
Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers



Abstract
1. Introduction
2. Membrane Steroid Receptors and Their Role in Hormone-Sensitive Cancers
2.1. Membrane Androgen Receptors
2.1.1. ZIP9
2.1.2. OXER1
2.1.3. GPRC6A
2.1.4. TRPM8
2.1.5. CaV1.2
2.2. Membrane Estrogen Receptors
2.2.1. GPER
2.2.2. NaV1.2
2.3. Membrane Progesterone Receptors
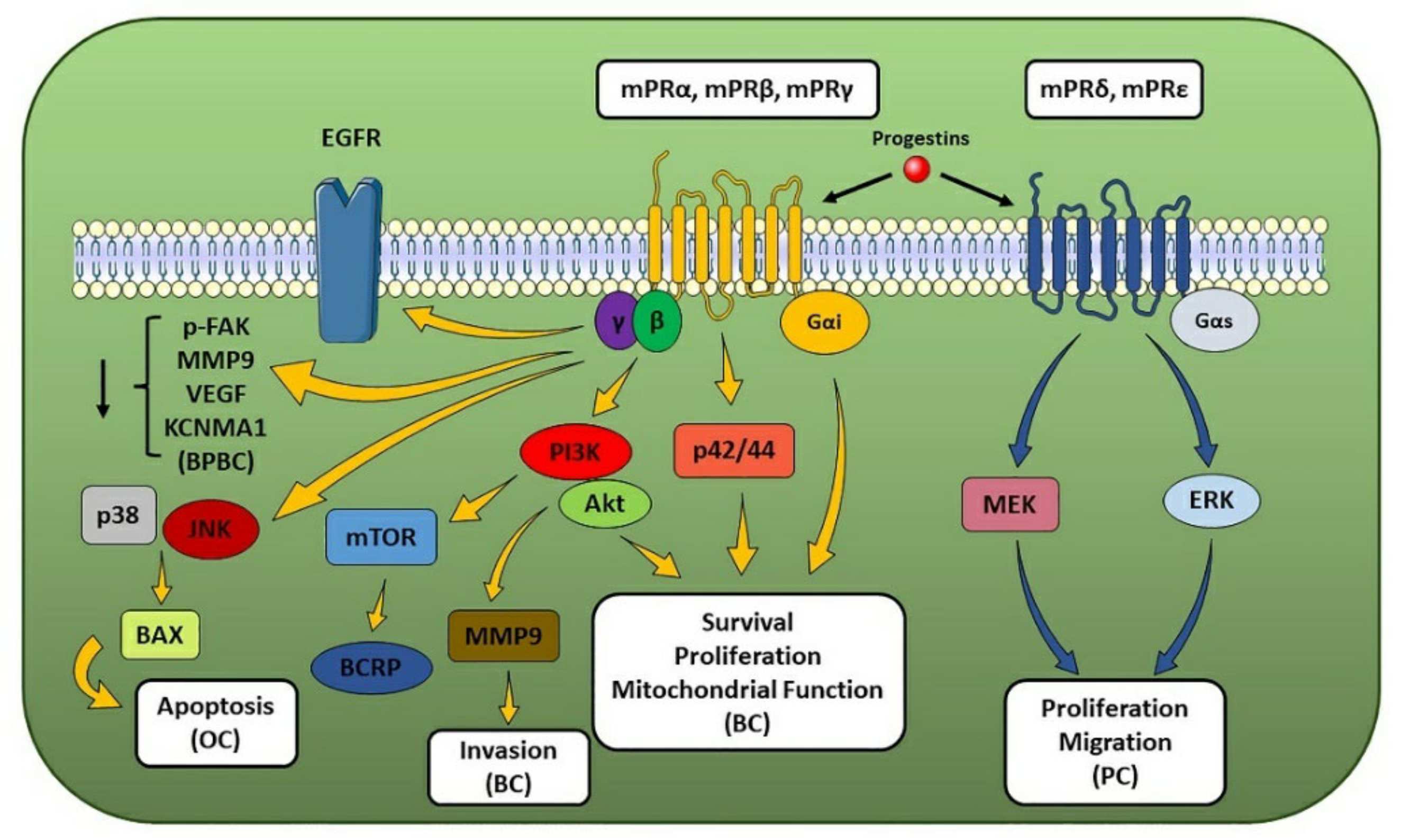
2.3.1. mPRs
2.3.2. MAPRs
3. Beyond mSRs Physiologic Role
3.1. mSRs as EDCs Targets
3.2. mSRs as Drug Targets
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Racchi, M.; Corsini, E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int. J. Mol. Sci. 2020, 21, 9229. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, J.; Corsini, E.; Williams, M.A.; Decker, W.; Manjili, M.H.; Otsuki, T.; Singh, N.; Al-Mulla, F.; Altemaimi, R.; Amedei, A.; et al. Chemical compounds from anthropogenic environment and immune evasion mechanisms: Potential interactions. Carcinogenesis 2015, 36, S111–S127. [Google Scholar] [CrossRef]
- Whirledge, S.; Cidlowski, J.A. Chapter 5-Steroid Hormone Action. In Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th ed.; Strauss, J., Barbieri, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 115–131. [Google Scholar]
- Levin, E.R. Translating extranuclear steroid receptor signaling to clinical medicine. Horm. Cancer 2014, 5, 140–145. [Google Scholar] [CrossRef][Green Version]
- Hammes, S.R.; Levin, E.R. Minireview: Recent advances in extranuclear steroid receptor actions. Endocrinology 2011, 152, 4489–4495. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Sainson, R.C.; Kim, J.K.; Hughes, C.C.; Levin, E.R. A Conserved Mechanism for Steroid Receptor Translocation to the Plasma Membrane. J. Biol. Chem. 2007, 282, 22278–22288. [Google Scholar] [CrossRef]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Treviño, L.S.; Gorelick, D.A. The Interface of Nuclear and Membrane Steroid Signaling. Endocrinology 2021, 162, bqab107. [Google Scholar] [CrossRef] [PubMed]
- Lichten, L.A.; Cousins, R.J. Mammalian Zinc Transporters: Nutritional and Physiologic Regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef]
- Thomas, P.; Converse, A.; Berg, H. ZIP9, a novel membrane androgen receptor and zinc transporter protein. Gen. Comp. Endocrinol. 2018, 257, 130–136. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Dong, J. Membrane androgen receptor characteristics of human ZIP9 (SLC39A) zinc transporter in prostate cancer cells: Androgen-specific activation and involvement of an inhibitory G protein in zinc and MAP kinase signaling. Mol. Cell. Endocrinol. 2017, 447, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Fukunaka, A.; Hagihara, M.; Watanabe, K.; Kamino, S.; Kambe, T.; Enomoto, S.; Hiromura, M. Essential Role of the Zinc Transporter ZIP9/SLC39A9 in Regulating the Activations of Akt and Erk in B-Cell Receptor Signaling Pathway in DT40 Cells. PLoS ONE 2013, 8, e58022. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Dong, J.; Berg, H. Identification and Characterization of Membrane Androgen Receptors in the ZIP9 Zinc Transporter Subfamily: II. Role of Human ZIP9 in Testosterone-Induced Prostate and Breast Cancer Cell Apoptosis. Endocrinology 2014, 155, 4250–4265. [Google Scholar] [CrossRef] [PubMed]
- Pascal, L.; Wang, Z. Unzipping Androgen Action Through ZIP9: A Novel Membrane Androgen Receptor. Endocrinology 2014, 155, 4120–4123. [Google Scholar] [CrossRef][Green Version]
- Bulldan, A.; Bartsch, J.W.; Konrad, L.; Scheiner-Bobis, G. ZIP9 but not the androgen receptor mediates testosterone-induced migratory activity of metastatic prostate cancer cells. Biochim. Biophys. Acta Bioenerg. 2018, 1865, 1857–1868. [Google Scholar] [CrossRef]
- Kalyvianaki, K.; Panagiotopoulos, A.A.; Malamos, P.; Moustou, E.; Tzardi, M.; Stathopoulos, E.N.; Ioannidis, G.S.; Marias, K.; Notas, G.; Theodoropoulos, P.A.; et al. Membrane androgen receptors (OXER1, GPRC6A AND ZIP9) in prostate and breast cancer: A comparative study of their expression. Steroids 2019, 142, 100–108. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Wang, C. Analysis of the prognostic significance of solute carrier (SLC) family 39 genes in breast cancer. Biosci. Rep. 2020, 40, BSR20200764. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.; Ko, S.; In, Y.-H.; Moon, H.-G.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.-H.; Ju, Y.S.; et al. Recurrent fusion transcripts detected by whole-transcriptome sequencing of 120 primary breast cancer samples. Genes Chromosom. Cancer 2015, 54, 681–691. [Google Scholar] [CrossRef]
