
Comprehensive Analysis of microRNAs in Human Adult Erythropoiesis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ex-Vivo Erythropoiesis
2.2. Small RNA Sequencing and Analysis of Differential Expression of miRNAs
2.3. miRNA Target Analysis
2.4. ChIP-Sequencing and ATAC-Seq Analysis
2.5. gRNA Design
2.6. Generation of Single and Dual sgRNA Lentiviral Expression Plasmids
2.7. Culture and Differentiation of HUDEP-2 Cells
2.8. Generation of Lentiviruses
2.9. Generation of Cas9-HUDEP-2 Cells
2.10. Lentiviral Transduction for miRNA Editing
2.11. Flow Cytometric Analysis
2.12. Real-Time PCR Analysis of miRNA Expression
2.13. Globin Chain Analysis by High-Performance Liquid Chromatography (HPLC)
3. Results
3.1. Ex-Vivo Erythropoiesis
3.2. Small RNA Sequencing of Cultured Erythroid Cells
3.3. Erythroid TF Occupancy and Chromatin Accessibility
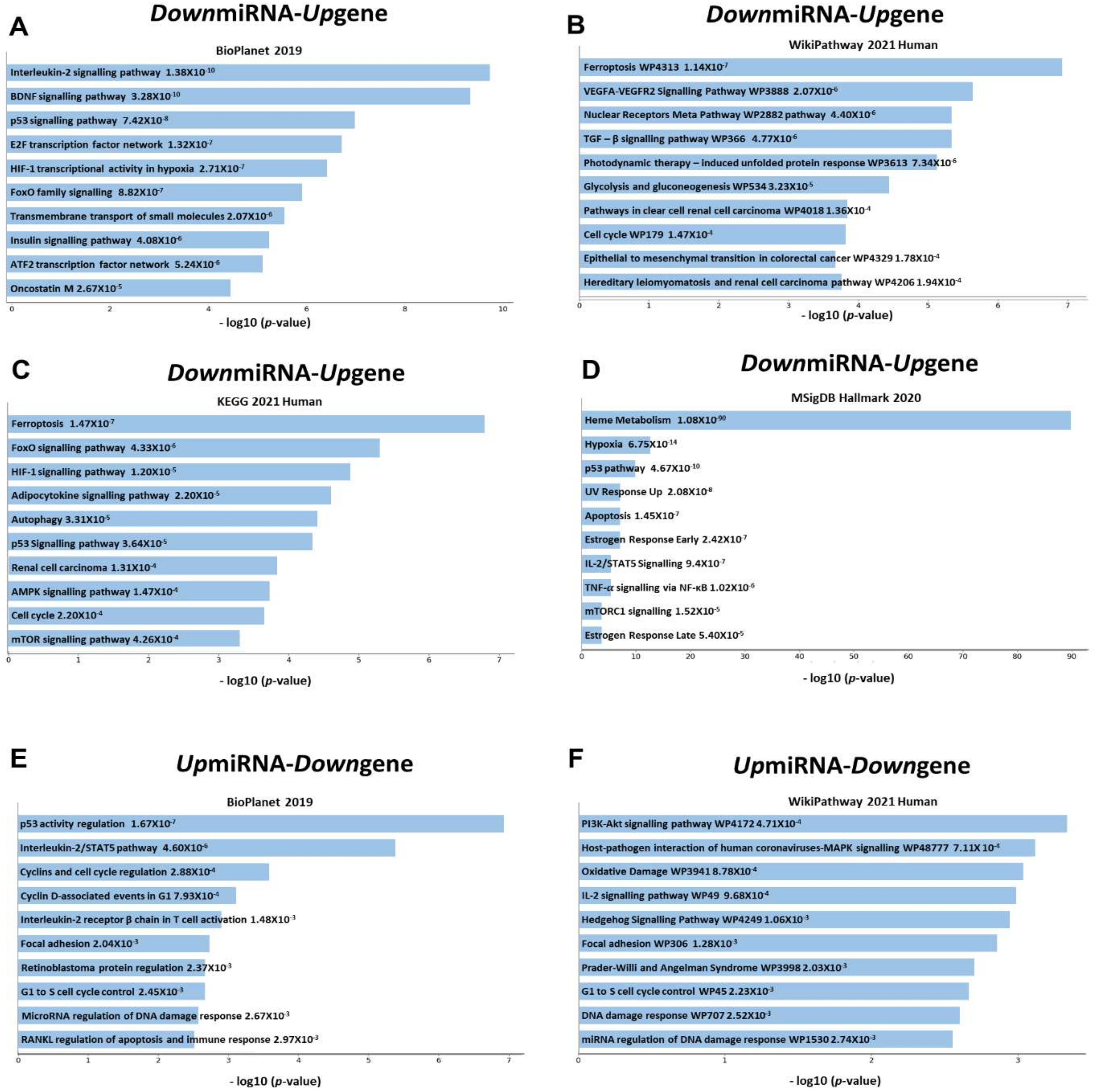
3.4. Pathway Analysis
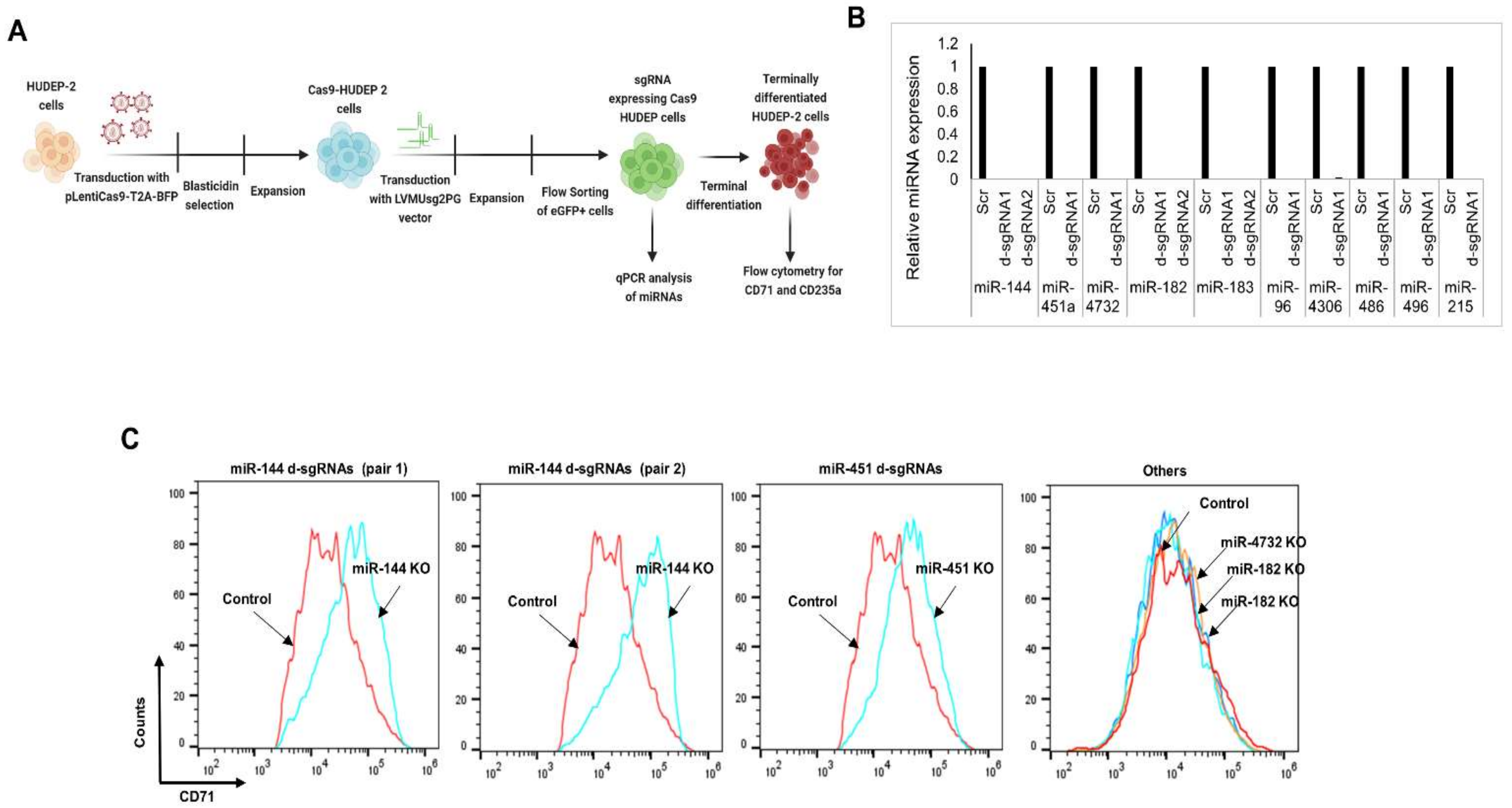
3.5. CRISPR-Cas9-Mediated Gene Editing of miRNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lodish, H.; Flygare, J.; Chou, S. From Stem Cell to Erythroblast: Regulation of Red Cell Production at Multiple Levels by Multiple Hormones. IUBMB Life 2010, 62, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hale, J.; Bhagia, P.; Xue, F.; Chen, L.; Jaffray, J.; Yan, H.; Lane, J.; Gallagher, P.G.; Mohandas, N.; et al. Isolation and Transcriptome Analyses of Human Erythroid Progenitors: BFU-E and CFU-E. Blood 2014, 124, 3636–3645. [Google Scholar] [CrossRef]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and Differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and Functional Characterization of Human Erythroblasts at Distinct Stages: Implications for Understanding of Normal and Disordered Erythropoiesis in Vivo. Blood 2019, 121, 3246–3254. [Google Scholar] [CrossRef]
- Gronowicz, G.; Swift, H.; Steck, T.L. Maturation of the Reticulocyte in Vitro. J. Cell Sci. 1984, 71, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From Stem Cell to Red Cell: Regulation of Erythropoiesis at Multiple Levels by Multiple Proteins, RNAs, and Chromatin Modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef]
- Pishesha, N.; Thiru, P.; Shi, J.; Eng, J.C.; Sankaran, V.G.; Lodish, H.F. Transcriptional Divergence and Conservation of Human and Mouse Erythropoiesis. Proc. Natl. Acad. Sci. USA 2014, 111, 4103–4108. [Google Scholar] [CrossRef] [PubMed]
- Merryweather-Clarke, S.; Higgs, D.R.; McGowan, S.J.; Soneji, A.T.; Roberts, D.J.; Buckle, V.J.; Waugh, C.; Taylor, S.; Clark, K.; Robson, K.J.H.; et al. Global Gene Expression Analysis of Human Erythroid Progenitors. Blood 2011, 117, e96–e108. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Das, S.; Byram, P.K.; Rahaman, M.; Dolai, T.K.; Chatterjee, A.; Chakravorty, N. MicroRNA Expression Patterns in HbE/β-Thalassemia Patients: The Passwords to Unlock Fetal Hemoglobin Expression in β-Hemoglobinopathies. Blood Cells Mol. Dis. 2021, 87, 102523. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Büsche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Füreder, W.; Bettelheim, P.; Pfeilstöcker, M.; et al. Normal and Pathological Erythropoiesis in Adults: From Gene Regulation to Targeted Treatment Concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shi, L. Long Non-Coding RNAs during Normal Erythropoiesis. Blood Sci. 2019, 1, 137–140. [Google Scholar] [CrossRef]
