
Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence

 , , and
, , and
Abstract
:1. Introduction
2. Mitochondrial Alterations Associated with Senescence
3. Alterations in Mitochondrial Metabolism Associated with Senescence
4. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence
- (i)
- Targeting ATM signal pathway.
- (ii)
- Targeting ROCK signal pathway.
- (iii)
- Targeting BRAF signal pathway.
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolden, J.E.; Lowe, S.W. 15—Cellular senescence. In The Molecular Basis of Cancer, 4th ed.; Mendelsohn, J., Gray, J.W., Howley, P.M., Israel, M.A., Thompson, C.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 229–238.e222. [Google Scholar]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar] [CrossRef]
- Hwang, E.; Yoon, G.; Kang, H. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 2009, 66, 2503–2524. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Torres, J.; Acín-Perez, R.; Cabezas-Sánchez, P.; Osorio, F.G.; Gonzalez-Gómez, C.; Megias, D.; Cámara, C.; López-Otín, C.; Enríquez, J.A.; Luque-García, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson–Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Din, S.; Konstandin, M.H.; Johnson, B.; Emathinger, J.; Völkers, M.; Toko, H.; Collins, B.; Ormachea, L.; Samse, K.; Kubli, D.A.; et al. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ. Res. 2014, 115, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [Green Version]
- Henze, K.; Martin, W. Evolutionary biology: Essence of mitochondria. Nature 2003, 426, 127–128. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlén, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nistér, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Strack, S.; Cribbs, J.T. Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J. Biol. Chem. 2012, 287, 10990–11001. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, O.S.; Qvit, N.; Haileselassie, B.; Shamloo, M.; Bernardi, P.; Mochly-Rosen, D. Interaction of mitochondrial fission factor with dynamin related protein 1 governs physiological mitochondrial function in vivo. Sci. Rep. 2018, 8, 14034. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Bba Gen. Subj. 2012, 1820, 595–600. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Kissová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Wilson, P.D.; Franks, L.M. The effect of age on mitochondrial ultrastructure. Gerontologia 1975, 21, 81–94. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Jendrach, M.; Pohl, S.; Vöth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 2006, 41, 674–682. [Google Scholar] [CrossRef]