- Hosoi, T.; Koguchi, Y.; Sugikawa, E.; Chikada, A.; Ogawa, K.; Tsuda, N.; Suto, N.; Tsunoda, S.; Taniguchi, T.; Ohnuki, T. Identification of a Novel Human Eicosanoid Receptor Coupled to Gi/o. J. Biol. Chem. 2002, 277, 31459–31465. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.E.; Rokach, J.; Powell, W.S. 5-Oxo-ETE and the OXE receptor. Prostaglandins Lipid Mediat. 2009, 89, 98–104. [Google Scholar] [CrossRef]
- Sarveswaran, S.; Ghosh, J. OXER1, a G protein-coupled oxoeicosatetraenoid receptor, mediates the survival-promoting effects of arachidonate 5-lipoxygenase in prostate cancer cells. Cancer Lett. 2013, 336, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Konya, V.; Blättermann, S.; Jandl, K.; Platzer, W.; Ottersbach, P.A.; Marsche, G.; Gütschow, M.; Kostenis, E.; Heinemann, A. A Biased Non-Gαi OXE-R Antagonist Demonstrates That Gαi Protein Subunit Is Not Directly Involved in Neutrophil, Eosinophil, and Monocyte Activation by 5-Oxo-ETE. J. Immunol. 2014, 192, 4774–4782. [Google Scholar] [CrossRef]
- Hosoi, T.; Sugikawa, E.; Chikada, A.; Koguchi, Y.; Ohnuki, T. TG1019/OXE, a Gαi/o-protein-coupled receptor, mediates 5-oxo-eicosatetraenoic acid-induced chemotaxis. Biochem. Biophys. Res. Commun. 2005, 334, 987–995. [Google Scholar] [CrossRef]
- Sarveswaran, S.; Thamilselvan, V.; Brodie, C.; Ghosh, J. Inhibition of 5-lipoxygenase triggers apoptosis in prostate cancer cells via down-regulation of protein kinase C-epsilon. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 2108–2117. [Google Scholar] [CrossRef]
- Langlois, A.; Chouinard, F.; Flamand, N.; Ferland, C.; Rola-Pleszczynski, M.; Laviolette, M. Crucial implication of protein kinase C (PKC)-, PKC-, ERK-1/2, and p38 MAPK in migration of human asthmatic eosinophils. J. Leukoc. Biol. 2009, 85, 656–663. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, J.T.; Rogers, L.C.; Paumi, C.M.; Hantgan, R.R.; Thomas, L.R.; Clay, C.E.; High, K.; Chen, Y.Q.; Willingham, M.C.; Smitherman, P.K.; et al. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2005, 1736, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Ghosh, J. Expression of 5-oxoETE receptor in prostate cancer cells: Critical role in survival. Biochem. Biophys. Res. Commun. 2006, 339, 93–98. [Google Scholar] [CrossRef]
- Grant, G.E.; Rubino, S.; Gravel, S.; Wang, X.; Patel, P.; Rokach, J.; Powell, W.S. Enhanced formation of 5-oxo-6,8,11,14-eicosatetraenoic acid by cancer cells in response to oxidative stress, docosahexaenoic acid and neutrophil-derived 5-hydroxy-6,8,11,14-eicosatetraenoic acid. Carcinogenesis 2011, 32, 822–828. [Google Scholar] [CrossRef]
- Kalyvianaki, K.; Gebhart, V.; Peroulis, N.; Panagiotopoulou, C.; Kiagiadaki, F.; Pediaditakis, I.; Aivaliotis, M.; Moustou, E.; Tzardi, M.; Notas, G.; et al. Antagonizing effects of membrane-acting androgens on the eicosanoid receptor OXER1 in prostate cancer. Sci. Rep. 2017, 7, srep44418. [Google Scholar] [CrossRef]
- Kampa, M.; Notas, G.; Castanas, E. Natural extranuclear androgen receptor ligands as endocrine disruptors of cancer cell growth. Mol. Cell. Endocrinol. 2017, 457, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Pelekanou, V.; Castanas, E.; Kampa, M. Conjugated and non-conjugated androgens differentially modulate specific early gene transcription in breast cancer in a cell-specific manner. Steroids 2010, 75, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Kampa, M.; Nifli, A.-P.; Charalampopoulos, I.; Alexaki, V.-I.; Theodoropoulos, P.A.; Stathopoulos, E.N.; Gravanis, A.; Castanas, E. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp. Cell Res. 2005, 307, 41–51. [Google Scholar] [CrossRef]
- Masi, M.; Garattini, E.; Bolis, M.; Di Marino, D.; Maraccani, L.; Morelli, E.; Grolla, A.A.; Fagiani, F.; Corsini, E.; Travelli, C.; et al. OXER1 and RACK1-associated pathway: A promising drug target for breast cancer progression. Onco-Genesis 2020, 9, 1–15. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, R.; Naz, F.; Chauhan, S.S.; Dinda, A.; Shukla, A.A.; Gill, K.; Kapoor, V.; Dey, S. Structure Based Design and Synthesis of Peptide Inhibitor of Human LOX-12: In Vitro and In Vivo Analysis of a Novel Therapeutic Agent for Breast Cancer. PLoS ONE 2012, 7, e32521. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, A.K.; Kumar, M.; Shekhar, S.; Rai, N.; Kaur, P.; Parshad, R.; Dey, S. Serum 5-LOX: A progressive protein marker for breast cancer and new approach for therapeutic target. Carcinogenesis 2016, 37, 912–917. [Google Scholar] [CrossRef] [PubMed]
- R2: Genomics Analysis and Visualization Platform. Available online: http://r2.amc.nl (accessed on 12 July 2021).