- Ludwig, L.S.; Lareau, C.A.; Bao, E.L.; Nandakumar, S.K.; Muus, C.; Ulirsch, J.C.; Chowdhary, K.; Buenrostro, J.D.; Mohandas, N.; An, X.; et al. Transcriptional States and Chromatin Accessibility Underlying Human Erythropoiesis. Cell Rep. 2019, 27, 3228–3240.e7. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of MiRNAs and SiRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Cassano, M.; Sampaolesi, M. Role of MiRNAs in Muscle Stem Cell Biology: Proliferation, Differentiation and Death. Curr. Pharm. Des. 2012, 18, 1718–1729. [Google Scholar] [CrossRef]
- Gurtan, A.M.; Sharp, P.A. The Role of MiRNAs in Regulating Gene Expression Networks. J. Mol. Biol. 2013, 425, 3582–3600. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, E.A.C. The Role of MicroRNAs in Cancer: No Small Matter. Eur. J. Cancer 2007, 43, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Galagali, H.; Kim, J.K. The Multifaceted Roles of MicroRNAs in Differentiation. Curr. Opin. Cell Biol. 2020, 67, 118–140. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Maki, M.; Ding, R.; Yang, Y.; Zhang, B.; Xiong, L. Genome-Wide Survey of Tissue-Specific MicroRNA and Transcription Factor Regulatory Networks in 12 Tissues. Sci. Rep. 2014, 4, 5150. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, A.A.; Fasano, J.D.; Martinez-Morilla, S.; Venkatesan, S.; Kupfer, G.; Hattangadi, S.M. MiR-181a Regulates Erythroid Enucleation via the Regulation of Xpo7 Expression. Haematologica 2018, 103, e341–e344. [Google Scholar] [CrossRef]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.-T. A Comprehensive Joint Analysis of the Long and Short RNA Transcriptomes of Human Erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.M.; Zhang, C.C.; Tao, Y.; Yao, H.; Qi, X.; Schwartz, R.J.; Jun-Shen Huang, L.; Olson, E.N. Defective Erythroid Differentiation in MiR-451 Mutant Mice Mediated by 14-3-3zeta. Genes Dev. 2010, 24, 1614–1619. [Google Scholar] [CrossRef]
- Zhang, L.; Flygare, J.; Wong, P.; Lim, B.; Lodish, H.F. MiR-191 Regulates Mouse Erythroblast Enucleation by down-Regulating Riok3 and Mxi1. Genes Dev. 2011, 25, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yu, J.; Yang, G.-H.; Wang, X.-S.; Zhang, J.-W. Regulation of Erythroid Differentiation by MiR-376a and Its Targets. Cell Res. 2011, 21, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Listowski, M.A.; Heger, E.; Bogusławska, D.M.; Machnicka, B.; Kuliczkowski, K.; Leluk, J.; Sikorski, A.F. MicroRNAs: Fine Tuning of Erythropoiesis. Cell. Mol. Biol. Lett. 2013, 18, 34–46. [Google Scholar] [CrossRef]
- Wang, D.; Si, S.; Wang, Q.; Luo, G.; Du, Q.; Liang, Q.; Guo, X.; Zhang, G.; Feng, J.; Leng, Z. MiR-27a Promotes Hemin-Induced Erythroid Differentiation of K562 Cells by Targeting CDC25B. Cell. Physiol. Biochem. 2018, 46, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Byon, J.C.H.; Papayannopoulou, T. MicroRNAs: Allies or Foes in Erythropoiesis? J. Cell. Physiol. 2012, 227, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Liu, Y.; Ji, P. Understanding Terminal Erythropoiesis: An Update on Chromatin Condensation, Enucleation, and Reticulocyte Maturation. Blood Rev. 2021, 46, 100740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sankaran, V.G.; Lodish, H.F. MicroRNAs in Erythroid and Megakaryocytic Differentiation and Megakaryocyte-Erythroid Progenitor Lineage Commitment. Leukemia 2012, 26, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Shang, X.; Chen, D.; Pang, D.; Zhao, C.; Xu, X. MicroRNA-2355-5p Regulates Γ-globin Expression in Human Erythroid Cells by Inhibiting KLF6. Br. J. Haematol. 2020, 193, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Lulli, V.; Romania, P.; Morsilli, O.; Cianciulli, P.; Gabbianelli, M.; Testa, U.; Giuliani, A.; Marziali, G. MicroRNA-486-3p Regulates γ-Globin Expression in Human Erythroid Cells by Directly Modulating BCL11A. PLoS ONE 2013, 8, e60436. [Google Scholar] [CrossRef] [PubMed]
