
FGFR1 Overexpression Induces Cancer Cell Stemness and Enhanced Akt/Erk-ER Signaling to Promote Palbociclib Resistance in Luminal A Breast Cancer Cells



Abstract
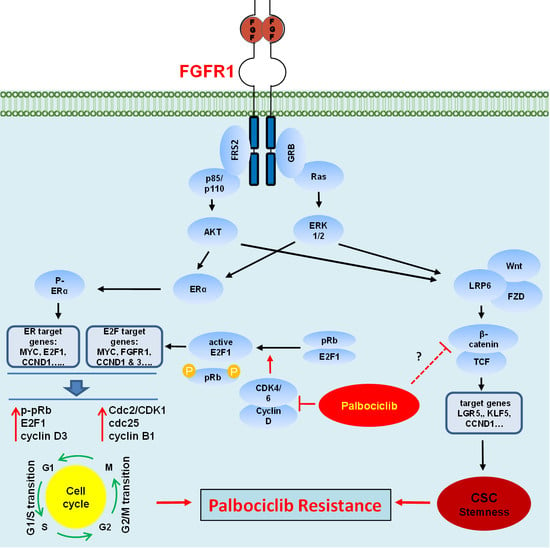
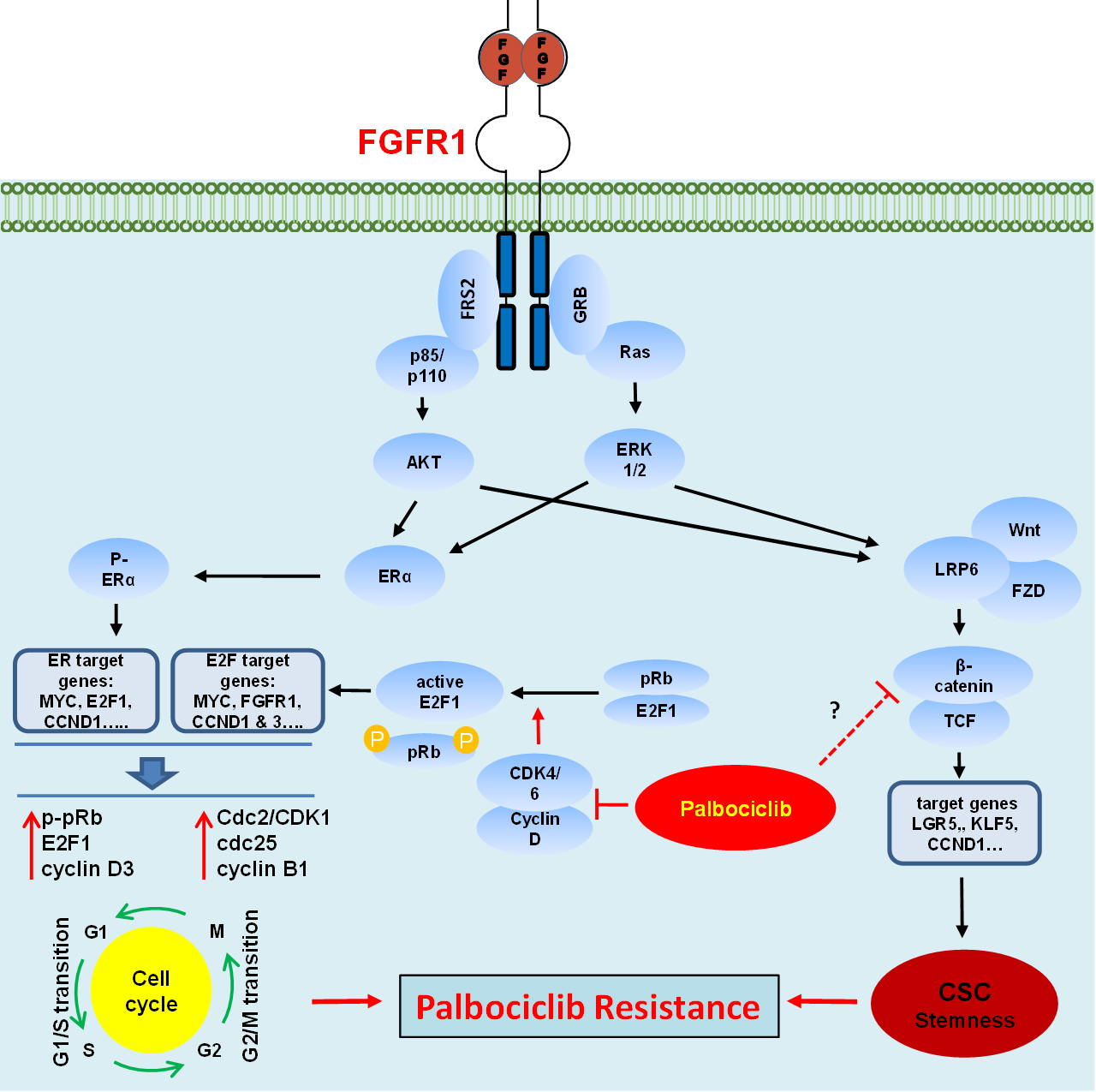
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Cell Proliferation Assay
2.4. Clonogenic Assays
2.5. Cell Cycle Analysis with Flow Cytometry
2.6. Transwell Migration and Invasion Assay
2.7. Tumorsphere Assays
2.8. Western Blot Analysis
2.9. Luciferase Assay
2.10. Quantitative RT-PCR (qRT-PCR)
2.11. Drug Synergy Analysis
2.12. Statistical Analysis
3. Results
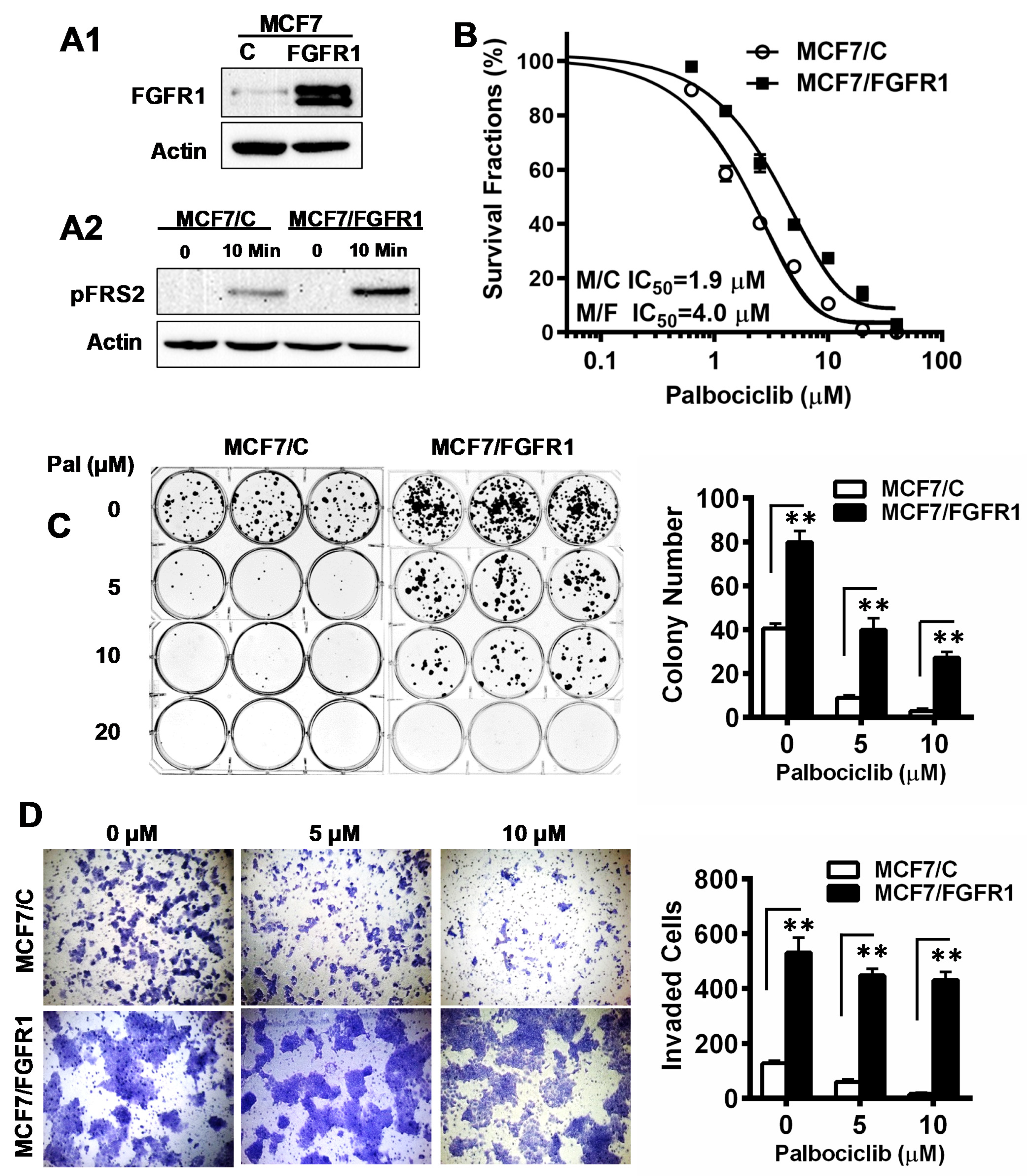
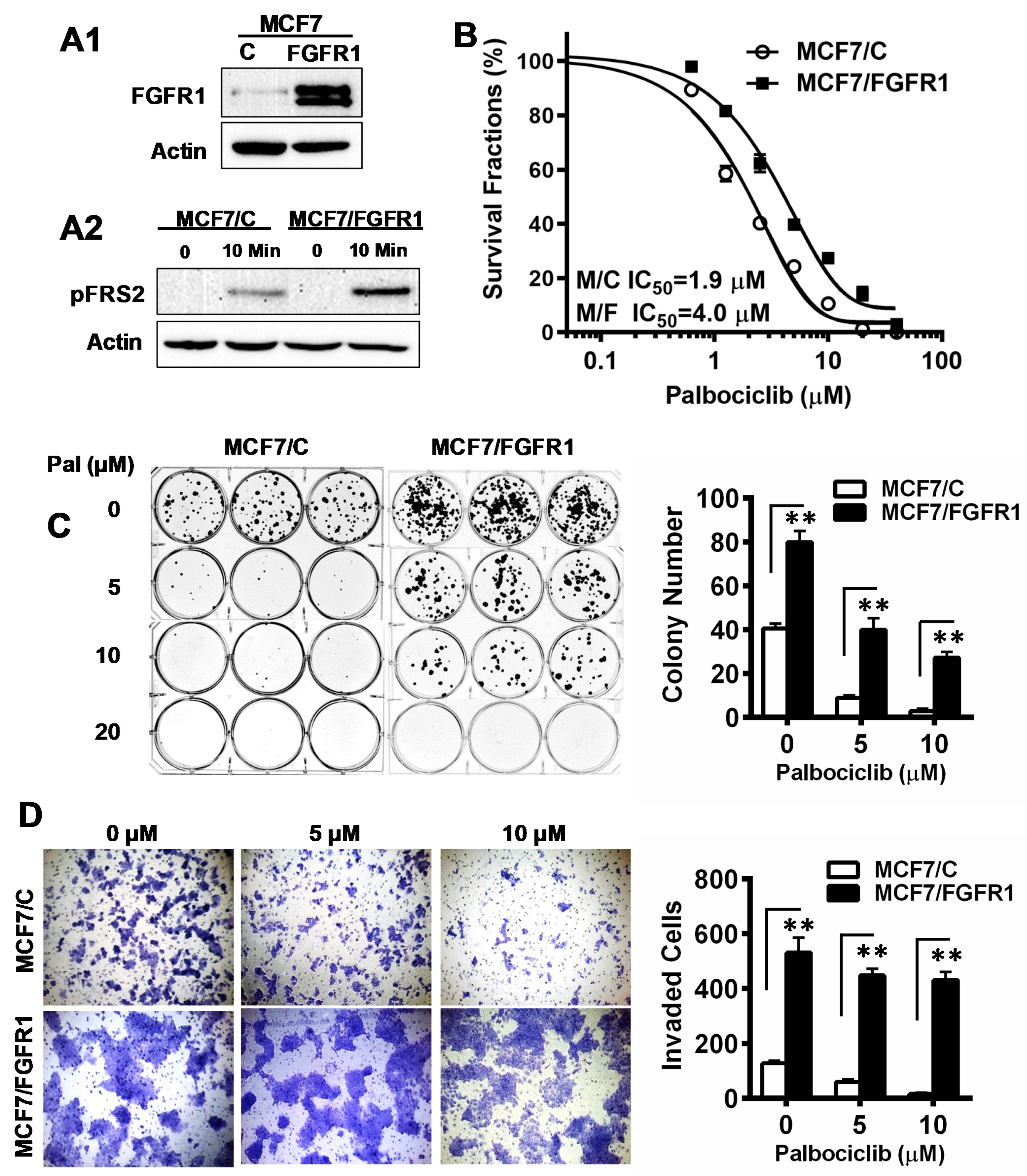
3.1. FGFR1 Overexpression Significantly Reduces Palbociclib-Induced Inhibition of Proliferation and Invasion of MCF-7 Cells
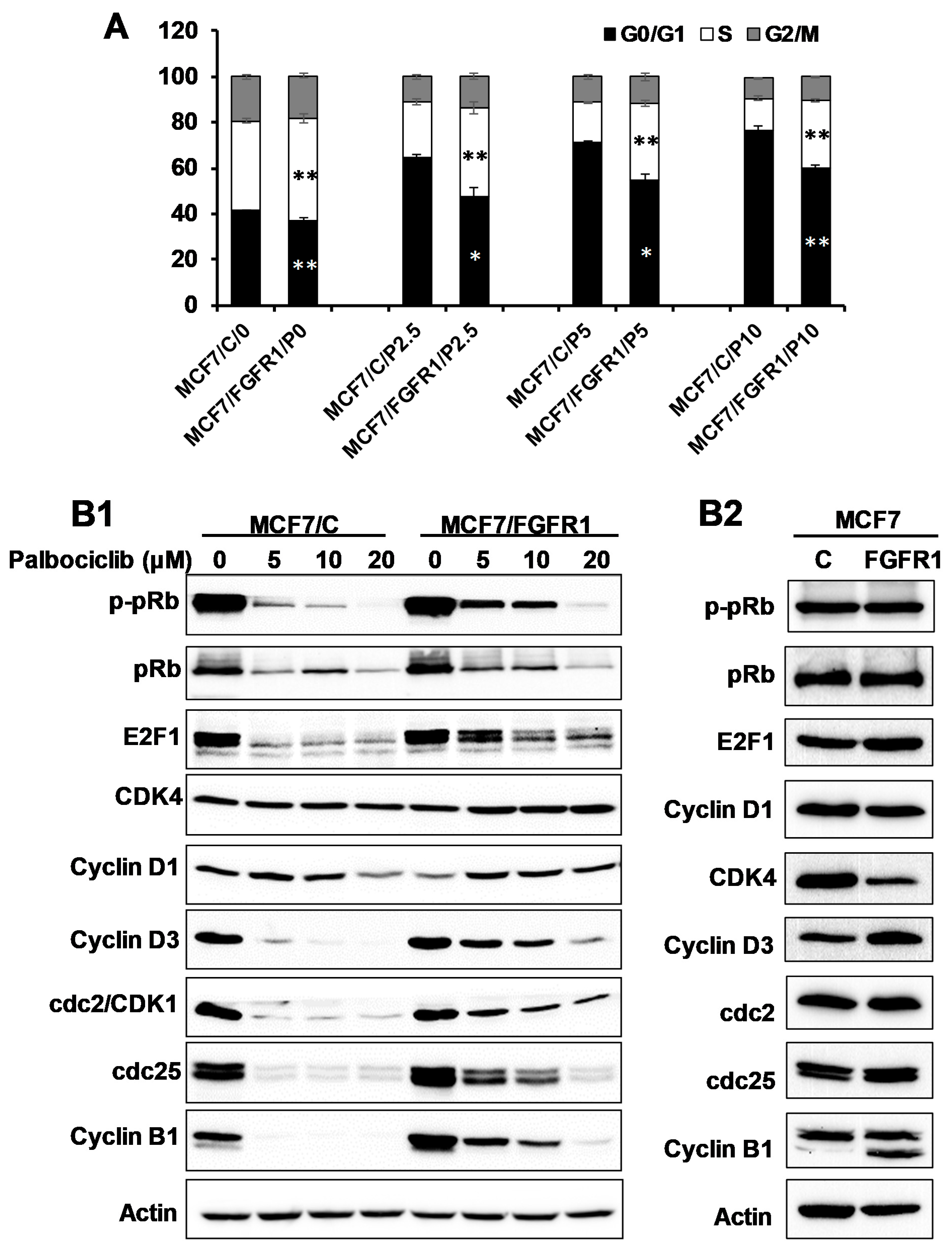
3.2. FGFR1 Abolishes Palbociclib-Induced Cell Cycle Arrest through Activation of Cell Cycle Regulators beyond Cyclin D-CDK4/6–pRb Pathway
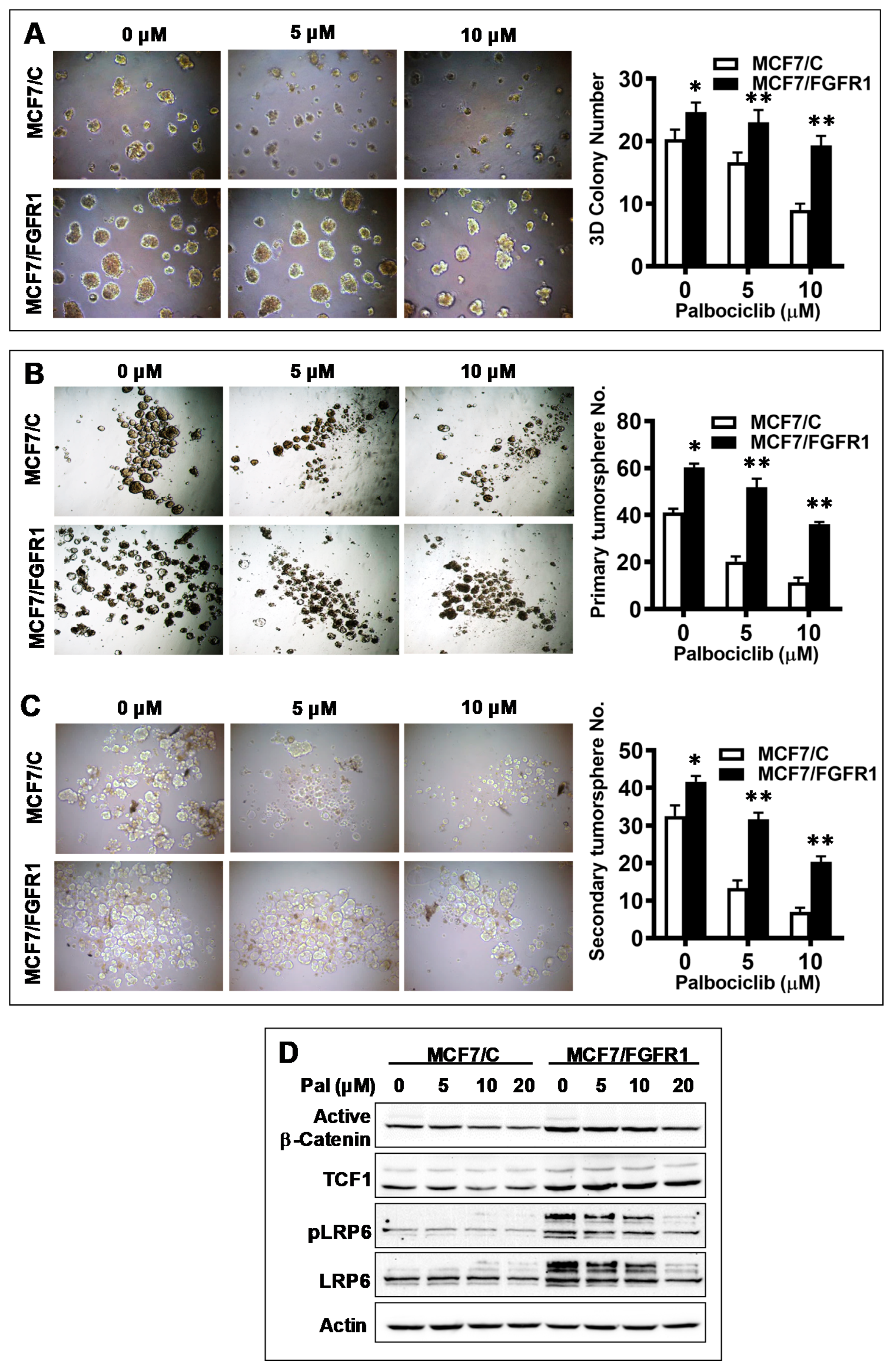
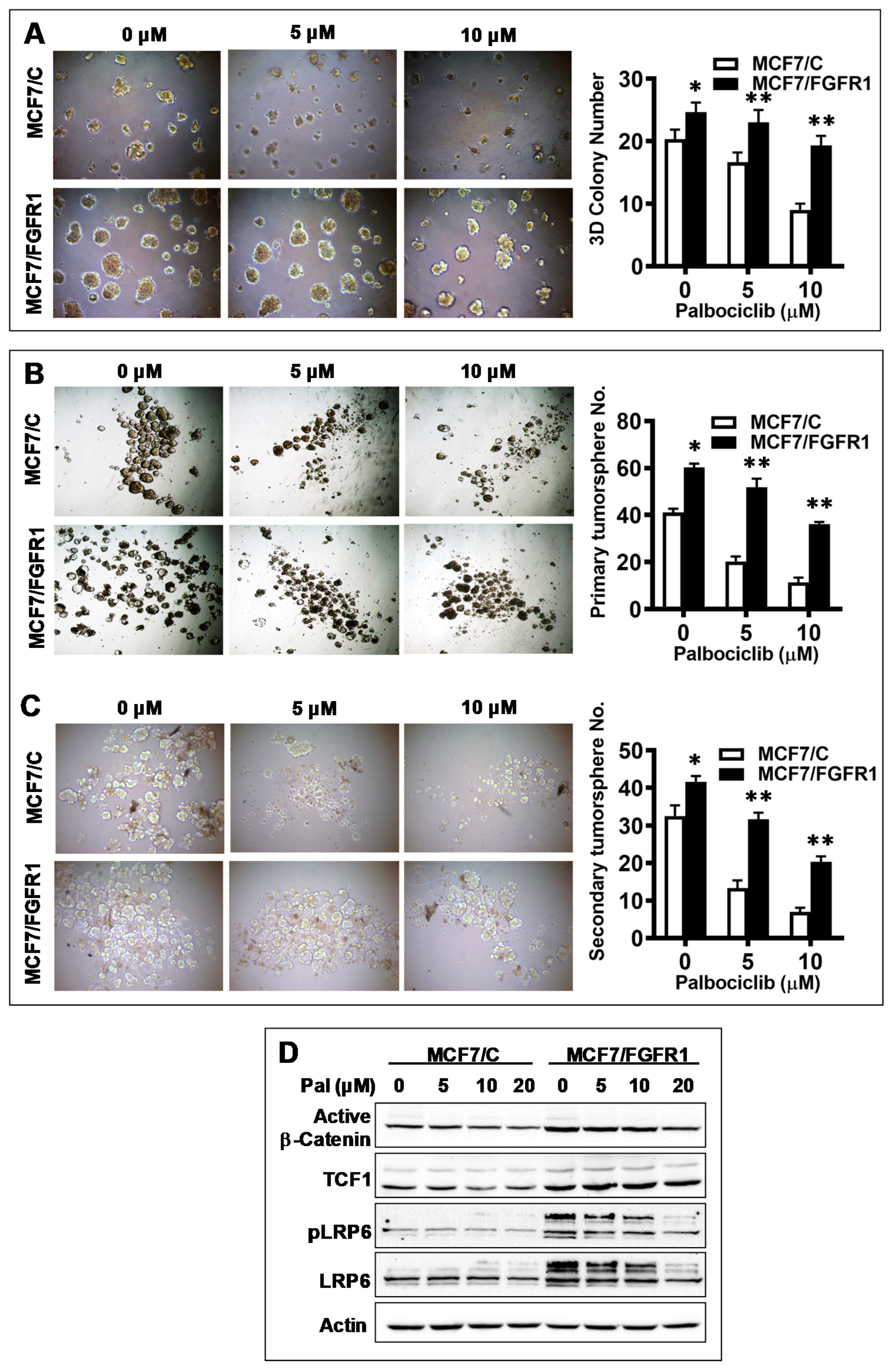
3.3. FGFR1 Overexpression-Induced Palbociclib Resistance Is Associated with Enhanced CSC Stemness and Upregulation of Wnt Signaling