- Yi, S.; Zhou, W. Tumorigenesis-related key genes in adolescents and young adults with HR(+)/HER2(−) breast cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 2701–2709. [Google Scholar]
- Pi, M.; Nishimoto, S.K.; Quarles, L.D. GPRC6A: Jack of all metabolism (or master of none). Mol. Metab. 2017, 6, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Pi, M.; Cox, J.V.; Nishimoto, S.K.; Quarles, L.D. CRISPR/Cas9 targeting of GPRC6A suppresses prostate cancer tumorigenesis in a human xenograft model. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef]
- Ye, R.; Pi, M.; Nooh, M.M.; Bahout, S.W.; Quarles, L.D. Human GPRC6A Mediates Testosterone-Induced Mitogen-Activated Protein Kinases and mTORC1 Signaling in Prostate Cancer Cells. Mol. Pharmacol. 2019, 95, 563–572. [Google Scholar] [CrossRef]
- Takata, R.; Akamatsu, S.; Kubo, M.; Takahashi, A.; Hosono, N.; Kawaguchi, T.; Tsunoda, T.; Inazawa, J.; Kamatani, N.; Ogawa, O.; et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat. Genet. 2010, 42, 751–754. [Google Scholar] [CrossRef]
- Long, Q.-Z.; Du, Y.-F.; Ding, X.-Y.; Li, X.; Song, W.-B.; Yang, Y.; Zhang, P.; Zhou, J.-P.; Liu, X.-G. Replication and Fine Mapping for Association of the C2orf43, FOXP4, GPRC6A and RFX6 Genes with Prostate Cancer in the Chinese Population. PLoS ONE 2012, 7, e37866. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Xu, Y.; Yang, K.; Shi, J.-J.; Zhang, X.; Yang, F.; Yuan, H.; Zhu, X.; Zhang, Y.-H.; Wang, J.-Y.; et al. Association of THADA, FOXP4, GPRC6A/RFX6 genes and 8q24 risk alleles with prostate cancer in Northern Chinese men. Off. J. Balk. Union Oncol. 2015, 20, 1223–1228. [Google Scholar]
- Qi, N.; Chen, Y.; Zeng, Y.; Bao, M.; Long, X.; Guo, Y.; Tan, A.; Gao, Y.; Zhang, H.; Yang, X.; et al. rs2274911 polymorphism in GPRC6A associated with serum E2 and PSA in a Southern Chinese male population. Gene 2020, 763, 145067. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhao, Y.-Y.; Yang, F.; Wang, J.-Y.; Shi, X.-H.; Zhu, X.-Q.; Xu, Y.; Wei, D.; Sun, L.; Zhang, Y.-G.; et al. Evidence for a role of GPRC6A in prostate cancer metastasis based on case-control and in vitro analyses. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2235–2248. [Google Scholar]
- Asuthkar, S.; Elustondo, P.A.; Demirkhanyan, L.; Sun, X.; Baskaran, P.; Velpula, K.K.; Thyagarajan, B.; Pavlov, E.V.; Zakharian, E. The TRPM8 Protein Is a Testosterone Receptor. J. Biol. Chem. 2015, 290, 2659–2669. [Google Scholar] [CrossRef] [PubMed]
- Asuthkar, S.; Demirkhanyan, L.; Sun, X.; Elustondo, P.A.; Krishnan, V.; Baskaran, P.; Velpula, K.K.; Thyagarajan, B.; Pavlov, E.V.; Zakharian, E. The TRPM8 Protein Is a Testosterone Receptor. J. Biol. Chem. 2015, 290, 2670–2688. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Yang, Z.; Zhu, G.; Chen, D.; Meng, Z. Menthol Inhibits the Proliferation and Motility of Prostate Cancer DU145 Cells. Pathol. Oncol. Res. 2012, 18, 903–910. [Google Scholar] [CrossRef]
- Liu, Y.; Mikrani, R.; He, Y.; Baig, M.M.F.A.; Abbas, M.; Naveed, M.; Tang, M.; Zhang, Q.; Li, C.; Zhou, X. TRPM8 channels: A review of distribution and clinical role. Eur. J. Pharmacol. 2020, 882, 173312. [Google Scholar] [CrossRef]
- Cunha, A.; Weigle, B.; Kiessling, A.; Bachmann, M.; Rieber, E.P. Tissue-specificity of prostate specific antigens: Comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006, 236, 229–238. [Google Scholar] [CrossRef]
- Lunardi, A.; Barbareschi, M.; Carbone, F.G.; Morelli, L.; Brunelli, M.; Fortuna, N.; Genovesi, S.; Alaimo, A. TRPM8 protein expression in hormone naïve local and lymph node metastatic prostate cancer. Pathologica 2021, 113, 95–101. [Google Scholar] [CrossRef]
- Henshall, S.M.; Afar, D.E.; Hiller, J.; Horvath, L.G.; Quinn, D.I.; Rasiah, K.K.; Gish, K.; Willhite, D.; Kench, J.G.; Gardiner-Garden, M.; et al. Survival analysis of genome-wide gene expression pro-files of prostate cancers identifies new prognostic targets of disease relapse. Cancer Res. 2003, 63, 4196–4203. [Google Scholar]
- Alaimo, A.; De Felice, D.; Genovesi, S.; Lorenzoni, M.; Lunardi, A. Tune the channel: TRPM8 targeting in prostate cancer. Oncoscience 2021, 8, 97–100. [Google Scholar] [CrossRef]
- Zhang, X.S.; Zhang, Y.; Wu, P.X.; Liu, S.J.; Zhou, J.Y.; Liu, S.X. Early diagnosis of prostate cancer by combined use of Trp-p8 expres-sion and PSA density of the transition zone. Zhonghua Nan Ke Xue 2015, 21, 724–728. [Google Scholar]
- Lunger, L.; Retz, M.; Bandur, M.; Souchay, M.; Vitzthum, E.; Jäger, M.; Weirich, G.; Schuster, T.; Autenrieth, M.; Kübler, H.; et al. KLK3 and TMPRSS2 for molecular lymph-node staging in prostate cancer patients undergoing radical prostatectomy. Prostate Cancer Prostatic Dis. 2021, 24, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Bai, V.U.; Murthy, S.; Chinnakannu, K.; Muhletaler, F.; Tejwani, S.; Barrack, E.R.; Kim, S.H.; Menon, M.; Veer Reddy, G.P. Androgen regulated TRPM8 expression: A potential mRNA marker for metastatic prostate cancer detection in body fluids. Int. J. Oncol. 2010, 36, 443–450. [Google Scholar]
- Huskova, Z.; Knillova, J.; Kolar, Z.; Vrbkova, J.; Kral, M.; Bouchal, J. The Percentage of Free PSA and Urinary Markers Distinguish Prostate Cancer from Benign Hyperplasia and Contribute to a More Accurate Indication for Prostate Biopsy. Biomedicines 2020, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High Expression of Transient Receptor Potential Channels in Human Breast Cancer Epithelial Cells and Tissues: Correlation with Pathological Parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Shuai, S.; Ding, D.; Li, R.; Luo, R. TRPM8 promotes aggressiveness of breast cancer cells by regulating EMT via activating AKT/GSK-3β pathway. Tumor Biol. 2014, 35, 8969–8977. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.; Jia, Z.; Zhao, W.; Zhou, C.; Zhang, R.; Ali, D.W.; Michalak, M.; Chen, X.-Z.; Tang, J. Transient Receptor Potential Melastatin 8 (TRPM8) Channel Regulates Proliferation and Migration of Breast Cancer Cells by Activating the AMPK-ULK1 Pathway to Enhance Basal Autophagy. Front. Oncol. 2020, 10, 2645. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Dhennin-Duthille, I.; Gautier, M.; Sevestre, H.; Ahidouch, A. Canaux cationiques TRP dans le cancer du sein: Expression, rôle et corrélation avec des paramètres cliniques. Bull. Cancer 2012, 99, 655–664. [Google Scholar] [CrossRef]