- Azzouzi, I.; Moest, H.; Winkler, J.; Fauchère, J.-C.; Gerber, A.P.; Wollscheid, B.; Stoffel, M.; Schmugge, M.; Speer, O. MicroRNA-96 Directly Inhibits γ-Globin Expression in Human Erythropoiesis. PLoS ONE 2011, 6, e22838. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Šćepanović, D.; Vergilio, J.-A.; Ji, P.; Kim, J.; Thiru, P.; Orkin, S.H.; Lander, E.S.; Lodish, H.F. MicroRNA-15a and -16-1 Act via MYB to Elevate Fetal Hemoglobin Expression in Human Trisomy 13. Proc. Natl. Acad. Sci. USA 2011, 108, LP1519–LP1524. [Google Scholar] [CrossRef]
- Li, B.; Zhu, X.; Ward, C.M.; Starlard-Davenport, A.; Takezaki, M.; Berry, A.; Ward, A.; Wilder, C.; Neunert, C.; Kutlar, A.; et al. MIR-144-Mediated NRF2 Gene Silencing Inhibits Fetal Hemoglobin Expression in Sickle Cell Disease. Exp. Hematol. 2019, 70, 85–96.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhao, L.; Zheng, J.; Wang, K.; Deng, H.; Liu, P.; Chen, L.; Mu, H. MicroRNA-144 Modulates Oxidative Stress Tolerance in SH-SY5Y Cells by Regulating Nuclear Factor Erythroid 2-Related Factor 2-Glutathione Axis. Neurosci. Lett. 2017, 655, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liu, Y.; Chen, Y.; Yin, C.; Chen, J.-J.; Liu, S. MiR-214 Protects Erythroid Cells against Oxidative Stress by Targeting ATF4 and EZH2. Free Radic. Biol. Med. 2016, 92, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; De Falco, L.; Asci, R.; Russo, R.; Colucci, S.; Gorrese, M.; Zollo, M.; Iolascon, A. Regulation of Divalent Metal Transporter 1 (DMT1) Non-IRE Isoform by the MicroRNA Let-7d in Erythroid Cells. Haematologica 2010, 95, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Mohammdai-Asl, J.; Ramezani, A.; Norozi, F.; Malehi, A.S.; Asnafi, A.A.; Far, M.A.J.; Mousavi, S.H.; Saki, N. MicroRNAs in Erythropoiesis and Red Blood Cell Disorders. Front. Biol. 2015, 10, 321–332. [Google Scholar] [CrossRef]
- Saki, N.; Abroun, S.; Soleimani, M.; Kavianpour, M.; Shahjahani, M.; Mohammadi-Asl, J.; Hajizamani, S. MicroRNA Expression in β-Thalassemia and Sickle Cell Disease: A Role in The Induction of Fetal Hemoglobin. Cell J. 2016, 17, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Bruchova, H.; Merkerova, M.; Josef, T. Prchal Aberrant Expression of MicroRNA in Polycythemia Vera. Haematologica 2008, 93, 1009–1016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lessard, S.; Beaudoin, M.; Orkin, S.H.; Bauer, D.E.; Lettre, G. 14q32 and Let-7 MicroRNAs Regulate Transcriptional Networks in Fetal and Adult Human Erythroblasts. Hum. Mol. Genet. 2018, 27, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Makki, M.S.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Deep Sequencing and Analyses of MiRNAs, IsomiRs and MiRNA Induced Silencing Complex (MiRISC)-Associated MiRNome in Primary Human Chondrocytes. Sci. Rep. 2017, 7, 15178. