Zhong, B.; Tan, B.; Liu, Y.; Shu, H.B. The ubiquitin-specific protease 17 is involved in virus-triggered type I IFN signaling. Cell Res. 2010, 20, 802–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.H.; Yeh, D.W.; Lai, C.Y.; Liu, Y.L.; Huang, L.R.; Lee, A.Y.; Jin, C.S.; Chuang, T.H. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 2018, 37, 6327–6340. [Google Scholar] [CrossRef]
- Jaworski, J.; de la Vega, M.; Fletcher, S.J.; McFarlane, C.; Greene, M.K.; Smyth, A.W.; Van Schaeybroeck, S.; Johnston, J.A.; Scott, C.J.; Rappoport, J.Z.; et al. USP17 is required for clathrin mediated endocytosis of epidermal growth factor receptor. Oncotarget 2014, 5, 6964–6975. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Park, H.H.; Kim, H.; Kim, H.N.; Kim, I.; Jeon, S.; Kim, W.; Bae, J.S.; Lee, W. Wnt5a and Wnt11 as acute respiratory distress syndrome biomarkers for severe acute respiratory syndrome coronavirus 2 patients. Eur. Respir. J. 2020, 56, 2001531. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Interplay of Opposing Effects of the WNT/β-Catenin Pathway and PPARγ and Implications for SARS-CoV2 Treatment. Front. Immunol. 2021, 12, 666693. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, H.; Dauphars, D.J.; He, Y.W. A Potential Role of Interleukin 10 in COVID-19 Pathogenesis. Trends Immunol. 2021, 42, 3–5. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Synowiec, A.; Jedrysik, M.; Branicki, W.; Klajmon, A.; Lei, J.; Owczarek, K.; Suo, C.; Szczepanski, A.; Wang, J.; Zhang, P.; et al. Identification of Cellular Factors Required for SARS-CoV-2 Replication. Cells 2021, 10, 3159. https://doi.org/10.3390/cells10113159
Synowiec A, Jedrysik M, Branicki W, Klajmon A, Lei J, Owczarek K, Suo C, Szczepanski A, Wang J, Zhang P, et al. Identification of Cellular Factors Required for SARS-CoV-2 Replication. Cells. 2021; 10(11):3159. https://doi.org/10.3390/cells10113159
Chicago/Turabian StyleSynowiec, Aleksandra, Malwina Jedrysik, Wojciech Branicki, Adrianna Klajmon, Jing Lei, Katarzyna Owczarek, Chen Suo, Artur Szczepanski, Jingru Wang, Pengyan Zhang, and et al. 2021. "Identification of Cellular Factors Required for SARS-CoV-2 Replication" Cells 10, no. 11: 3159. https://doi.org/10.3390/cells10113159
APA StyleSynowiec, A., Jedrysik, M., Branicki, W., Klajmon, A., Lei, J., Owczarek, K., Suo, C., Szczepanski, A., Wang, J., Zhang, P., Labaj, P. P., & Pyrc, K. (2021). Identification of Cellular Factors Required for SARS-CoV-2 Replication. Cells, 10(11), 3159. https://doi.org/10.3390/cells10113159