- Benzi, G.; Pastoris, O.; Marzatico, F.; Villa, R.F.; Dagani, F.; Curti, D. The mitochondrial electron transfer alteration as a factor involved in the brain aging. Neurobiol. Aging 1992, 13, 361–368. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; Castelluccio, C.; Fato, R.; Formiggini, G.; Genova, M.L.; Marchetti, M.; Pich, M.M.; Pallotti, F.; Parenti Castelli, G.; et al. Mitochondrial complex I defects in aging. Mol. Cell. Biochem. 1997, 174, 329–333. [Google Scholar] [CrossRef]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Ishii, T.; Miyazawa, M.; Onouchi, H.; Yasuda, K.; Hartman, P.S.; Ishii, N. Model animals for the study of oxidative stress from complex II. Biochim. Biophys. Acta 2013, 1827, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.S.; Byun, H.O.; Cho, H.; Kim, B.K.; Yoon, G. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J. Biol. Chem. 2003, 278, 51577–51586. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.; Kim, H.J.; Yoon, Y.S.; Cho, H.; Lim, I.K.; Lee, J.H. Iron chelation-induced senescence-like growth arrest in hepatocyte cell lines: Association of transforming growth factor beta1 (TGF-beta1)-mediated p27Kip1 expression. Biochem. J. 2002, 366, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.O.; Jung, H.J.; Kim, M.J.; Yoon, G. PKCδ phosphorylation is an upstream event of GSK3 inactivation-mediated ROS generation in TGF-β1-induced senescence. Free Radic. Res. 2014, 48, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.O.; Jung, H.J.; Seo, Y.H.; Lee, Y.K.; Hwang, S.C.; Hwang, E.S.; Yoon, G. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) β1-induced senescence. Exp. Cell Res. 2012, 318, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chadwick, A.; Dart, C.; Kamishima, T.; Quayle, J. Bioenergetic profile of human coronary artery smooth muscle cells and effect of metabolic intervention. PLoS ONE 2017, 12, e0177951. [Google Scholar] [CrossRef] [Green Version]