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering MicroRNAs from Deep Sequencing Data Using MiRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Reid, J.G.; Gunaratne, P.H. Expression Profiling of MicroRNAs by Deep Sequencing. Brief. Bioinform. 2009, 10, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 Cell Types Reveals Evidence for the Expression of Numerous Novel Primate- and Tissue-Specific MicroRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The Impact of MicroRNAs on Protein Output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting MicroRNA Genes to the Core Transcriptional Regulatory Circuitry of Embryonic Stem Cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef]
- Liu, N.; Xu, S.; Yao, Q.; Zhu, Q.; Kai, Y.; Hsu, J.Y.; Sakon, P.; Pinello, L.; Yuan, G.-C.; Bauer, D.E.; et al. Transcription Factor Competition at the γ-Globin Promoters Controls Hemoglobin Switching. Nat. Genet. 2021, 53, 511–520. [Google Scholar] [CrossRef]
- Xu, J.; Shao, Z.; Glass, K.; Bauer, D.E.; Pinello, L.; van Handel, B.; Hou, S.; Stamatoyannopoulos, J.A.; Mikkola, H.K.A.; Yuan, G.-C.; et al. Combinatorial Assembly of Developmental Stage-Specific Enhancers Controls Gene Expression Programs during Human Erythropoiesis. Dev. Cell 2012, 23, 796–811. [Google Scholar] [CrossRef]
- Basak, A.; Munschauer, M.; Lareau, C.A.; Montbleau, K.E.; Ulirsch, J.C.; Hartigan, C.R.; Schenone, M.; Lian, J.; Wang, Y.; Huang, Y.; et al. Control of Human Hemoglobin Switching by LIN28B-Mediated Regulation of BCL11A Translation. Nat. Genet. 2020, 52, 138–145. [Google Scholar] [CrossRef]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of Immortalized Human Erythroid Progenitor Cell Lines Able to Produce Enucleated Red Blood Cells. PLoS ONE 2013, 8, e59890. [Google Scholar] [CrossRef] [PubMed]
- Stocks, M.B.; Mohorianu, I.; Beckers, M.; Paicu, C.; Moxon, S.; Thody, J.; Dalmay, T.; Moulton, V. The UEA SRNA Workbench (Version 4.4): A Comprehensive Suite of Tools for Analyzing MiRNAs and SRNAs. Bioinformatics 2018, 34, 3382–3384. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating High Confidence MicroRNAs Using Deep Sequencing Data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Zheng, R.; Wan, C.; Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Chen, C.-H.; Brown, M.; Zhang, X.; Meyer, C.A.; et al. Cistrome Data Browser: Expanded Datasets and New Tools for Gene Regulatory Analysis. Nucleic Acids Res. 2019, 47, D729–D735. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V.P.; Yan, H.; Lezon-Geyda, K.; An, X.; Hale, J.; Hillyer, C.D.; Mohandas, N.; Gallagher, P.G. A Unique Epigenomic Landscape Defines Human Erythropoiesis. Cell Rep. 2019, 28, 2996–3009.e7. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved Vectors and Genome-Wide Libraries for CRISPR Screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Danzer, C.; Rechsteiner, M.; Lehmann, H.; Brandt, L.P.; Hejhal, T.; Catalano, A.; Busenhart, P.; Gonçalves, A.F.; Brandt, S.; et al. A Versatile Modular Vector System for Rapid Combinatorial Mammalian Genetics. J. Clin. Investig. 2015, 125, 1603–1619. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, J.; Satchwell, T.J.; Meinders, M.; Daniels, D.E.; Regan, F.; Thornton, N.M.; Wilson, M.C.; Dobbe, J.G.G.; Streekstra, G.J.; Trakarnsanga, K.; et al. Enhancement of Red Blood Cell Transfusion Compatibility Using CRISPR-Mediated Erythroblast Gene Editing. EMBO Mol. Med. 2018, 10, e8454. [Google Scholar] [CrossRef]