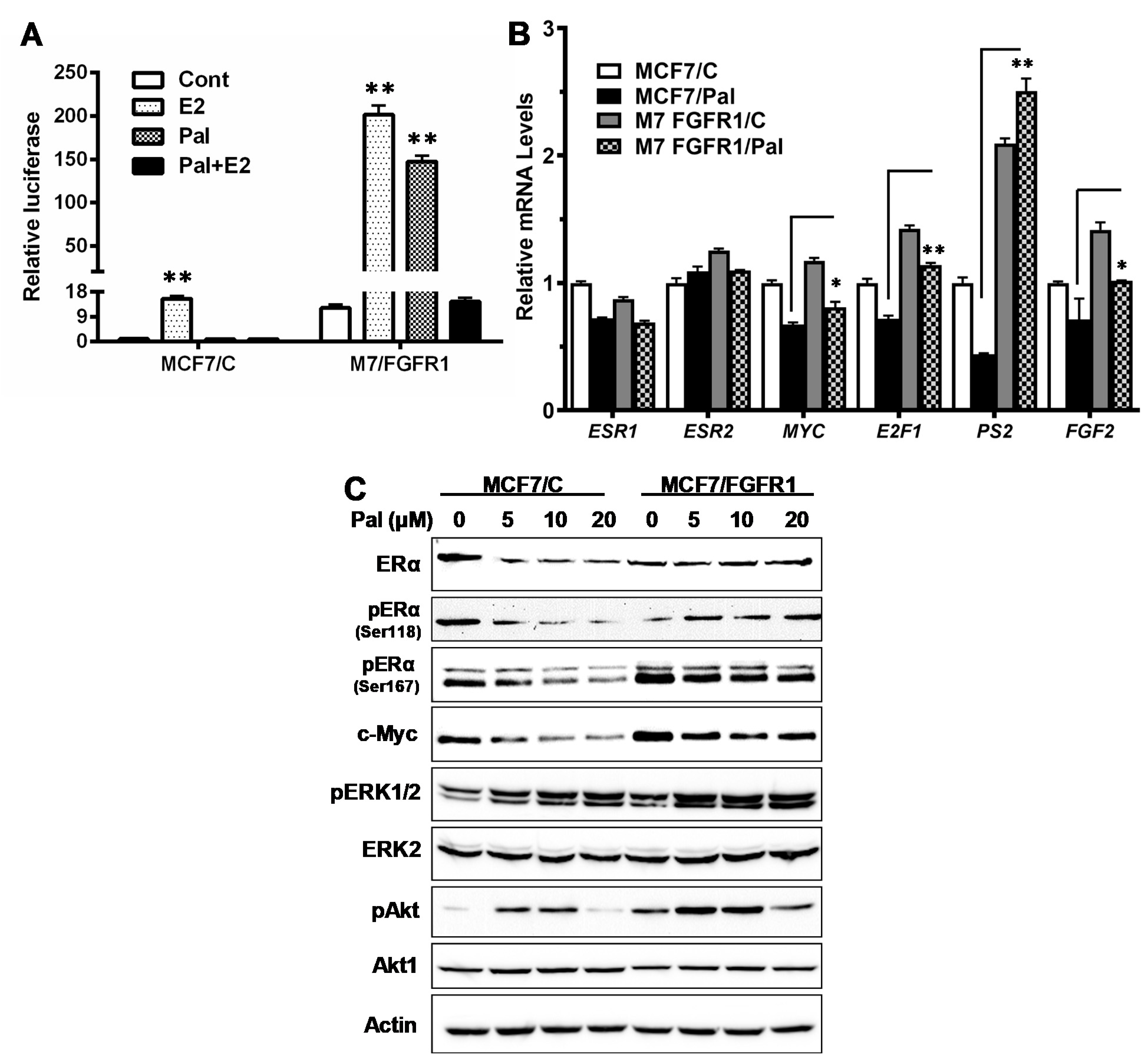
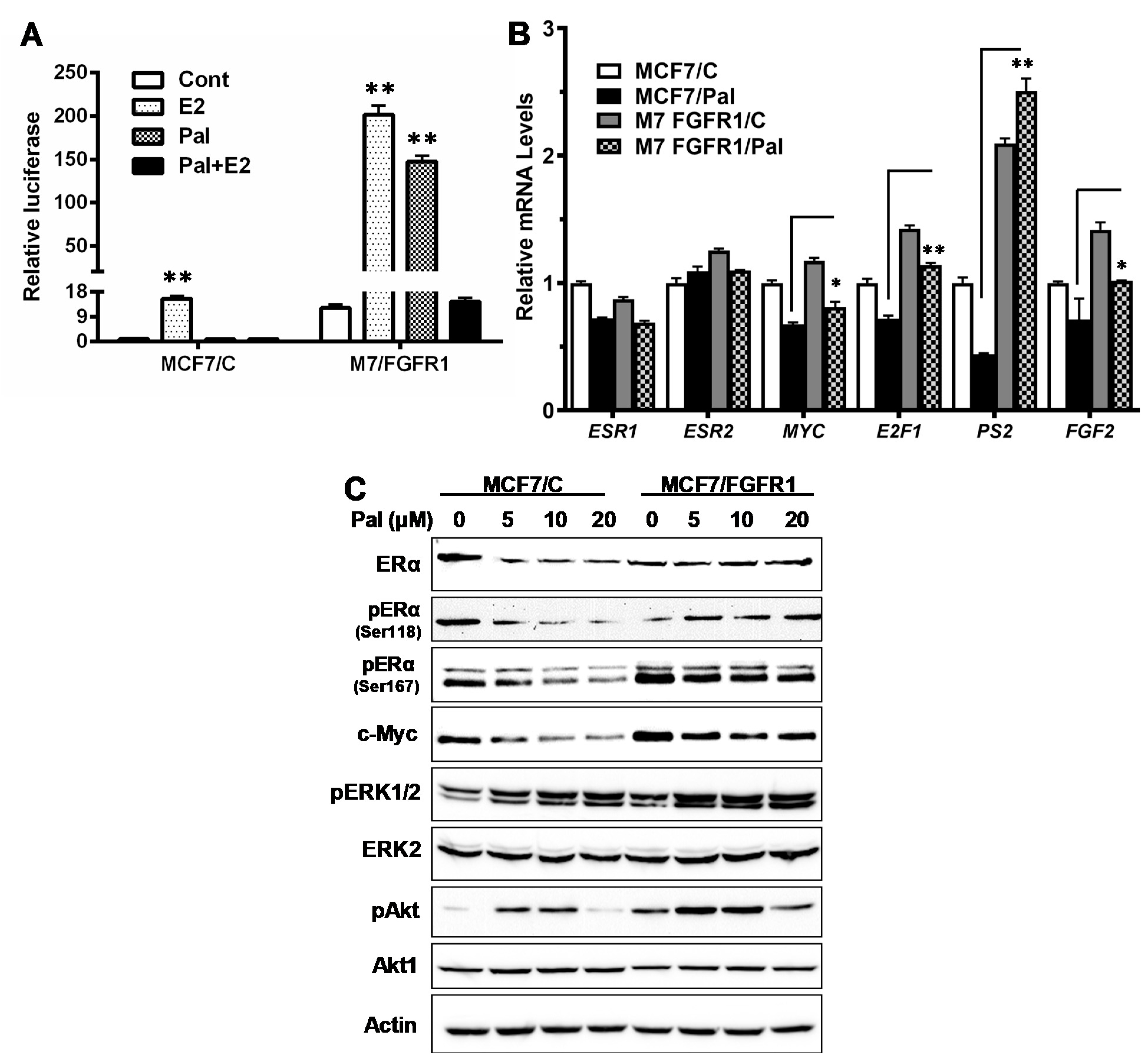
3.4. FGFR1 Activates ER-Mediated Transcription through Induction of ERα Phosphorylation by Activated Akt and ERK1/2 in MCF-7/FGFR1 Cells
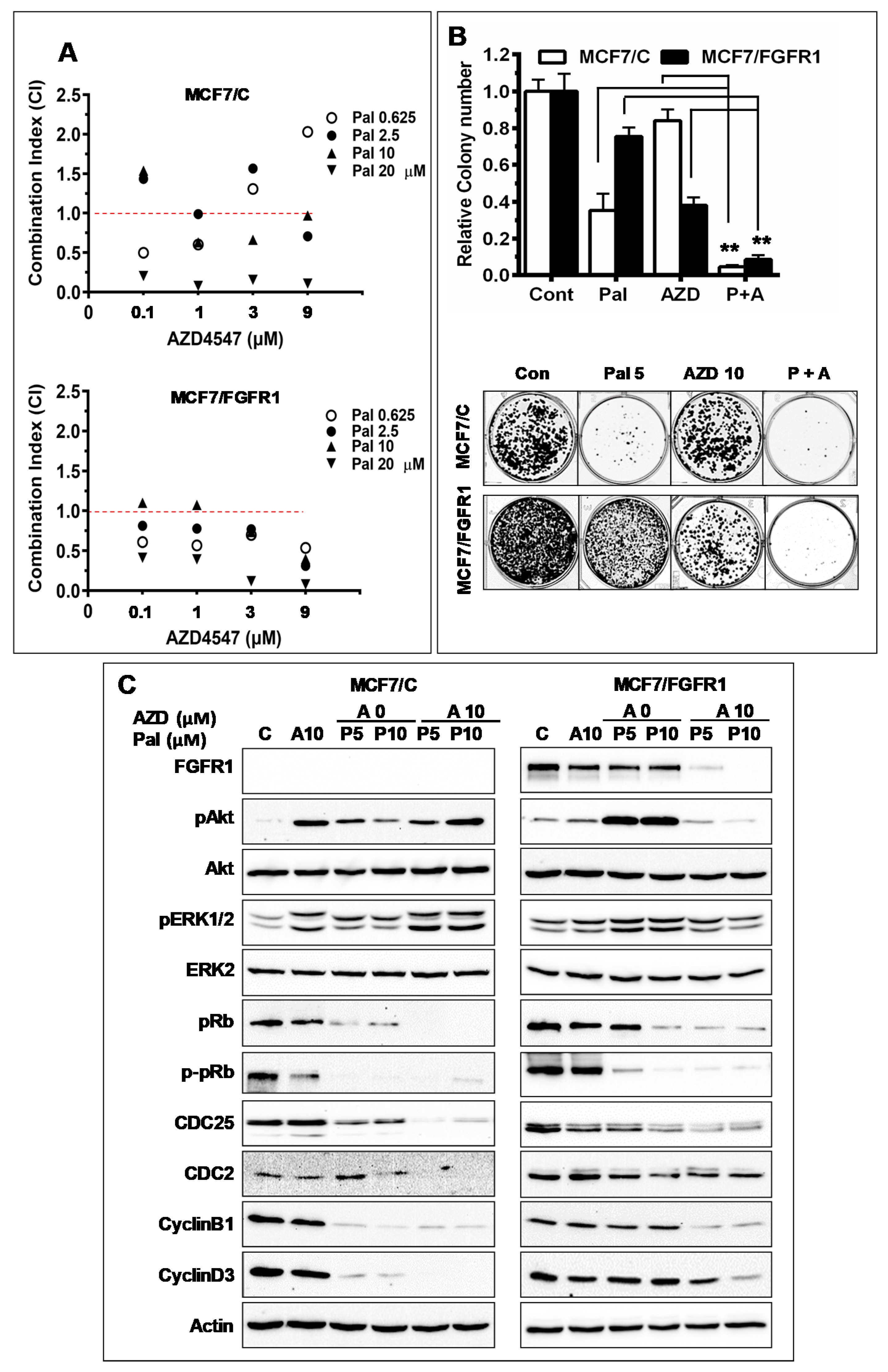
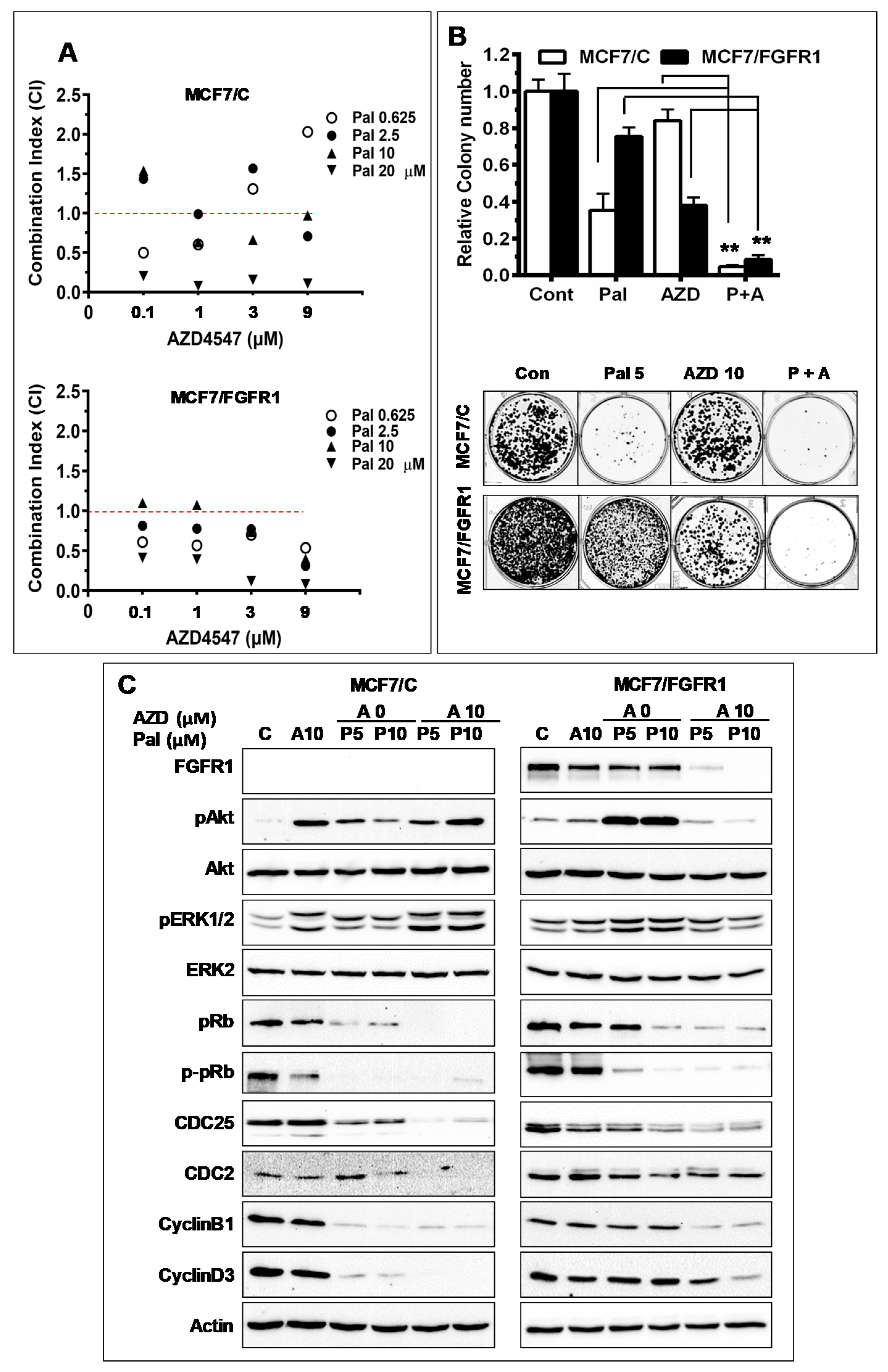
3.5. Combination with FGFR-Targeting Agent AZD4547 Enhances Palbociclib-Induced Inhibition of FGFR1 Overexpressing Cancer Cells
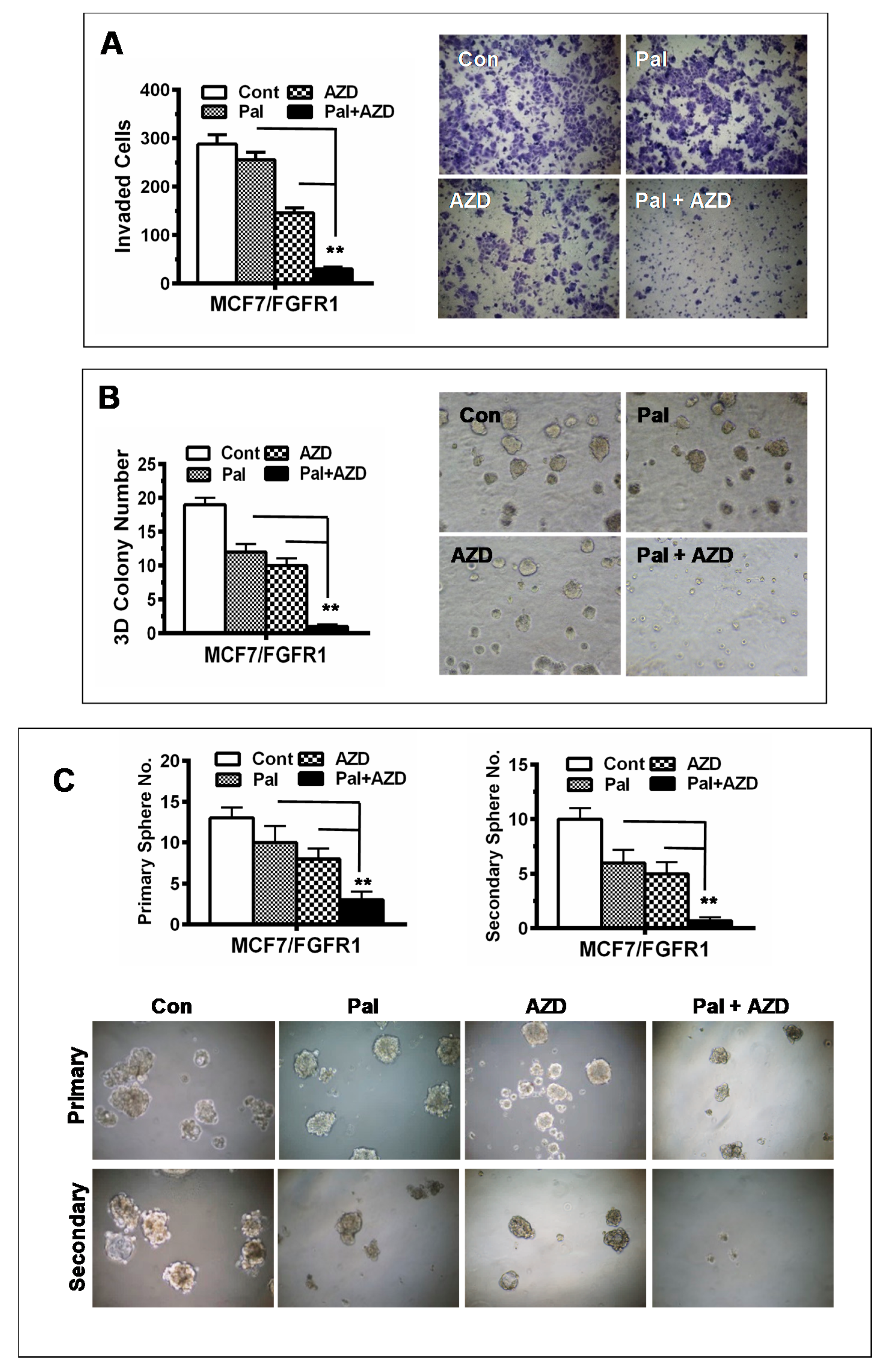
3.6. Combination with AZD4547 Abrogates FGFR1 Overexpression-Associated Promotion of Invasion and CSC Stemness in Palbociclib-Treated Cells
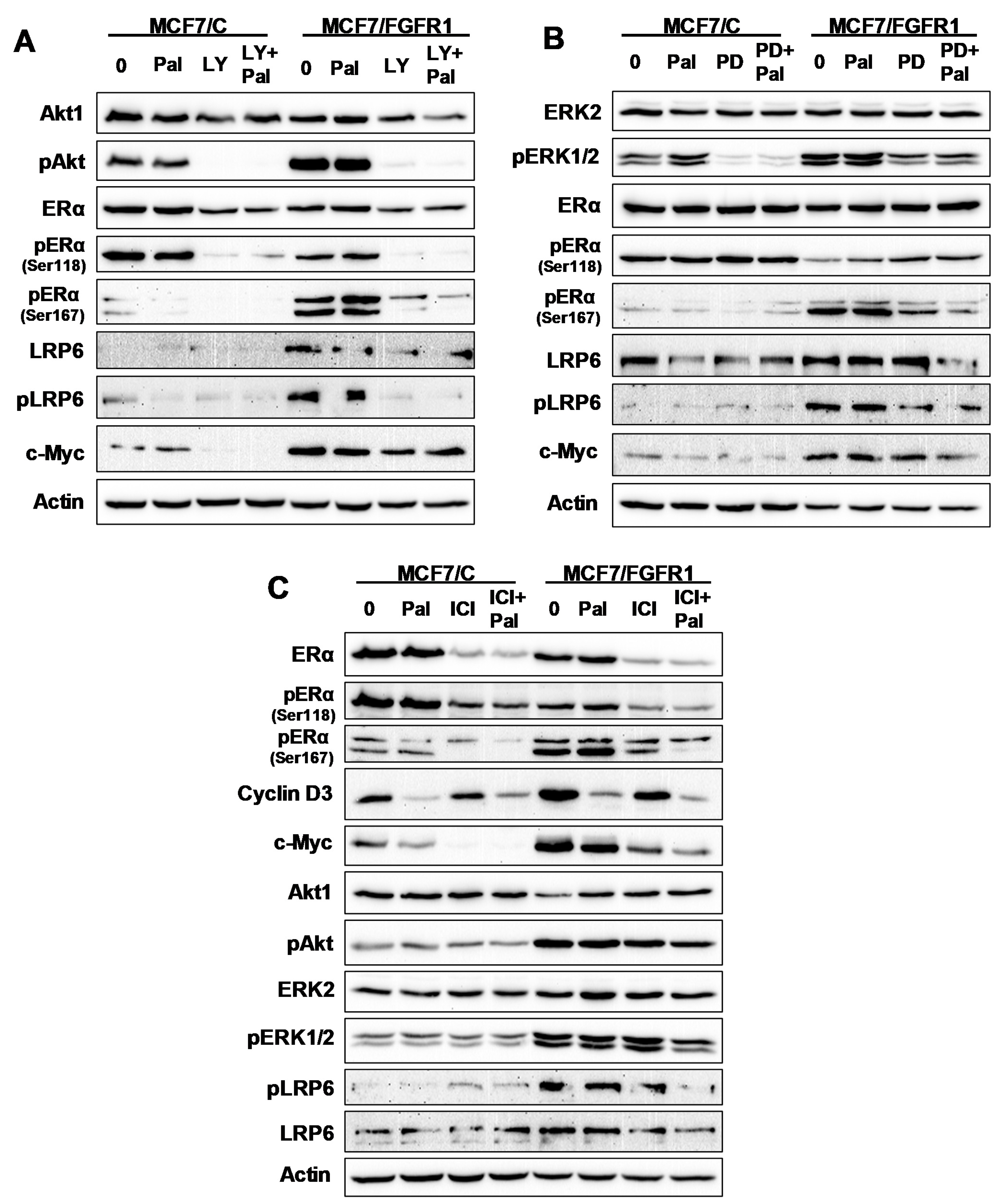
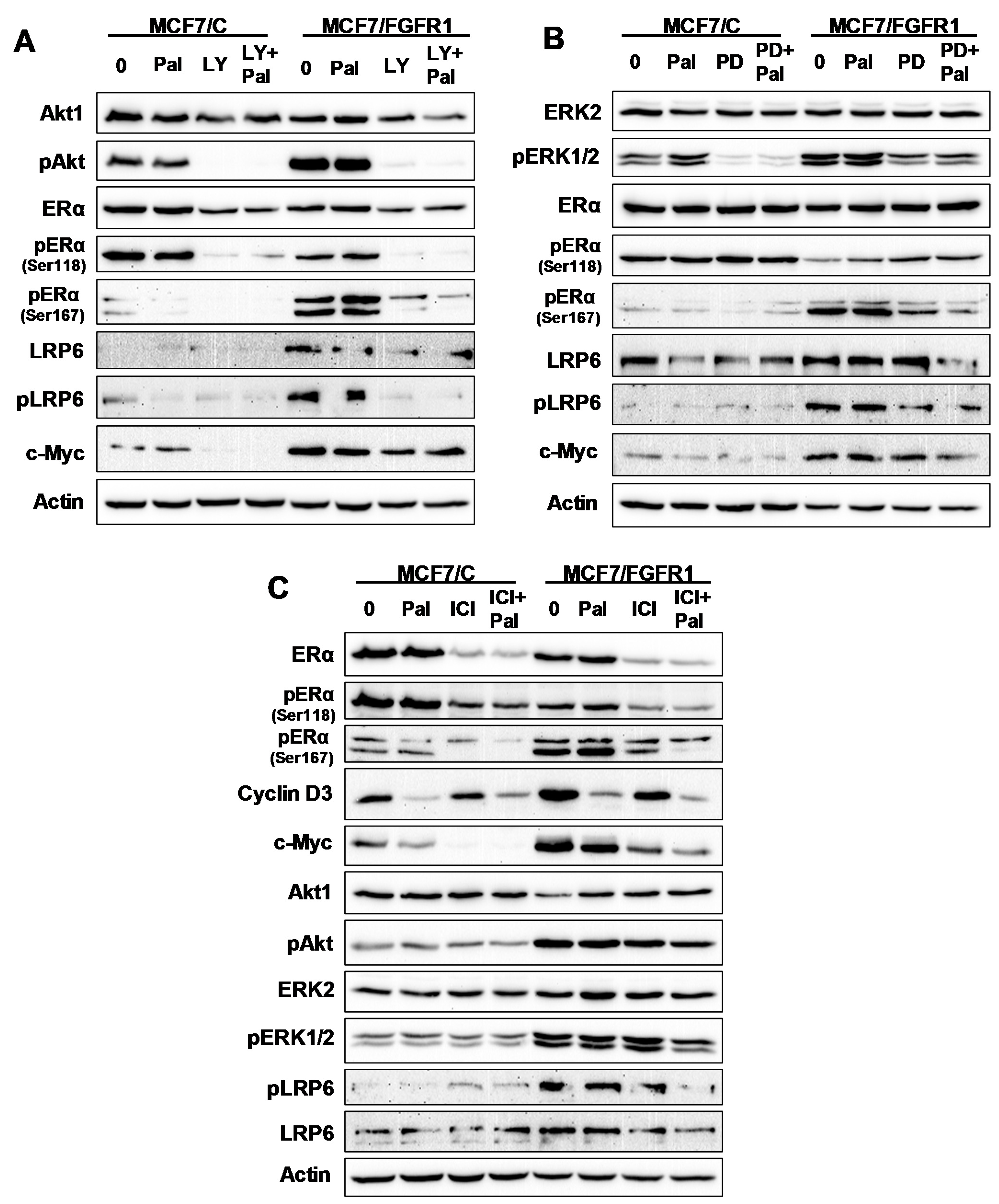
3.7. The Effect of Inhibitor Targeting PI3K/Akt, MAPK/Erk, ERα, and LRP6 Pathways on FGFR1 Overexpression-Induced Signal Transduction
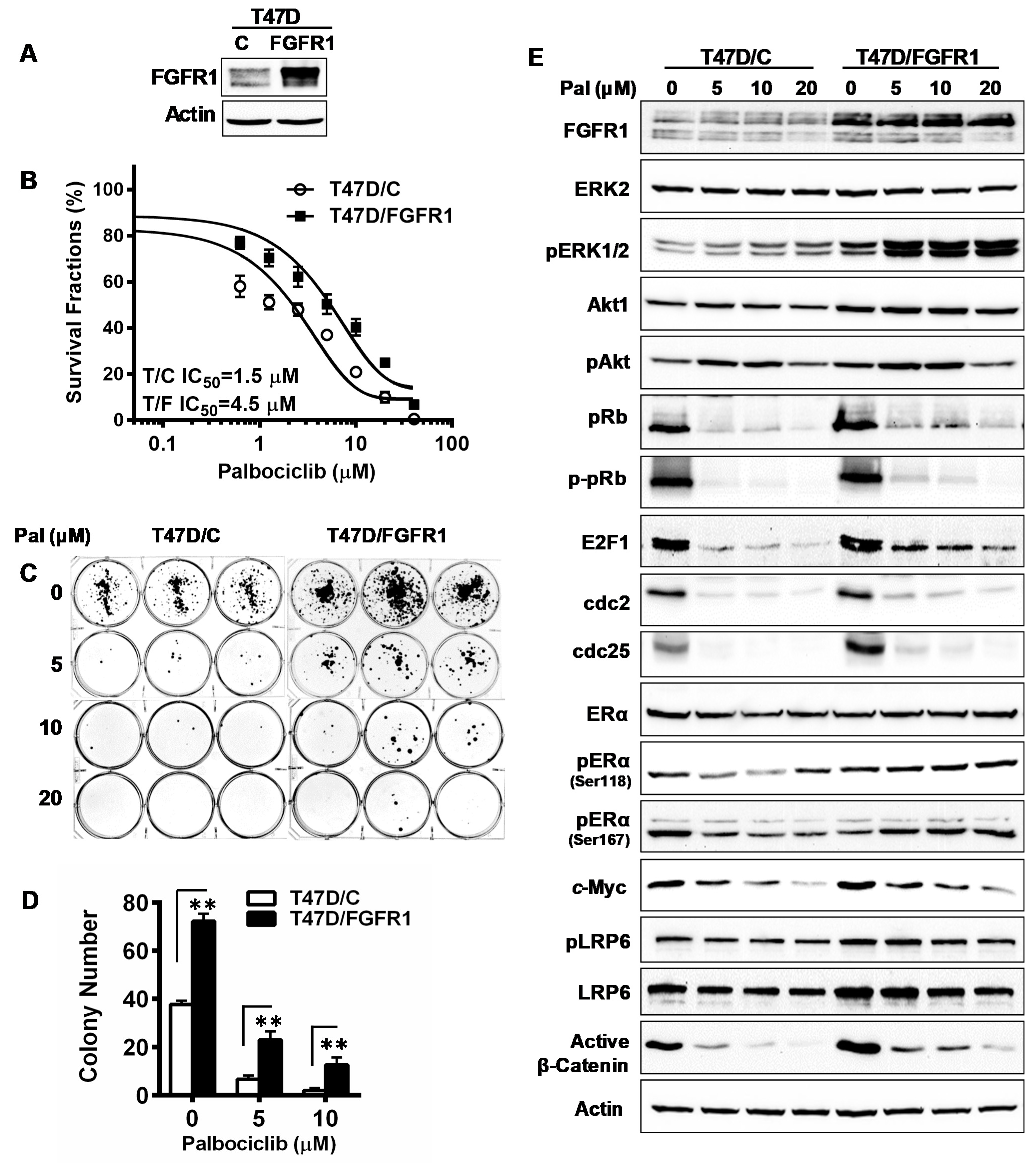
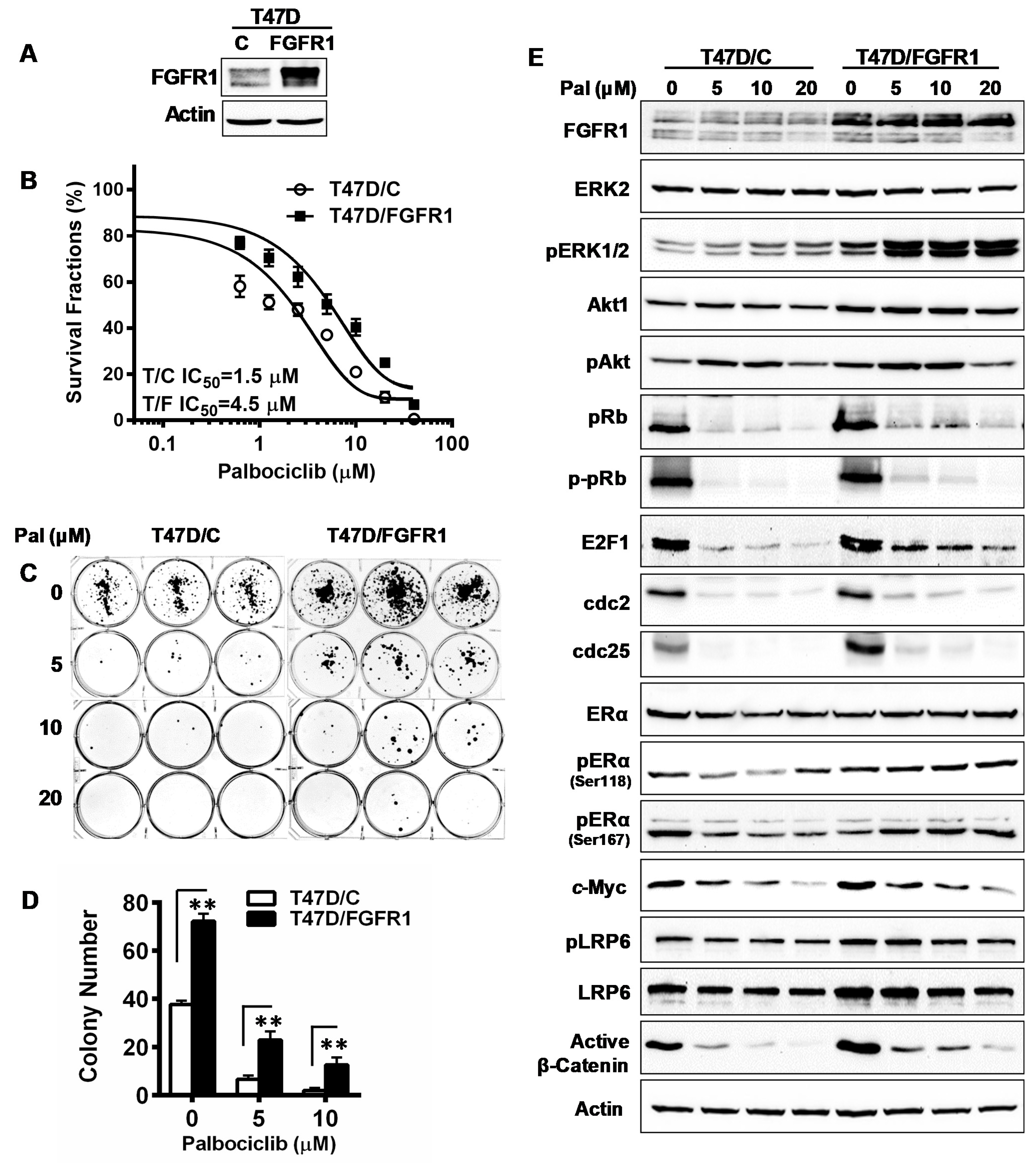
3.8. Overexpression of FGFR1 Induces Palbociclib Resistance in T47D Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Lynce, F.; Shajahan-Haq, A.N.; Swain, S.M. CDK4/6 inhibitors in breast cancer therapy: Current practice and future opportunities. Pharmacol. Ther. 2018, 