- Bittles, A.H.; Harper, N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci. Rep. 1984, 4, 751–756. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Stöckl, P.; Hütter, E.; Eigenbrodt, E.; Jansen-Dürr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Goodell, S.; Cortopassi, G. Analysis of oxygen consumption and mitochondrial permeability with age in mice. Mech. Ageing Dev. 1998, 101, 245–256. [Google Scholar] [CrossRef]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol. Aging 2014, 35, 975–989. [Google Scholar] [CrossRef] [Green Version]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Jensen, M.B.; Jasper, H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, A.; Madreiter-Sokolowski, C.; Stryeck, S.; Abdellatif, M. Targeting the mitochondria-proteostasis axis to delay aging. Front. Cell Dev. Biol. 2021, 9, 656201. [Google Scholar] [CrossRef]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Tachibana, T.; Ishikawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta 2014, 1840, 1404–1412. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Brandt, U. Energy converting NADH: Quinone oxidoreductase (complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Treberg, J.R.; Brand, M.D. Chapter 3—Mechanisms of mitochondrial free radical production and their relationship to the aging process. In Handbook of the Biology of Aging, 7th ed.; Masoro, E.J., Austad, S.N., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 47–61. [Google Scholar]
- Song, J.; Pfanner, N.; Becker, T. Assembling the mitochondrial ATP synthase. Proc. Natl. Acad. Sci. USA 2018, 115, 2850–2852. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological chaperones and coenzyme Q10 treatment improves mutant β-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and signaling in mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca(2+) transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Barros, M.H.; Bandy, B.; Tahara, E.B.; Kowaltowski, A.J. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 49883–49888. [Google Scholar] [CrossRef] [Green Version]
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef]
- Ohya, Y.; Umemoto, N.; Tanida, I.; Ohta, A.; Iida, H.; Anraku, Y. Calcium-sensitive cls mutants of Saccharomyces cerevisiae showing a Pet- phenotype are ascribable to defects of vacuolar membrane H(+)-ATPase activity. J. Biol. Chem. 1991, 266, 13971–13977. [Google Scholar] [CrossRef]
- Barcelos, I.P.d.; Haas, R.H. CoQ10 and aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Alcázar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Hirano, M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 183–188. [Google Scholar] [CrossRef]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Olgun, A.; Akman, S.; Serdar, M.A.; Kutluay, T. Oxidative phosphorylation enzyme complexes in caloric restriction. Exp. Gerontol. 