- Pratt, S.J.P.; Lee, R.M.; Chang, K.T.; Hernández-Ochoa, E.O.; Annis, D.A.; Ory, E.C.; Thompson, K.N.; Bailey, P.C.; Mathias, T.J.; Ju, J.A.; et al. Mechanoactivation of NOX2-generated ROS elicits persistent TRPM8 Ca2+ signals that are inhibited by oncogenic KRas. Proc. Natl. Acad. Sci. USA 2020, 117, 26008–26019. [Google Scholar] [CrossRef]
- Zamponi, G.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.G.; Sanderson, J.L.; Gorski, J.A.; Scott, J.D.; Catterall, W.A.; Sather, W.A.; Dell’Acqua, M.L. AKAP-Anchored PKA Maintains Neuronal L-type Calcium Channel Activity and NFAT Transcriptional Signaling. Cell Rep. 2014, 7, 1577–1588. [Google Scholar] [CrossRef]
- Qin, J.; Nag, S.; Wang, W.; Zhou, J.; Zhang, W.-D.; Wang, H.; Zhang, R. NFAT as cancer target: Mission possible? Biochim. Biophys. Acta Bioenerg. 2014, 1846, 297–311. [Google Scholar] [CrossRef]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef]
- Déliot, N.; Constantin, B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.; Peres, C.G.; Vaz, C.V.; Gomes, I.M.; Figueira, M.I.; Cairrão, E.; Verde, I.; Maia, C.J.; Socorro, S. 5α-Dihydrotestosterone regulates the expression of L-type calcium channels and calcium-binding protein regucalcin in human breast cancer cells with suppression of cell growth. Med. Oncol. 2015, 32, 228. [Google Scholar] [CrossRef] [PubMed]
- Scragg, J.L.; Dallas, M.L.; Peers, C. Molecular requirements for L-type Ca2+ channel blockade by testosterone. Cell Calcium 2007, 42, 11–15. [Google Scholar] [CrossRef]
- Soltysik, K.; Czekaj, P. Membrane estrogen receptors—Is it an alternative way of estrogen action? J. Physiol. Pharmacol. 2013, 64, 129–142. [Google Scholar]
- Taheri, M.; Shoorei, H.; Dinger, M.E.; Ghafouri-Fard, S. Perspectives on the Role of Non-Coding RNAs in the Regulation of Expression and Function of the Estrogen Receptor. Cancers 2020, 12, 2162. [Google Scholar] [CrossRef]
- Kampa, M.; Notas, G.; Pelekanou, V.; Troullinaki, M.; Andrianaki, M.; Azariadis, K.; Kampouri, E.; Lavrentaki, K.; Castanas, E. Early membrane initiated transcriptional effects of estrogens in breast cancer cells: First pharmacological evidence for a novel membrane estrogen receptor element (ERx). Steroids 2012, 77, 959–967. [Google Scholar] [CrossRef]
- Elappano, R.; Episano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef]
- Marjon, N.A.; Hu, C.; Hathaway, H.J.; Prossnitz, E.R. G Protein–Coupled Estrogen Receptor Regulates Mammary Tumorigenesis and Metastasis. Mol. Cancer Res. 2014, 12, 1644–1654. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen Signaling through the Transmembrane G Protein-Coupled Receptor GPR30. Annu. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Hugo, E.R.; Brandebourg, T.D.; Woo, J.G.; Loftus, J.; Alexander, J.W.; Ben-Jonathan, N. Bisphenol A at Environmentally Relevant Doses Inhibits Adiponectin Release from Human Adipose Tissue Explants and Adipocytes. Environ. Health Perspect. 2008, 116, 1642–1647. [Google Scholar] [CrossRef]
- Shi, H.; Kumar, S.P.D.S.; Liu, X. G Protein-Coupled Estrogen Receptor in Energy Homeostasis and Obesity Pathogenesis. Prog. Mol. Biol. Transl. Sci. 2013, 114, 193–250. [Google Scholar] [CrossRef]
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in vivo? Front. Endocrinol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Hathaway, H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef]
- Jung, J. Role of G Protein-Coupled Estrogen Receptor in Cancer Progression. Toxicol. Res. 2019, 35, 209–214. [Google Scholar] [CrossRef]
- Talia, M.; De Francesco, E.M.; Rigiracciolo, D.C.; Muoio, M.G.; Muglia, L.; Belfiore, A.; Maggiolini, M.; Sims, A.H.; Lappano, R. The G Protein-Coupled Estrogen Receptor (GPER) Expression Correlates with Pro-Metastatic Pathways in ER-Negative Breast Cancer: A Bioinformatics Analysis. Cells 2020, 9, 622. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, S.; Wang, Z.; Feng, X.; Liu, P.; Lv, X.-B.; Kun-Liang, G.; Yu, F.-X.; Sun, Y.; Yuan, H.; et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J. Clin. Investig. 2015, 125, 2123–2135. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, L.; Liang, S.; Liu, Z.-C.; Zhao, Z.-Y.; Yang, J.; Wang, D.; Yang, D.-Q. GPER stabilizes F-actin cytoskeleton and activates TAZ via PLCβ-PKC and Rho/ROCK-LIMK-Cofilin pathway. Biochem. Biophys. Res. Commun. 2019, 516, 976–982. [Google Scholar] [CrossRef]
- Yang, H.; Wang, C.; Liao, H.; Wang, Q. Activation of GPER by E2 promotes proliferation, invasion and migration of breast cancer cells by regulating the miR-124/CD151 pathway. Oncol. Lett. 2021, 21, 1–9. [Google Scholar] [CrossRef]
- Tao, S.; He, H.; Chen, Q.; Yue, W. GPER mediated estradiol reduces miR-148a to promote HLA-G expression in breast cancer. Biochem. Biophys. Res. Commun. 2014, 451, 74–78. [Google Scholar] [CrossRef]
- Tao, S.; He, H.; Chen, Q. Estradiol induces HOTAIR levels via GPER-mediated miR-148a inhibition in breast cancer. J. Transl. Med. 2015, 13, 1–8. [Google Scholar] [CrossRef]
- Magruder, H.T.; Quinn, J.A.; Schwartzbauer, J.E.; Reichner, J.; Huang, A.; Filardo, E.J. The G Protein-Coupled Estrogen Receptor-1, GPER-1, Promotes Fibrillogenesis via a Shc-Dependent Pathway Resulting in Anchorage-Independent Growth. Horm. Cancer 2014, 5, 390–404. [Google Scholar] [CrossRef][Green Version]
- Santolla, M.F.; Avino, S.; Pellegrino, M.A.; De Francesco, E.M.; De Marco, P.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Rigiracciolo, D.C.; Scarpelli, A.; et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. 2015, 6, e1834. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhu, Q.; Liu, M.; Tu, G.; Li, Q.; Yuan, J.; Wen, S.; Yang, G. GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int. J. Oncol. 2017, 51, 1191–1198. [Google Scholar] [CrossRef]
- Zekas, E.; Prossnitz, E.R. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer 2015, 15, 1–12. [Google Scholar] [CrossRef]
- Chan, Y.; Lai, A.C.; Lin, R.; Wang, Y.; Wang, Y.; Chang, W.-W.; Wu, H.; Lin, Y.; Wu, J.; Yu, J.; et al. GPER-induced signaling is essential for the survival of breast cancer stem cells. Int. J. Cancer 2020, 146, 1674–1685. [Google Scholar] [CrossRef]
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell. Endocrinol. 2020, 506, 110762. [Google Scholar] [CrossRef]
- Ignatov, T.; Treeck, O.; Kalinski, T.; Ortmann, O.; Ignatov, A. GPER-1 expression is associated with a decreased response rate to primary tamoxifen therapy of breast cancer patients. Arch. Gynecol. Obstet. 2020, 301, 565–571. [Google Scholar] [CrossRef]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef] [PubMed]