191, 65–73. [Google Scholar] [CrossRef]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Gomatou, G.; Trontzas, I.; Ioannou, S.; Drizou, M.; Syrigos, N.; Kotteas, E. Mechanisms of resistance to cyclin-dependent kinase 4/6 inhibitors. Mol. Biol. Rep. 2021, 48, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; An, H.J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Xu, B.; Krie, A.; De, P.; Williams, C.; Elsey, R.; Klein, J.; Leyland-Jones, B. Utilizing Tumor and Plasma Liquid Biopsy in Treatment Decision Making for an Estrogen Receptor-Positive Advanced Breast Cancer Patient. Cureus 2017, 9, e1408. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. BCR 2016, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 2020, 10, 1174–1193. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [Google Scholar] [CrossRef]
- Mao, P.; Cohen, O.; Kowalski, K.J.; Kusiel, J.G.; Buendia-Buendia, J.E.; Cuoco, M.S.; Exman, P.; Wander, S.A.; Waks, A.G.; Nayar, U.; et al. Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER(+) Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5974–5989. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Harding, B.; Aspuria, P.J.; Agadjanian, H.; Cheon, D.J.; Mizuno, T.; Greenberg, D.; Allen, J.R.; Spurka, L.; Funari, V.; Spiteri, E.; et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015, 6, 696–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Kim, J.E.; Kim, Y.J.; Lim, J.M.; Kim, S.; Kim, J.W.; Lee, K.H.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J.; et al. Cyclin E overexpression confers resistance to the CDK4/6 specific inhibitor palbociclib in gastric cancer cells. Cancer Lett. 2018, 430, 123–132. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. BCR 2020, 22, 89. [Google Scholar] [CrossRef]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Pond, A.C.; Bin, X.; Batts, T.; Roarty, K.; Hilsenbeck, S.; Rosen, J.M. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells 2013, 31, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Penault-Llorca, F.; Bertucci, F.; Adélaïde, J.; Parc, P.; Coulier, F.; Jacquemier, J.; Birnbaum, D.; deLapeyrière, O. Expression of FGF and FGF receptor genes in human breast cancer. Int. J. Cancer 1995, 61, 170–176. [Google Scholar] [CrossRef]