2002, 37, 639–645. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.-H. Effects of resveratrol and SIRT1 on PGC-1α activity and mitochondrial biogenesis: A reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Brunetti, D.; Bottani, E.; Segala, A.; Marchet, S.; Rossi, F.; Orlando, F.; Malavolta, M.; Carruba, M.O.; Lamperti, C.; Provinciali, M.; et al. Targeting multiple mitochondrial processes by a metabolic modulator prevents sarcopenia and cognitive decline in SAMP8 mice. Front. Pharmacol. 2020, 11, 1171. [Google Scholar] [CrossRef]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide riboside-the current state of research and therapeutic uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.-H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burmistrova, O.; Olias-Arjona, A.; Lapresa, R.; Jimenez-Blasco, D.; Eremeeva, T.; Shishov, D.; Romanov, S.; Zakurdaeva, K.; Almeida, A.; Fedichev, P.O.; et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Rep. 2019, 9, 11670. [Google Scholar] [CrossRef] [Green Version]
- Weimer, S.; Priebs, J.; Kuhlow, D.; Groth, M.; Priebe, S.; Mansfeld, J.; Merry, T.L.; Dubuis, S.; Laube, B.; Pfeiffer, A.F.; et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 2014, 5, 3563. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Cimolai, M.C.; Alvarez, S.; Bode, C.; Bugger, H. Mitochondrial mechanisms in septic cardiomyopathy. Int. J. Mol. Sci. 2015, 16, 17763–17778. [Google Scholar] [CrossRef]
- Kuk, M.U.; Kim, J.W.; Lee, Y.S.; Cho, K.A.; Park, J.T.; Park, S.C. Alleviation of senescence via ATM Inhibition in accelerated aging models. Mol. Cells 2019, 42, 210. [Google Scholar] [CrossRef]
- Park, J.T.; Kang, H.T.; Park, C.H.; Lee, Y.S.; Cho, K.A.; Park, S.C. A crucial role of ROCK for alleviation of senescence-associated phenotype. Exp. Gerontol. 2018, 106, 8–15. [Google Scholar] [CrossRef]
- Hussain, M.R.M.; Baig, M.; Mohamoud, H.S.A.; Ulhaq, Z.; Hoessli, D.C.; Khogeer, G.S.; Al-Sayed, R.R.; Al-Aama, J.Y. BRAF gene: From human cancers to developmental syndromes. Saudi J. Biol. Sci. 2015, 22, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Kuk, M.U.; Choy, H.E.; Park, S.C.; Park, J.T. Mitochondrial metabolic reprograming via BRAF inhibition ameliorates senescence. Exp. Gerontol. 2019, 126, 110691. [Google Scholar] [CrossRef] [PubMed]