- Weißenborn, C.; Ignatov, T.; Poehlmann, A.; Wege, A.K.; Costa, S.D.; Zenclussen, A.; Ignatov, A. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 663–671. [Google Scholar] [CrossRef]
- Martin, S.; Lebot, M.N.; Sukkarn, B.; Ball, G.; Green, A.; Rakha, E.A.; Ellis, I.; Storr, S.J. Low expression of G protein-coupled oestrogen receptor 1 (GPER) is associated with adverse survival of breast cancer patients. Oncotarget 2018, 9, 25946–25956. [Google Scholar] [CrossRef]
- Tutzauer, J.; Sjöström, M.; Bendahl, P.-O.; Rydén, L.; Fernö, M.; Leeb-Lundberg, L.M.F.; Alkner, S. Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PLoS ONE 2020, 15, e0231786. [Google Scholar] [CrossRef]
- Weissenborn, C.; Ignatov, T.; Nass, N.; Kalinski, T.; Costa, S.D.; Zenclussen, A.C.; Ignatov, A. GPER Promoter Methylation Controls GPER Expression in Breast Cancer Patients. Cancer Investig. 2017, 35, 1–8. [Google Scholar] [CrossRef]
- Hernández-Silva, C.D.; Villegas-Pineda, J.C.; Pereira-Suárez, A.L. Expression and Role of the G Protein-Coupled Estrogen Receptor (GPR30/GPER) in the Development and Immune Response in Female Reproductive Cancers. Front. Endocrinol. 2020, 11, 544. [Google Scholar] [CrossRef]
- Ignatov, T.; Modl, S.; Thulig, M.; Weißenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J. Ovarian Res. 2013, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Fraungruber, P.; Kaltofen, T.; Heublein, S.; Kuhn, C.; Mayr, D.; Burges, A.; Mahner, S.; Rathert, P.; Jeschke, U.; Trillsch, F. G Protein-Coupled Estrogen Receptor Correlates with Dkk2 Expression and Has Prognostic Impact in Ovarian Cancer Patients. Front. Endocrinol. 2021, 12, 18. [Google Scholar] [CrossRef]
- Heublein, S.; Mayr, D.; Vrekoussis, T.; Friese, K.; Hofmann, S.S.; Jeschke, U.; Lenhard, M. The G-Protein Coupled Estrogen Receptor (GPER/GPR30) is a Gonadotropin Receptor Dependent Positive Prognosticator in Ovarian Carcinoma Patients. PLoS ONE 2013, 8, e71791. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lv, X.; Jiang, C.; Davis, J.S. The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Am. J. Transl. Res. 2012, 4, 390–402. [Google Scholar] [PubMed]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013, 4, e869. [Google Scholar] [CrossRef] [PubMed]
- Schüler-Toprak, S.; Skrzypczak, M.; Ignatov, T.; Ignatov, A.; Ortmann, O.; Treeck, O. G protein-coupled estrogen receptor 1 (GPER-1) and agonist G-1 inhibit growth of ovarian cancer cells by activation of anti-tumoral transcriptome responses: Impact of GPER-1 mRNA on survival. J. Cancer Res. Clin. Oncol. 2020, 146, 3175–3188. [Google Scholar] [CrossRef]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells 2021, 10, 619. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, H.; Wen, H.; Jiang, X.; Cao, X.; Zhang, G.; Liu, G. The novel estrogen receptor GPER regulates the migration and invasion of ovarian cancer cells. Mol. Cell. Biochem. 2013, 378, 1–7. [Google Scholar] [CrossRef]
- Liu, H.; Yan, Y.; Wen, H.; Jiang, X.; Cao, X.; Zhang, G.; Liu, G. A novel estrogen receptor GPER mediates proliferation induced by 17β-estradiol and selective GPER agonist G-1 in estrogen receptor α (ERα)-negative ovarian cancer cells. Cell Biol. Int. 2014, 38, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, X.; Zhao, Y.; Wen, H.; Liu, G. Role of GPER on proliferation, migration and invasion in ligand-independent manner in human ovarian cancer cell line SKOV3. Cell Biochem. Funct. 2015, 33, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Suzawa, M.; Blind, R.; Tobias, S.C.; Bulun, S.E.; Scanlan, T.S.; Ingraham, H.A. Stimulating the GPR30 Estrogen Receptor with a Novel Tamoxifen Analogue Activates SF-1 and Promotes Endometrial Cell Proliferation. Cancer Res. 2009, 69, 5415–5423. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Romeo, E.; De Marco, P.; De Francesco, E.M.; Abonante, S.; Maggiolini, M. GPER mediates the Egr-1 expression induced by 17β-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res. Treat. 2011, 133, 1025–1035. [Google Scholar] [CrossRef]
- Filigheddu, N.; Sampietro, S.; Chianale, F.; Porporato, P.E.; Gaggianesi, M.; Gregnanin, I.; Rainero, E.; Ferrara, M.; Perego, B.; Riboni, F.; et al. Diacylglycerol kinase α mediates 17-β-estradiol-induced proliferation, motility, and anchorage-independent growth of Hec-1A endometrial cancer cell line through the G protein-coupled estrogen receptor GPR30. Cell. Signal. 2011, 23, 1988–1996. [Google Scholar] [CrossRef]
- Deng, J.; Wang, W.; Yu, G.; Ma, X. MicroRNA-195 inhibits epithelial-mesenchymal transition by targeting G protein-coupled estrogen receptor 1 in endometrial carcinoma. Mol. Med. Rep. 2019, 20, 4023–4032. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Chen, Z.; Wang, W. MicroRNA-424 suppresses estradiol-induced cell proliferation via targeting GPER in endometrial cancer cells. Cell. Mol. Boil. 2015, 61, 96–101. [Google Scholar]
- Lau, K.-M.; Ma, F.M.-T.; Xia, J.T.; Chan, Q.K.Y.; Ng, C.-F.; To, K.-F. Activation of GPR30 stimulates GTP-binding of Gαi1 protein to sustain activation of Erk1/2 in inhibition of prostate cancer cell growth and modulates metastatic properties. Exp. Cell Res. 2017, 350, 199–209. [Google Scholar] [CrossRef]
- Chimento, A.; De Luca, A.; Nocito, M.C.; Avena, P.; La Padula, D.; Zavaglia, L.; Pezzi, V. Role of GPER-Mediated Signaling in Testicular Functions and Tumorigenesis. Cells 2020, 9, 2115. [Google Scholar] [CrossRef]
- Chevalier, N.; Hinault, C.; Clavel, S.; Paul-Bellon, R.; Fenichel, P. GPER and Testicular Germ Cell Cancer. Front. Endocrinol. 2021, 11, 1084. [Google Scholar] [CrossRef]
- Kotula-Balak, M.; Gorowska-Wojtowicz, E.; Milon, A.; Pawlicki, P.; Tworzydlo, W.; Płachno, B.J.; Krakowska, I.; Hejmej, A.; Wolski, J.K.; Bilinska, B. Towards understanding leydigioma: Do G protein-coupled estrogen receptor and peroxisome proliferator–activated receptor regulate lipid metabolism and steroidogenesis in Leydig cell tumors? Protoplasma 2020, 257, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Börjesson, S.I.; Elinder, F. Structure, Function, and Modification of the Voltage Sensor in Voltage-Gated Ion Channels. Cell Biophys. 2008, 52, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Fiske, J.L.; Fomin, V.P.; Brown, M.L.; Duncan, R.L.; Sikes, R.A. Voltage-sensitive ion channels and cancer. Cancer Metastasis Rev. 2006, 25, 493–500. [Google Scholar] [CrossRef]