- Busk, P.K. A Tool for Design of Primers for MicroRNA-Specific Quantitative RT-QPCR. BMC Bioinform. 2014, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Nath, A.; Thamodaran, V.; Ijee, S.; Palani, D.; Rajendiran, V.; Venkatesan, V.; Datari, P.; Pai, A.A.; Janet, N.B.; et al. Direct Generation of Immortalized Erythroid Progenitor Cell Lines from Peripheral Blood Mononuclear Cells. Cells 2021, 10, 523. [Google Scholar] [CrossRef]
- Pope, S.H.; Fibach, E.; Sun, J.; Chin, K.; Rodgers, G.P. Two-Phase Liquid Culture System Models Normal Human Adult Erythropoiesis at the Molecular Level. Eur. J. Haematol. 2000, 64, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Giarratana, M.-C.; Kobari, L.; Lapillonne, H.; Chalmers, D.; Kiger, L.; Cynober, T.; Marden, M.C.; Wajcman, H.; Douay, L. Ex Vivo Generation of Fully Mature Human Red Blood Cells from Hematopoietic Stem Cells. Nat. Biotechnol. 2005, 23, 69–74. [Google Scholar] [CrossRef]
- Migliaccio, A.R.; Masselli, E.; Varricchio, L.; Whitsett, C. Ex-Vivo Expansion of Red Blood Cells: How Real for Transfusion in Humans? Blood Rev. 2012, 26, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.R.; Palis, J. Blood in a Dish: In Vitro Synthesis of Red Blood Cells. Drug Discov. Today. Dis. Mech. 2011, 8, e3–e8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.; Tan, Y.S.; Cheng, W.-C.; Kingsbury, T.J.; Heimfeld, S.; Civin, C.I. MIR144 and MIR451 Regulate Human Erythropoiesis via RAB14. Br. J. Haematol. 2015, 168, 583–597. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Simmini, S.; Abreu-Goodger, C.; Bartonicek, N.; di Giacomo, M.; Bilbao-Cortes, D.; Horos, R.; von Lindern, M.; Enright, A.J.; O’Carroll, D. The MiR-144/451 Locus Is Required for Erythroid Homeostasis. J. Exp. Med. 2010, 207, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Tang, D. (Ed.) Ferroptosis in Health and Disease, 1st ed.; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-030-26780-3. [Google Scholar]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017, 12, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Yi, B.; Ma, R.; Zhang, X.; Zhao, H.; Xi, Y. CRISPR/Cas9, a Novel Genomic Tool to Knock down MicroRNA in Vitro and in Vivo. Sci. Rep. 2016, 6, 22312. [Google Scholar] [CrossRef]
- Yudovich, D.; Bäckström, A.; Schmiderer, L.; Žemaitis, K.; Subramaniam, A.; Larsson, J. Combined Lentiviral- and RNA-Mediated CRISPR/Cas9 Delivery for Efficient and Traceable Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Sci. Rep. 2020, 10, 22393. [Google Scholar] [CrossRef] [PubMed]
- Moir-Meyer, G.; Cheong, P.L.; Olijnik, A.-A.; Brown, J.; Knight, S.; King, A.; Kurita, R.; Nakamura, Y.; Gibbons, R.J.; Higgs, D.R.; et al. Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods Protoc. 2018, 1, 28. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.E.; Magis, W.; Vu, J.; Heo, S.-J.; Wartiovaara, K.; Walters, M.C.; Kurita, R.; Nakamura, Y.; Boffelli, D.; Martin, D.I.K.; et al. CRISPR-Cas9 Interrogation of a Putative Fetal Globin Repressor in Human Erythroid Cells. PLoS ONE 2019, 14, e0208237. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e19. [Google Scholar] [CrossRef]
- Tsai, F.-Y.; Orkin, S.H. Transcription Factor GATA-2 Is Required for Proliferation/Survival of Early Hematopoietic Cells and Mast Cell Formation, But Not for Erythroid and Myeloid Terminal Differentiation. Blood 1997, 89, 3636–3643. [Google Scholar] [CrossRef]