- Marcu, R.; Wiczer, B.M.; Neeley, C.K.; Hawkins, B.J. Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD+/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell Biol. 2014, 34, 2890–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca(2+) Uptake and the fine-tuning of aerobic metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef]
- Jahangir, A.; Sagar, S.; Terzic, A. Aging and cardioprotection. J. Appl. Physiol. 2007, 103, 2120–2128. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A selective and cell-permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after hypoxia/reoxygenation injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef]
- De Jesús García-Rivas, G.; Guerrero-Hernández, A.; Guerrero-Serna, G.; Rodríguez-Zavala, J.S.; Zazueta, C. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J. 2005, 272, 3477–3488. [Google Scholar] [CrossRef]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.M.; Liang, D.D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [Green Version]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where metabolism meets senescence: Focus on endothelial cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.M.; Hong, S.M.; Lee, Y.-K.; Min, S.; Yoon, G. Metabolic features and regulation in cell senescence. BMB Rep. 2019, 52, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Campisi, J. From ancient pathways to aging cells—Connecting metabolism and cellular senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Alteration | Outcome(s) | Experimental Model and References |
|---|---|---|
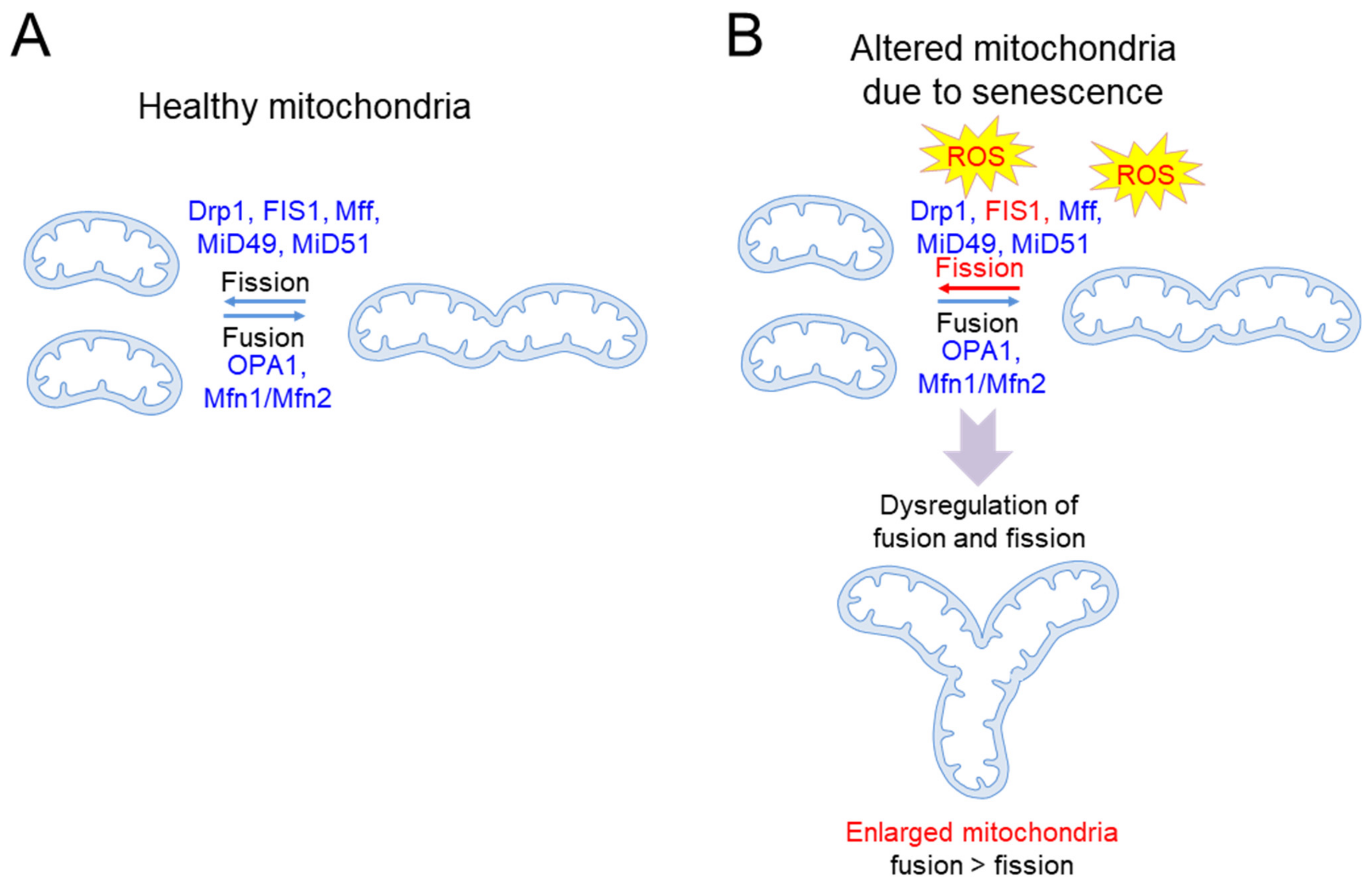
| Alteration in mitochondrial morphology | Formation of giant mitochondria featuring highly interconnected networks | Human fibroblasts [20] |
| A significant increase in the proportion of giant mitochondria | 30-month-old C57/BL mice [22] | |
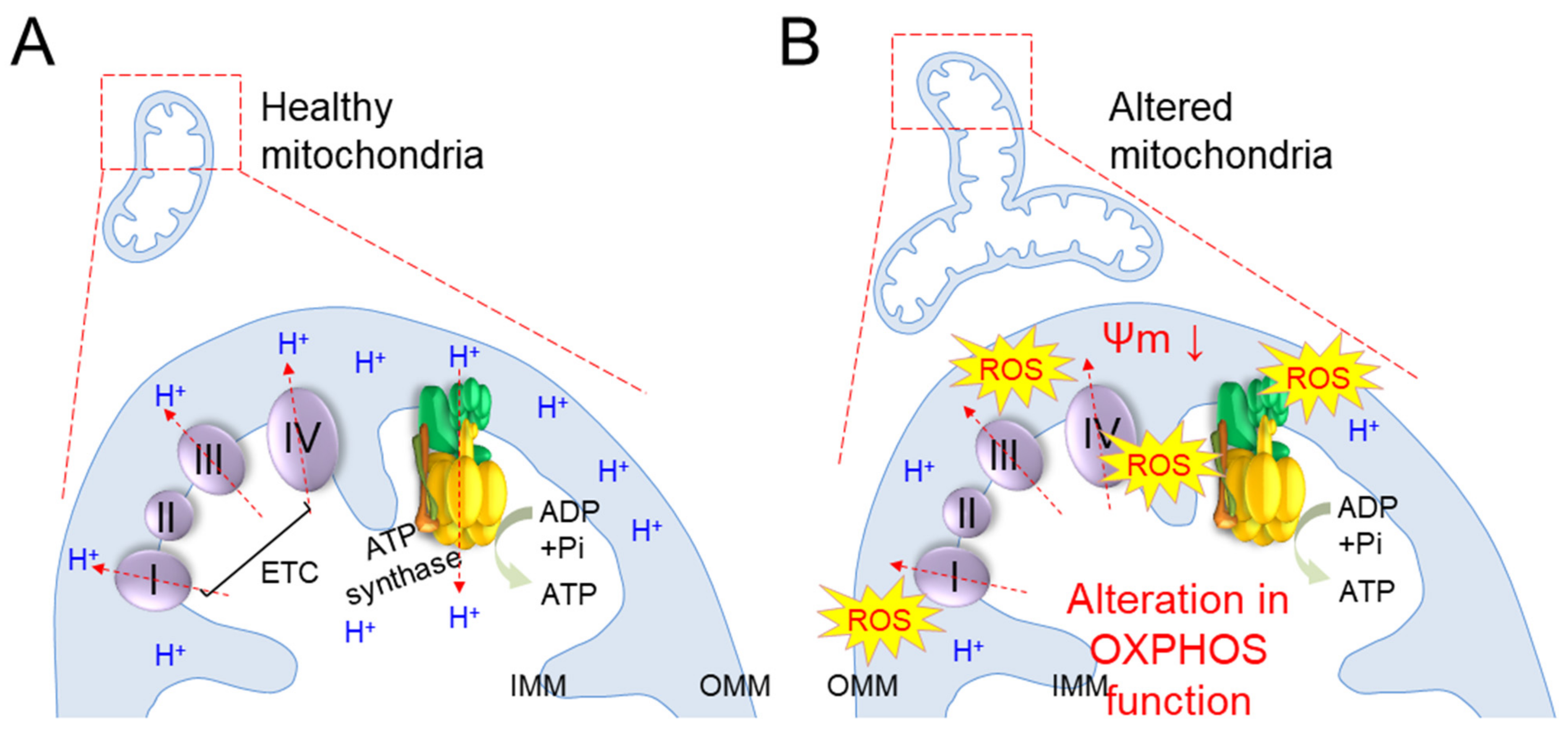
| Alteration in mitochondrial function | Large aggregates of mitochondria with low ΔΨm and impaired ATP production | Senescent endothelial cells [24] |
| The oxidative stress induced by rotenone and antimycin A deteriorates mitochondrial function | Human fibroblasts [25] | |
| Alteration in OXPHOS function | Deterioration of the ETC complexes in liver, brain and muscle tissues Decrease in mitochondrial respiratory function | 20-, 60-, or 100-week-old Wistar rat [26] Tissues from aged rats [27] 2-, 12-, 18-, or 24-month-old C57BL6 mice [28] |
| A mouse model of senescence produced by mev-1 (ortholog of the complex II) mutation exhibits deterioration of OXPHOS accompanying precocious age-dependent corneal physiological changes | Tet-mev-1 conditional transgenic mice [29] | |