- Jean-Yves, L.; Halima, O.-A.; Olivier, S.; Pierre, B.; Ahmed, A.; Christophe, V. Voltage-Gated Ion Channels, New Targets in Anti-Cancer Research. Recent Pat. Anti-Cancer Drug Discov. 2007, 2, 189–202. [Google Scholar] [CrossRef]
- Shan, B.; Dong, M.; Tang, H.; Wang, N.; Zhang, J.; Yan, C.; Jiao, X.; Zhang, H.; Wang, C. Voltage-gated sodium channels were differentially expressed in human normal prostate, benign prostatic hyperplasia and prostate cancer cells. Oncol. Lett. 2014, 8, 345–350. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, R.; Shen, Y.; Cai, J.; Lei, M.; Wang, L.-Y. Expression of voltage-gated sodium channel α subunit in human ovarian cancer. Oncol. Rep. 2010, 23, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Kow, L.-M.; Pfaff, D.W. Rapid estrogen actions on ion channels: A survey in search for mechanisms. Steroids 2016, 111, 46–53. [Google Scholar] [CrossRef]
- Sula, A.; Hollingworth, D.; Ng, L.C.; Larmore, M.; DeCaen, P.G.; Wallace, B. A tamoxifen receptor within a voltage-gated sodium channel. Mol. Cell 2021, 81, 1160–1169. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Dong, J.; Groenen, P.; Kelder, J.; De Vlieg, J.; Zhu, Y.; Tubbs, C. Steroid and G Protein Binding Characteristics of the Seatrout and Human Progestin Membrane Receptor α Subtypes and Their Evolutionary Origins. Endocrinology 2007, 148, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.S.; Klein, K.; Zanger, U.M. Membrane Associated Progesterone Receptors: Promiscuous Proteins with Pleiotropic Functions—Focus on Interactions with Cytochromes P450. Front. Pharmacol. 2017, 8, 159. [Google Scholar] [CrossRef]
- Zhu, Y.; Bond, J.; Thomas, P. Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P. characteristics of membrane progesterone alpha (mPR?) and progesterone membrane receptor component 1 (PGMRC1) and their roles in mediating rapid progestin actions. Front. Neuroendocrinol. 2008, 29, 292–312. [Google Scholar] [CrossRef]
- Pang, Y.; Dong, J.; Thomas, P. Characterization, Neurosteroid Binding and Brain Distribution of Human Membrane Progesterone Receptors δ and ϵ (mPRδ and mPRϵ) and mPRδ Involvement in Neurosteroid Inhibition of Apoptosis. Endocrinology 2013, 154, 283–295. [Google Scholar] [CrossRef]
- Dosiou, C.; Hamilton, A.E.; Pang, Y.; Overgaard, M.T.; Tulac, S.; Dong, J.; Thomas, P.; Guidice, L.C. Expression of membrane proges-terone receptors (mPRs) on human T lymphocytes and Jurkat cells and activation of G proteins by progesterone. J. Endocrinol. 2008, 196, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Dressing, G.E.; Thomas, P. Identification of membrane progestin receptors in human breast cancer cell lines and biopsies and their potential involvement in breast cancer. Steroids 2007, 72, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.F.; Thomas, P. Progesterone signals through membrane progesterone receptors (mPRs) in MDA-MB-468 and mPR-transfected MDA-MB-231 breast cancer cells which lack full-length and C-terminal truncated isoforms of the nuclear progesterone receptor. Steroids 2011, 76, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Li, W.; You, S. Progesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells by a mem-brane progesterone receptor alpha mediated pathway. Breast Cancer Res. 2010, 12, R34. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Thomas, P.; Lange, C.A. Signaling events mediated by membrane progesterone receptors (mPRs) in ovarian cancer cells. Horm. Cancer 2010, 1, 167–176. [Google Scholar] [CrossRef]
- Wu, X.; Sun, L.; Wang, X.; Su, P.; Li, Z.; Zhang, C.; Wang, Y.; Gao, P.; Ma, R. Breast Cancer Invasion and Metastasis by mPRα Through the PI3K/Akt Signaling Pathway. Pathol. Oncol. Res. 2015, 22, 471–476. [Google Scholar] [CrossRef]
- Xie, M.; Zhu, X.; Liu, Z.; Shrubsole, M.; Varma, V.; Mayer, I.A.; Dai, Q.; Chen, Q.; You, S. Membrane Progesterone Receptor Alpha as a Potential Prognostic Biomarker for Breast Cancer Survival: A Retrospective Study. PLoS ONE 2012, 7, e35198. [Google Scholar] [CrossRef]
- Dressing, G.E.; Alyea, R.; Pang, Y.; Thomas, P. Membrane Progesterone Receptors (mPRs) Mediate Progestin Induced Antimorbidity in Breast Cancer Cells and Are Expressed in Human Breast Tumors. Horm. Cancer 2012, 3, 101–112. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, J.; Li, W.; Zhang, C.; Su, P.; Wang, Y.; Sun, W.; Wang, X.; Li, L.; Wu, X. Rapamycin Antagonizes BCRP-Mediated Drug Resistance Through the PI3K/Akt/mTOR Signaling Pathway in mPRα-Positive Breast Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, W.; Zhang, H.; Hu, Y.; Yu, L.; Zhang, Y.; Zhang, Y.; Wang, S.; Wang, P.; Xia, W. Progesterone suppresses triple-negative breast cancer growth and metastasis to the brain via membrane progesterone receptor α. Int. J. Mol. Med. 2017, 40, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhou, L.; Chen, X.; Gainey, L.O.; Xiao, J.; Nanes, M.S.; Hou, A.; You, S.; Chen, Q. Progesterone and Src Family Inhibitor PP1 Synergistically Inhibit Cell Migration and Invasion of Human Basal Phenotype Breast Cancer Cells. BioMed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Romero-Sánchez, M.; Peiper, S.C.; Evans, B.; Wang, Z.; Catasús, L.; Ribe, A.; Prat, J.; Giri, J.G. Expression profile of heptahelical putative membrane progesterone receptors in epithelial ovarian tumors. Hum. Pathol. 2008, 39, 1026–1033. [Google Scholar] [CrossRef]
- Charles, N.J.; Thomas, P.; Lange, C.A. Expression of Membrane Progesterone Receptors (mPR/PAQR) in Ovarian Cancer Cells: Implications for Progesterone-Induced Signaling Events. Horm. Cancer 2010, 1, 167–176. [Google Scholar] [CrossRef]
- Sinreih, M.; Knific, T.; Thomas, P.; Grazio, S.F.; Rižner, T.L. Membrane progesterone receptors β and γ have potential as prognostic biomarkers of endometrial cancer. J. Steroid Biochem. Mol. Biol. 2018, 178, 303–311. [Google Scholar] [CrossRef]
- Li, B.; Lin, Z.; Liang, Q.; Hu, Y.; Xu, W.-F. PAQR6 expression enhancement suggests a worse prognosis in prostate cancer patients. Open Life Sci. 2018, 13, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Nakayama, Y.; Konishi, M.; Terasawa, K.; Ohta, M.; Itoh, N.; Fujimoto, M. Functions of MAPR (membrane-associated progesterone receptor) family members as heme/steroid-binding proteins. Curr. Protein Pept. Sci. 2012, 13, 687–696. [Google Scholar] [CrossRef]