- Shi, Y.J.; Tsang, J.Y.; Ni, Y.B.; Chan, S.K.; Chan, K.F.; Tse, G.M. FGFR1 is an adverse outcome indicator for luminal A breast cancers. Oncotarget 2016, 7, 5063–5073. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Genetic alterations of FGF receptors: An emerging field in clinical cancer diagnostics and therapeutics. Expert Rev. Anticancer Ther. 2010, 10, 1375–1379. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Ma, Z.; Cheng, Q.; Wu, Y.; Parris, A.B.; Kong, L.; Yang, X. FGFR1 overexpression renders breast cancer cells resistant to metformin through activation of IRS1/ERK signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118877. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kabotyanski, E.B.; Dou, Y.; Reineke, L.C.; Zhang, P.; Zhang, X.H.; Malovannaya, A.; Jung, S.Y.; Mo, Q.; Roarty, K.P.; et al. FGFR1-Activated Translation of WNT Pathway Components with Structured 5’ UTRs Is Vulnerable to Inhibition of EIF4A-Dependent Translation Initiation. Cancer Res. 2018, 78, 4229–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- McDermott, S.C.; Rodriguez-Ramirez, C.; McDermott, S.P.; Wicha, M.S.; Nör, J.E. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018, 9, 25148–25165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, E.; Chen, T.; Kommajosyula, N.; Chen, Z.; Herter-Sprie, G.S.; Cornell, L.; Wong, K.K.; Shapiro, G.I. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 2018, 60, 31572. [Google Scholar] [CrossRef] [Green Version]