- Ludwig, L.S.; Lareau, C.A.; Bao, E.L.; Nandakumar, S.K.; Muus, C.; Ulirsch, J.C.; Chowdhary, K.; Buenrostro, J.D.; Mohandas, N.; An, X.; et al. Transcriptional Regulation of Erythropoiesis: An Affair Involving Multiple Partners. Oncogene 2002, 89, 3636–3643. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Kluiver, J.; Gibcus, J.H.; Hettinga, C.; Adema, A.; Richter, M.K.S.; Halsema, N.; Slezak-Prochazka, I.; Ding, Y.; Kroesen, B.-J.; van den Berg, A. Rapid Generation of MicroRNA Sponges for MicroRNA Inhibition. PLoS ONE 2012, 7, e29275. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. MicroRNA Sponges: Progress and Possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef]
- Medina, P.P.; Slack, F.J. Inhibiting MicroRNA Function in Vivo. Nat. Methods 2009, 6, 37–38. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-Scale Deletion Screening of Human Long Non-Coding RNAs Using a Paired-Guide RNA CRISPR–Cas9 Library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [PubMed]
- Bruchova, H.; Yoon, D.; Agarwal, A.M.; Mendell, J.; Prchal, J.T. Regulated Expression of MicroRNAs in Normal and Polycythemia Vera Erythropoiesis. Exp. Hematol. 2007, 35, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Choong, M.; Yang, H.; McNiece, I. MicroRNA Expression Profiling during Human Cord Blood-Derived CD34 Cell Erythropoiesis. Exp. Hematol. 2007, 35, 551–564. [Google Scholar] [CrossRef]
- Felli, N.; Pedini, F.; Romania, P.; Biffoni, M.; Morsilli, O.; Castelli, G.; Santoro, S.; Chicarella, S.; Sorrentino, A.; Peschle, C.; et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica 2009, 94, 479–486. [Google Scholar] [CrossRef]
- Papagiannopoulos, C.I.; Theodoroula, N.F.; Vizirianakis, I.S. MiR-16-5p Promotes Erythroid Maturation of Erythroleukemia Cells by Regulating Ribosome Biogenesis. Pharmaceuticals 2021, 14, 137. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Pierce, S.; Bueso-Ramos, C.; Calin, G.; Boyano-Adánez, M.D.C.; García-Manero, G. Overexpression of MiR-125a in Myelodysplastic Syndrome CD34+ Cells Modulates NF-ΚB Activation and Enhances Erythroid Differentiation Arrest. PLoS ONE 2014, 9, e93404. [Google Scholar] [CrossRef]
- Zhai, P.-F.; Wang, F.; Su, R.; Lin, H.-S.; Jiang, C.-L.; Yang, G.-H.; Yu, J.; Zhang, J.-W. The Regulatory Roles of MicroRNA-146b-5p and Its Target Platelet-Derived Growth Factor Receptor α (PDGFRA) in Erythropoiesis and Megakaryocytopoiesis. J Biol. Chem. 2014, 289, 22600–22613. [Google Scholar] [CrossRef]
- Kirschner, M.B.; Edelman, J.J.B.; Kao, S.C.-H.; Vallely, M.P.; van Zandwijk, N.; Reid, G. The Impact of Hemolysis on Cell-Free MicroRNA Biomarkers. Front. Genet. 2013, 4, 94. [Google Scholar] [CrossRef]
- Juzenas, S.; Lindqvist, C.M.; Ito, G.; Dolshanskaya, Y.; Halfvarson, J.; Franke, A.; Hemmrich-Stanisak, G. Depletion of Erythropoietic MiR-486-5p and MiR-451a Improves Detectability of Rare MicroRNAs in Peripheral Blood-Derived Small RNA Sequencing Libraries. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, C.; Zhou, W.; Shi, Y.; Guo, P.; Liu, Y.; Wang, J.; Zhang, C.-Y.; Zhang, C. Elevation of Circulating MiR-210-3p in High-Altitude Hypoxic Environment. Front. Physiol. 2016, 7, 84. [Google Scholar] [CrossRef]
- Walker, A.L.; Steward, S.; Howard, T.A.; Mortier, N.; Smeltzer, M.; Wang, Y.-D.; Ware, R.E. Epigenetic and Molecular Profiles of Erythroid Cells after Hydroxyurea Treatment in Sickle Cell Anemia. Blood 2011, 118, 5664–5670. [Google Scholar] [CrossRef]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key Regulators of Stem Cells. Nat. Rev. Mol. Cell. Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef]