- Ruan, X.; Zhang, Y.; Mueck, A.O.; Willibald, M.; Seeger, H.; Fehm, T.; Brucker, S.; Neubauer, H. Increased expression of progesterone receptor membrane component 1 is associated with aggressive phenotype and poor prognosis in ER-positive and negative breast cancer. Menopause 2017, 24, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-T.; May, E.W.S.; Chang, J.-F.; Hu, R.-Y.; Wang, L.H.-C.; Chan, H.-L. PGRMC1 contributes to doxorubicin-induced chemoresistance in MES-SA uterine sarcoma. Cell. Mol. Life Sci. 2015, 72, 2395–2409. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, S.Y.; Choi, H.S.; Kim, M.K.; Lee, H.M.; Jang, Y.-J.; Ryu, C.J. Progesterone Receptor Membrane Component 1 suppresses the p53 and Wnt/β-catenin pathways to promote human pluripotent stem cell self-renewal. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Thejer, B.M.; Adhikary, P.P.; Kaur, A.; Teakel, S.L.; Van Oosterum, A.; Seth, I.; Pajic, M.; Hannan, K.M.; Pavy, M.; Poh, P.; et al. PGRMC1 phosphorylation affects cell shape, motility, glycolysis, mitochondrial form and function, and tumor growth. BMC Mol. Cell Biol. 2020, 21, 1–24. [Google Scholar] [CrossRef]
- Neubauer, H.; Ma, Q.; Zhou, J.; Yu, Q.; Ruan, X.; Seeger, H.; Fehm, T.; Mueck, A.O. Possible role of PGRMC1 in breast cancer development. Climacteric 2013, 16, 509–513. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Rohe, H.J.; Twist, K.E.; Mattingly, M.N.; Craven, R.J. Progesterone Receptor Membrane Component 1 (Pgrmc1): A Heme-1 Domain Protein That Promotes Tumorigenesis and Is Inhibited by a Small Molecule. J. Pharmacol. Exp. Ther. 2010, 333, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Wu, A.; Yang, H. Expression of progesterone receptor membrane component-1 is associated with the malignant pheno-types of breast cancer. Nan Fang Yi Ke Da Xue Xue Bao 2012, 32, 635–638. [Google Scholar]
- Cai, G.; Yang, X.; Ruan, X.; Wang, J.; Fang, Y.; Wei, Y.; Zhang, Y.; Gu, M.; Mueck, A.O. Association of circulating Progesterone Receptor Membrane Component-1 (PGRMC1) with PGRMC1 expression in breast tumour tissue and with clinical breast tumour characteristics. Maturitas 2020, 140, 64–71. [Google Scholar] [CrossRef]
- Ruan, X.; Cai, G.; Wei, Y.; Gu, M.; Zhang, Y.; Zhao, Y.; Mueck, A.O. Association of circulating Progesterone Receptor Membrane Component-1 (PGRMC1) with breast tumor characteristics and comparison with known tumor markers. Menopause 2020, 27, 183–193. [Google Scholar] [CrossRef]
- Clark, N.C.; Friel, A.; Pru, C.A.; Zhang, L.; Shioda, T.; Rueda, B.R.; Peluso, J.J.; Pru, J.K. Progesterone receptor membrane component 1 promotes survival of human breast cancer cells and the growth of xenograft tumors. Cancer Biol. Ther. 2016, 17, 262–271. [Google Scholar] [CrossRef]
- Neubauer, H.; Adam, G.; Seeger, H.; Mueck, A.O.; Solomayer, E.; Wallwiener, D.; Cahill, M.; Fehm, T. Membrane-initiated effects of progesterone on proliferation and activation of VEGF in breast cancer cells. Climacteric 2009, 12, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Lee, Y.H.; Jo, S.L.; Heo, J.H.; Kim, G.; Lee, G.-S.; An, B.-S.; Baek, I.-J.; Hong, E.-J. Absence of progesterone receptor membrane component 1 reduces migration and metastasis of breast cancer. Cell Commun. Signal. 2021, 19, 1–11. [Google Scholar] [CrossRef]
- Gu, M.; Wang, L.; Yang, C.; Li, X.; Jia, C.; Croteau, S.; Ruan, X.; Hardy, P. Micro-RNA-181a suppresses progestin-promoted breast cancer cell growth. Maturitas 2018, 114, 60–66. [Google Scholar] [CrossRef]
- Cantonero, C.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. PGRMC1 Inhibits Progesterone-Evoked Proliferation and Ca2+ Entry Via STIM2 in MDA-MB-231 Cells. Int. J. Mol. Sci. 2020, 21, 7641. [Google Scholar] [CrossRef]
- Pedroza, D.A.; Rajamanickam, V.; Subramani, R.; Bencomo, A.; Galvez, A.; Lakshmanaswamy, R. Progesterone receptor membrane component 1 promotes the growth of breast cancers by altering the phosphoproteome and augmenting EGFR/PI3K/AKT signalling. Br. J. Cancer 2020, 123, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Asperger, H.; Stamm, N.; Gierke, B.; Pawlak, M.; Hofmann, U.; Zanger, U.M.; Marton, A.; Katona, R.L.; Buhala, A.; Vizler, C.; et al. Progesterone receptor membrane component 1 regulates lipid homeostasis and drives oncogenic signaling resulting in breast cancer progression. Breast Cancer Res. 2020, 22, 1–16. [Google Scholar] [CrossRef]
- Pedroza, D.A.; Subramani, R.; Tiula, K.; Do, A.; Rashiraj, N.; Galvez, A.; Chatterjee, A.; Bencomo, A.; Rivera, S.; Lakshmanaswamy, R. Crosstalk between progesterone receptor membrane component 1 and estrogen receptor α promotes breast cancer cell proliferation. Lab. Investig. 2021, 101, 733–744. [Google Scholar] [CrossRef]
- Ponikwicka-Tyszko, D.; Chrusciel, M.; Pulawska, K.; Bernaczyk, P.; Sztachelska, M.; Guo, P.; Li, X.; Toppari, J.; Huhtaniemi, I.T.; Wolczynski, S.; et al. Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice. Cancers 2020, 12, 3263. [Google Scholar] [CrossRef]
- Friel, A.; Zhang, L.; Pru, C.A.; Clark, N.C.; McCallum, M.L.; Blok, L.J.; Shioda, T.; Peluso, J.J.; Rueda, B.R.; Pru, J.K. Progesterone receptor membrane component 1 deficiency attenuates growth while promoting chemosensitivity of human endometrial xenograft tumors. Cancer Lett. 2015, 356, 434–442. [Google Scholar] [CrossRef]
- Peluso, J.J. Progesterone signaling mediated through progesterone receptor membrane component-1 in ovarian cells with special emphasis on ovarian cancer. Steroids 2011, 76, 903–909. [Google Scholar] [CrossRef][Green Version]
- Zhu, X.; Han, Y.; Fang, Z.; Wu, W.; Ji, M.; Teng, F.; Zhu, W.; Yang, X.; Jia, X.; Zhang, C. Progesterone protects ovarian cancer cells from cisplatin-induced inhibitory effects through progesterone receptor membrane component 1/2 as well as AKT signaling. Oncol. Rep. 2013, 30, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Kasubuchi, M.; Terasawa, K.; Kimura, I. Perspectives on Membrane-associated Progesterone Receptors as Prospective Therapeutic Targets. Curr. Drug Targets 2016, 17, 1189–1197. [Google Scholar] [CrossRef]
- Schwickert, A.; Weghake, E.; Brüggemann, K.; Engbers, A.; Brinkmann, B.; Kemper, B.; Seggewiß, J.; Stock, C.; Ebnet, K.; Kiesel, L.; et al. microRNA miR-142-3p Inhibits Breast Cancer Cell Invasiveness by Synchronous Targeting of WASL, Integrin Alpha V, and Additional Cytoskeletal Elements. PLoS ONE 2015, 10, e0143993. [Google Scholar] [CrossRef] [PubMed]