- Maehara, O.; Suda, G.; Natsuizaka, M.; Ohnishi, S.; Komatsu, Y.; Sato, F.; Nakai, M.; Sho, T.; Morikawa, K.; Ogawa, K.; et al. Fibroblast growth factor-2-mediated FGFR/Erk signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis 2017, 38, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Parris, A.B.; Zhao, Q.; Howard, E.W.; Zhao, M.; Ma, Z.; Yang, X. Buformin inhibits the stemness of erbB-2-overexpressing breast cancer cells and premalignant mammary tissues of MMTV-erbB-2 transgenic mice. J. Exp. Clin. Cancer Res. CR 2017, 36, 28. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Howard, E.W.; Parris, A.B.; Ma, Z.; Xing, Y.; Yang, X. Bisphenol AF promotes estrogen receptor-positive breast cancer cell proliferation through amphiregulin-mediated crosstalk with receptor tyrosine kinase signaling. PLoS ONE 2019, 14, e0216469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Parris, A.B.; Howard, E.W.; Zhao, M.; Ma, Z.; Guo, Z.; Xing, Y.; Yang, X. FGFR inhibitor, AZD4547, impedes the stemness of mammary epithelial cells in the premalignant tissues of MMTV-ErbB2 transgenic mice. Sci. Rep. 2017, 7, 11306. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Lu, Y.; Jansen, V.M.; Bauer, J.A.; Hanker, A.; González-Ericsson, P.; Lee, K.-M.; Nixon, M.J.; Guerrero-Zotano, A.L.; Schwarz, L.J.; et al. Abstract GS6-05: Gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2018, 78, 4. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.; Le Peuch, C.; Labbé, J.C.; Meyer, A.N.; Donoghue, D.J.; Dorée, M. Initial activation of cyclin-B1-cdc2 kinase requires phosphorylation of cyclin B1. EMBO Rep. 2002, 3, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piasecka, D.; Braun, M.; Kitowska, K.; Mieczkowski, K.; Kordek, R.; Sadej, R.; Romanska, H. FGFs/FGFRs-dependent signalling in regulation of steroid hormone receptors—Implications for therapy of luminal breast cancer. J. Exp. Clin. Cancer Res. CR 2019, 38, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turczyk, L.; Kitowska, K.; Mieszkowska, M.; Mieczkowski, K.; Czaplinska, D.; Piasecka, D.; Kordek, R.; Skladanowski, A.C.; Potemski, P.; Romanska, H.M.; et al. FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia 2017, 19, 791–804. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Q.; Ma, Z.; Shi, Y.; Parris, A.B.; Kong, L.; Yang, X. FGFR1 Overexpression Induces Cancer Cell Stemness and Enhanced Akt/Erk-ER Signaling to Promote Palbociclib Resistance in Luminal A Breast Cancer Cells. Cells 2021, 10, 3008. https://doi.org/10.3390/cells10113008
Cheng Q, Ma Z, Shi Y, Parris AB, Kong L, Yang X. FGFR1 Overexpression Induces Cancer Cell Stemness and Enhanced Akt/Erk-ER Signaling to Promote Palbociclib Resistance in Luminal A Breast Cancer Cells. Cells. 2021; 10(11):3008. https://doi.org/10.3390/cells10113008
Chicago/Turabian StyleCheng, Qiong, Zhikun Ma, Yujie Shi, Amanda B. Parris, Lingfei Kong, and Xiaohe Yang. 2021. "FGFR1 Overexpression Induces Cancer Cell Stemness and Enhanced Akt/Erk-ER Signaling to Promote Palbociclib Resistance in Luminal A Breast Cancer Cells" Cells 10, no. 11: 3008. https://doi.org/10.3390/cells10113008
APA StyleCheng, Q., Ma, Z., Shi, Y., Parris, A. B., Kong, L., & Yang, X. (2021). FGFR1 Overexpression Induces Cancer Cell Stemness and Enhanced Akt/Erk-ER Signaling to Promote Palbociclib Resistance in Luminal A Breast Cancer Cells. Cells, 10(11), 3008. https://doi.org/10.3390/cells10113008