- Kretov, D.A.; Walawalkar, I.A.; Mora-Martin, A.; Shafik, A.M.; Moxon, S.; Cifuentes, D. Ago2-Dependent Processing Allows MiR-451 to Evade the Global MicroRNA Turnover Elicited during Erythropoiesis. Mol. Cell 2020, 78, 317–328.e6. [Google Scholar] [CrossRef]
- Sun, L.; Yu, Y.; Niu, B.; Wang, D. Red Blood Cells as Potential Repositories of MicroRNAs in the Circulatory System. Fron. Genet. 2020, 11, 442. [Google Scholar] [CrossRef]
- Fang, X.; Shen, F.; Lechauve, C.; Xu, P.; Zhao, G.; Itkow, J.; Wu, F.; Hou, Y.; Wu, X.; Yu, L.; et al. MiR-144/451 Represses the LKB1/AMPK/MTOR Pathway to Promote Red Cell Precursor Survival during Recovery from Acute Anemia. Haematologica 2018, 103, 406–416. [Google Scholar] [CrossRef]
- Lee, Y.T.; de Vasconcellos, J.F.; Yuan, J.; Byrnes, C.; Noh, S.-J.; Meier, E.R.; Kim, K.S.; Rabel, A.; Kaushal, M.; Muljo, S.A.; et al. LIN28B-Mediated Expression of Fetal Hemoglobin and Production of Fetal-like Erythrocytes from Adult Human Erythroblasts Ex Vivo. Blood 2013, 122, 1034–1041. [Google Scholar] [CrossRef]
- Kretov, D.A.; Shafik, A.M.; Cifuentes, D. Assessing MiR-451 Activity and Its Role in Erythropoiesis. Methods Mol. Biol. 2018, 1680, 179–190. [Google Scholar] [CrossRef]
- O’Carroll, D.; Mecklenbrauker, I.; Das, P.P.; Santana, A.; Koenig, U.; Enright, A.J.; Miska, E.A.; Tarakhovsky, A. A Slicer-Independent Role for Argonaute 2 in Hematopoiesis and the MicroRNA Pathway. Genes Dev. 2007, 21, 1999–2004. [Google Scholar] [CrossRef]
- Gnanapragasam, M.N.; McGrath, K.E.; Catherman, S.; Xue, L.; Palis, J.; Bieker, J.J.; O’Carroll, D.; Mecklenbrauker, I.; Das, P.P.; Santana, A.; et al. EKLF/KLF1-Regulated Cell Cycle Exit Is Essential for Erythroblast Enucleation. Blood 2016, 21, 1631–1641. [Google Scholar] [CrossRef]
- Held, M.A.; Greenfest-Allen, E.; Jachimowicz, E.; Stoeckert, C.J.; Stokes, M.P.; Wood, A.W.; Wojchowski, D.M. Phospho-Proteomic Discovery of Novel Signal Transducers Including Thioredoxin-Interacting Protein as Mediators of Erythropoietin-Dependent Human Erythropoiesis. Exp. Hematol. 2020, 84, 29–44. [Google Scholar] [CrossRef]
- Hu, W.; Yuan, B.; Lodish, H.F. Cpeb4-Mediated Translational Regulatory Circuitry Controls Terminal Erythroid Differentiation. Dev. Cell 2014, 30, 660–672. [Google Scholar] [CrossRef]
- Tanimura, N.; Liao, R.; Wilson, G.M.; Dent, M.R.; Cao, M.; Burstyn, J.N.; Hematti, P.; Liu, X.; Zhang, Y.; Zheng, Y.; et al. GATA/Heme Multi-Omics Reveals a Trace Metal-Dependent Cellular Differentiation Mechanism. Dev. Cell 2018, 46, 581–594.e4. [Google Scholar] [CrossRef]
- Undi, R.B.; Kandi, R.; Gutti, R.K. MicroRNAs as Haematopoiesis Regulators. Adv. Hematol. 2013, 2013, 695754. [Google Scholar] [CrossRef]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef]
- Maeda, T.; Ito, K.; Merghoub, T.; Poliseno, L.; Hobbs, R.M.; Wang, G.; Dong, L.; Maeda, M.; Dore, L.C.; Zelent, A.; et al. LRF Is an Essential Downstream Target of GATA1 in Erythroid Development and Regulates BIM-Dependent Apoptosis. Dev. Cell 2009, 17, 527–540. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Ludwig, L.S.; Sicinska, E.; Xu, J.; Bauer, D.E.; Eng, J.C.; Patterson, H.C.; Metcalf, R.A.; Natkunam, Y.; Orkin, S.H.; et al. Cyclin D3 Coordinates the Cell Cycle during Differentiation to Regulate Erythrocyte Size and Number. Genes Dev. 2012, 26, 2075–2087. [Google Scholar] [CrossRef]