| Decrease in complex II activity sustains the disruption of ΔΨm with significantly reduced intracellular ATP levels prior to the acquisition of the senescence phenotype | Chang cells [30] Hepatocyte cell lines [31] | |
| TGF-β1-mediated inhibition of complex IV directly triggers the senescence arrest in mink lung epithelial cells | Mink lung epithelial cells [32,33,34] | |
| Decreasing dependence on OXPHOS but increasing dependence on glycolysis | Glycolysis is upregulated to generate additional ATP to compensate for the loss of energy production in dysfunctional mitochondria | Human coronary artery smooth muscle cells [35] |
| The increase in glucose consumption and lactic acid production | Human fibroblasts [36] | |
| Significant transitions to more glycolytic states | Human fibroblasts [37] | |
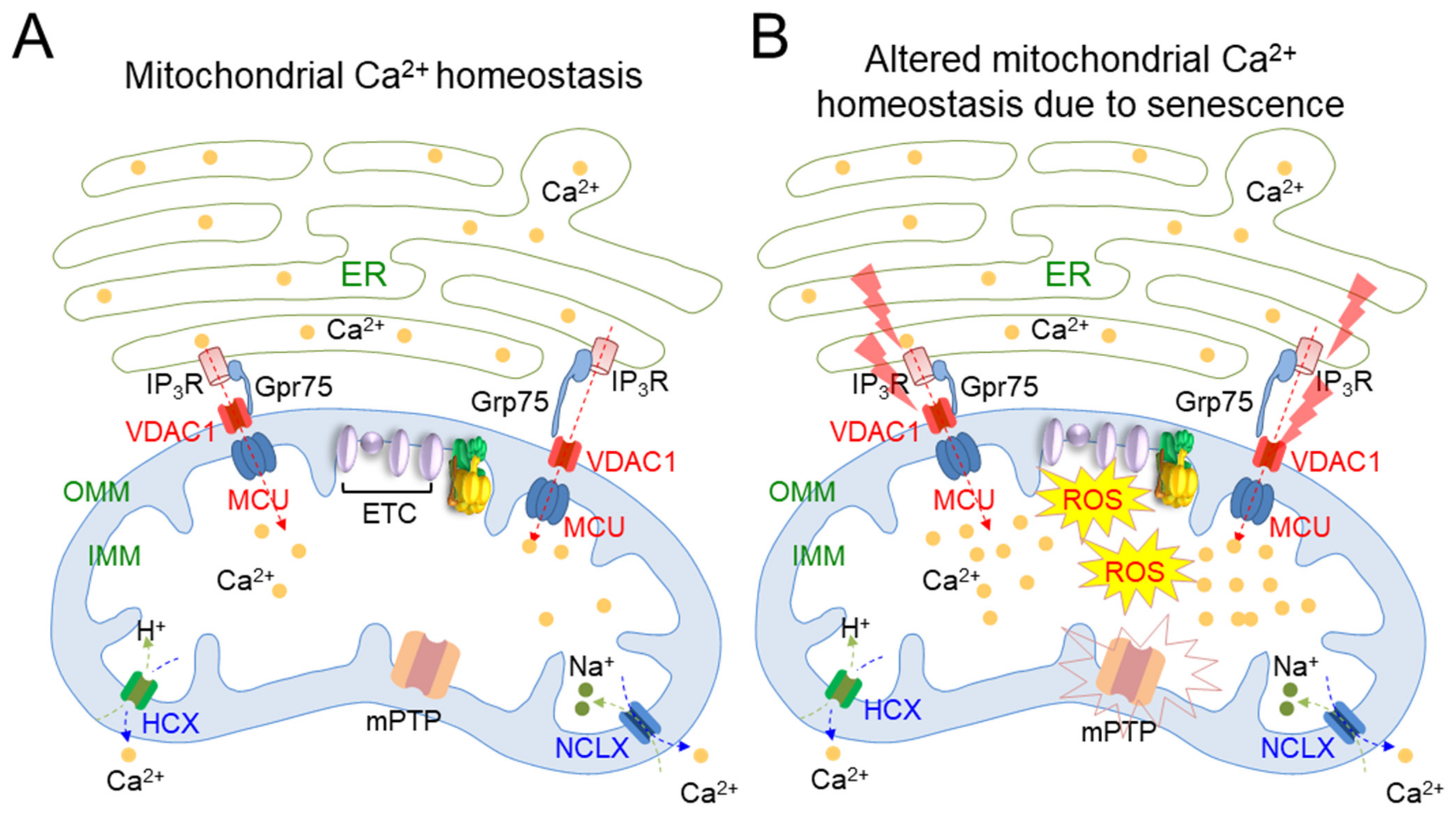
| Alteration in mitochondrial Ca2+ homeostasis | Senescence triggers IP3R to release Ca2+ from the ER and causes VDAC/MCU channels to initiate inward flow of Ca2+ | Human endothelial cells and human fibroblasts [38] |
| Mitochondria overloaded with Ca2+ causes the collapse of electron transport in the ETC | Human endothelial cells and human fibroblasts [38] | |
| Sustained opening of the mitochondrial transition pore (mPTP) | 36-month-old C57BL/6J mice [39] | |
| Then, mPTP opening causes a rapid collapse in ΔΨm and swelling of mitochondria | Neural progenitor cells [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.H.; Park, J.Y.; Lee, H.; Song, E.S.; Kuk, M.U.; Joo, J.; Oh, S.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells 2021, 10, 3003. https://doi.org/10.3390/cells10113003
Lee YH, Park JY, Lee H, Song ES, Kuk MU, Joo J, Oh S, Kwon HW, Park JT, Park SC. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells. 2021; 10(11):3003. https://doi.org/10.3390/cells10113003
Chicago/Turabian StyleLee, Yun Haeng, Ji Yun Park, Haneur Lee, Eun Seon Song, Myeong Uk Kuk, Junghyun Joo, Sekyung Oh, Hyung Wook Kwon, Joon Tae Park, and Sang Chul Park. 2021. "Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence" Cells 10, no. 11: 3003. https://doi.org/10.3390/cells10113003
APA StyleLee, Y. H., Park, J. Y., Lee, H., Song, E. S., Kuk, M. U., Joo, J., Oh, S., Kwon, H. W., Park, J. T., & Park, S. C. (2021). Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells, 10(11), 3003. https://doi.org/10.3390/cells10113003