- Uras, I.Z.; Scheicher, R.M.; Kollmann, K.; Glösmann, M.; Prchal-Murphy, M.; Tigan, A.S.; Fux, D.A.; Altamura, S.; Neves, J.; Muckenthaler, M.U.; et al. Cdk6 Contributes to Cytoskeletal Stability in Erythroid Cells. Haematologica 2017, 102, 995–1005. [Google Scholar] [CrossRef]
- Magnusson, M.; Brun, A.C.M.; Miyake, N.; Larsson, J.; Ehinger, M.; Bjornsson, J.M.; Wutz, A.; Sigvardsson, M.; Karlsson, S. HOXA10 Is a Critical Regulator for Hematopoietic Stem Cells and Erythroid/Megakaryocyte Development. Blood 2007, 109, 3687–3696. [Google Scholar] [CrossRef]
- Lacronique, V.; Varlet, P.; Mayeux, P.; Porteu, A.; Gisselbrecht, S.; Kahn, A.; Lacombe, C. Bcl-2 Targeted Overexpression into the Erythroid Lineage of Transgenic Mice Delays but Does Not Prevent the Apoptosis of Erythropoietin-Deprived Erythroid Progenitors. Blood 1997, 90, 3050–3056. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Total Reads | log2FC | |||
|---|---|---|---|---|---|
| PID3 vs. PIID9 | PID3 vs. PIID11 | PID3 vs. PIID14 | |||
| hsa-miR-495-3p | 728 | −3.07 | −5.62 | −7.66 | Downregulated |
| hsa-miR-573 | 557 | −0.75 | −2.23 | −6.94 | |
| hsa-miR-370-3p | 8244 | −3.67 | −5.21 | −6.15 | |
| hsa-miR-134-5p | 1873 | −3.20 | −4.57 | −5.99 | |
| hsa-miR-543 | 3293 | −4.52 | −7.59 | −5.36 | |
| hsa-miR-4425 | 520 | −2.63 | −3.34 | −5.30 | |
| hsa-miR-493-5p | 3908 | −3.39 | −5.22 | −5.15 | |
| hsa-miR-654-3p | 1477 | −3.59 | −5.45 | −4.58 | |
| hsa-miR-409-3p | 6051 | −3.06 | −5.10 | −4.26 | |
| hsa-miR-323a-3p | 552 | −3.84 | −5.63 | −3.71 | |
| hsa-miR-1-3p | 44,708 | −3.83 | −5.01 | −3.57 | |
| hsa-miR-486-5p | 7,025,604 | 3.44 | 4.45 | 5.08 | |
| hsa-miR-183-3p | 1725 | 5.04 | 6.34 | 5.26 | |
| hsa-miR-144-5p | 275,363 | 4.64 | 5.61 | 5.56 | Upregulated |
| hsa-miR-96-5p | 68,467 | 5.04 | 5.70 | 6.10 | |
| hsa-miR-183-5p | 435,443 | 4.54 | 5.12 | 6.61 | |
| hsa-miR-182-5p | 975,481 | 4.71 | 6.04 | 6.98 | |
| hsa-miR-4732-5p | 697 | 4.92 | 6.94 | 7.00 | |
| hsa-miR-375-3p | 606 | 5.29 | 6.36 | 7.05 | |
| hsa-miR-144-3p | 270,635 | 5.47 | 6.96 | 7.71 | |
| hsa-miR-4732-3p | 1288 | 3.90 | 6.80 | 8.81 | |
| hsa-miR-451a | 669,963 | 6.99 | 9.70 | 10.21 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nath, A.; Rayabaram, J.; Ijee, S.; Bagchi, A.; Chaudhury, A.D.; Roy, D.; Chambayil, K.; Singh, J.; Nakamura, Y.; Velayudhan, S.R. Comprehensive Analysis of microRNAs in Human Adult Erythropoiesis. Cells 2021, 10, 3018. https://doi.org/10.3390/cells10113018
Nath A, Rayabaram J, Ijee S, Bagchi A, Chaudhury AD, Roy D, Chambayil K, Singh J, Nakamura Y, Velayudhan SR. Comprehensive Analysis of microRNAs in Human Adult Erythropoiesis. Cells. 2021; 10(11):3018. https://doi.org/10.3390/cells10113018
Chicago/Turabian StyleNath, Aneesha, Janakiram Rayabaram, Smitha Ijee, Abhirup Bagchi, Anurag Dutta Chaudhury, Debanjan Roy, Karthik Chambayil, Jyoti Singh, Yukio Nakamura, and Shaji R. Velayudhan. 2021. "Comprehensive Analysis of microRNAs in Human Adult Erythropoiesis" Cells 10, no. 11: 3018. https://doi.org/10.3390/cells10113018
APA StyleNath, A., Rayabaram, J., Ijee, S., Bagchi, A., Chaudhury, A. D., Roy, D., Chambayil, K., Singh, J., Nakamura, Y., & Velayudhan, S. R. (2021). Comprehensive Analysis of microRNAs in Human Adult Erythropoiesis. Cells, 10(11), 3018. https://doi.org/10.3390/cells10113018