- Fahlén, M.; Zhang, H.; Lofgren, L.; Masironi, B.; Von Schoultz, E.; Von Schoultz, B.; Sahlin, L. Expression of Progesterone and Androgen Receptors in the Breast of Premenopausal Women, Considering Menstrual Phase. Anticancer. Res. 2018, 38, 1499–1510. [Google Scholar] [CrossRef]
- Causey, M.W.; Huston, L.J.; Harold, D.M.; Charaba, C.J.; Ippolito, D.L.; Hoffer, Z.S.; Brown, T.A.; Stallings, J. Transcriptional Analysis of Novel Hormone Receptors PGRMC1 and PGRMC2 as Potential Biomarkers of Breast Adenocarcinoma Staging. J. Surg. Res. 2011, 171, 615–622. [Google Scholar] [CrossRef]
- Albrecht, C.; Huck, V.; Wehling, M.; Wendler, A. In vitro inhibition of SKOV-3 cell migration as a distinctive feature of progesterone receptor membrane component type 2 versus type. Steroids 2012, 77, 1543–1550. [Google Scholar] [CrossRef]
- Han, K.-H.; Lee, S.-H.; Ha, S.-A.; Kim, H.K.; Lee, C.; Kim, D.-H.; Gong, K.H.; Yoo, J.; Kim, S.; Kim, J.W. The functional and structural characterization of a novel oncogene GIG47 involved in the breast tumorigenesis. BMC Cancer 2012, 12, 274. [Google Scholar] [CrossRef]
- Ohta, H.; Kimura, I.; Konishi, M.; Itoh, N. Neudesin as a unique secreted protein with multi-functional roles in neural functions, energy metabolism, and tumorigenesis. Front. Mol. Biosci. 2015, 2, 24. [Google Scholar] [CrossRef]
- Bruce, A.; Rybak, A.P. CYB5D2 Requires Heme-Binding to Regulate HeLa Cell Growth and Confer Survival from Chemotherapeutic Agents. PLoS ONE 2014, 9, e86435. [Google Scholar] [CrossRef]
- Xie, Y.; Shen, Y.T.; Kapoor, A.; Ojo, D.; Wei, F.; De Melo, J.; Lin, X.; Wong, N.; Yan, J.; Tao, L.; et al. CYB5D2 displays tumor suppression activities towards cervical cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 556–565. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. Novel mechanism of endocrine disruption by fungicides through binding to the membrane androgen receptor, ZIP9 (SLC39A9), and antagonizing rapid testosterone induction of the intrinsic apoptotic pathway. Steroids 2019, 149, 108415. [Google Scholar] [CrossRef] [PubMed]
- Périan, S.; Vanacker, J.-M. GPER as a Receptor for Endocrine-Disrupting Chemicals (EDCs). Front. Endocrinol. 2020, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Qie, Y.; Qin, W.; Zhao, K.; Liu, C.; Zhao, L.; Guo, L.-H. Environmental Estrogens and Their Biological Effects through GPER Mediated Signal Pathways. Environ. Pollut. 2021, 278, 116826. [Google Scholar] [CrossRef]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef]
- Xu, F.; Wang, X.; Wu, N.; He, S.; Yi, W.; Xiang, S.; Zhang, P.; Xie, X.; Ying, C. Bisphenol A induces proliferative effects on both breast cancer cells and vascular endothelial cells through a shared GPER-dependent pathway in hypoxia. Environ. Pollut. 2017, 231, 1609–1620. [Google Scholar] [CrossRef]
- Cao, L.; Ren, X.-M.; Liang-Hong, G.; Zhang, J.; Qin, W.-P.; Yang, Y.; Wan, B.; Guo, L.-H. Bisphenol AF and Bisphenol B Exert Higher Estrogenic Effects than Bisphenol A via G Protein-Coupled Estrogen Receptor Pathway. Environ. Sci. Technol. 2017, 51, 11423–11430. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Jiang, G.; Wu, Y.; Li, J.; Liang, W.; Chen, L.; Su, Q.; Li, W.; Du, J.; Wong, C.K.; et al. GPER/Hippo-YAP signal is involved in Bisphenol S induced migration of triple negative breast cancer (TNBC) cells. J. Hazard. Mater. 2018, 355, 1–9. [Google Scholar] [CrossRef]
- Lei, B.; Sun, S.; Zhang, X.; Feng, C.; Xu, J.; Wen, Y.; Huang, Y.; Wu, M.; Yu, Y. Bisphenol AF exerts estrogenic activity in MCF-7 cells through activation of Erk and PI3K/Akt signals via GPER signaling pathway. Chemosphere 2019, 220, 362–370. [Google Scholar] [CrossRef]
- Lei, B.; Tang, Q.; Sun, S.; Zhang, X.; Huang, Y.; Xu, L. Insight into the mechanism of tetrachlorobisphenol A (TCBPA)-induced proliferation of breast cancer cells by GPER-mediated signaling pathways. Environ. Pollut. 2021, 275, 116636. [Google Scholar] [CrossRef]
- Castillo-Sanchez, R.; Ramirez-Ricardo, J.; Martinez-Baeza, E.; Cortes-Reynosa, P.; Candanedo-Gonzales, F.; Gomez, R.; Salazar, E.P. Bisphenol A induces focal adhesions assembly and activation of FAK, Src and ERK2 via GPER in MDA-MB-231 breast cancer cells. Toxicol. Vitr. 2020, 66, 104871. [Google Scholar] [CrossRef]
- Chevalier, N.; Bouskine, A.; Fenichel, P. Bisphenol A promotes testicular seminoma cell proliferation through GPER/GPR. Int. J. Cancer 2012, 130, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Gorowska-Wojtowicz, E.; Duliban, M.; Kudrycka, M.; Dutka, P.; Pawlicki, P.; Milon, A.; Zarzycka, M.; Placha, W.; Kotula-Balak, M.; Ptak, A.; et al. Leydig cell tumorigenesis—Implication of G-protein coupled membrane estrogen receptor, peroxisome proliferator-activated receptor and xenoestrogen exposure. In vivo and in vitro appraisal. Tissue Cell 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Galbiati, V.; Maddalon, A.; Iulini, M.; Kenda, M.; Dolenc, M.S.; Marinovich, M.; Racchi, M.; Corsini, E. Effect of estrogen-active compounds on the expression of RACK1 and immunological implications. Arch. Toxicol. 2020, 94, 2081–2095. [Google Scholar] [CrossRef]
- Buoso, E.; Kenda, M.; Masi, M.; Linciano, P.; Galbiati, V.; Racchi, M.; Dolenc, M.S.; Corsini, E. Effects of Bisphenols on RACK1 Expression and Their Immunological Implications in THP-1 Cells. Front. Pharmacol. 2021, 12, 2585. [Google Scholar] [CrossRef]
- Racchi, M.; Buoso, E.; Ronfani, M.; Serafini, M.M.; Galasso, M.; Lanni, C.; Corsini, E. Role of Hormones in the Regulation of RACK1 Expression as a Signaling Checkpoint in Immunosenescence. Int. J. Mol. Sci. 2017, 18, 1453. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Cirillo, F.; Lappano, R.; Bruno, L.; Rizzuti, B.; Grande, F.; Guzzi, R.; Briguori, S.; Miglietta, A.M.; Nakajima, M.; Di Martino, M.T.; et al. AHR and GPER mediate the stimulatory effects induced by 3-methylcholanthrene in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef]
- Tokumoto, T.; Tokumoto, M.; Thomas, P. Interactions of Diethylstilbestrol (DES) and DES Analogs with Membrane Progestin Receptor-α and the Correlation with Their Nongenomic Progestin Activities. Endocrinology 2007, 148, 3459–3467. [Google Scholar] [CrossRef]
- Seeger, H.; Ruan, X.; Neubauer, H.; Brucker, S.; Mueck, A.O. Membrane-initiated effects of Serelys® on proliferation and apoptosis of human breast cancer cells. Gynecol. Endocrinol. 2018, 34, 353–356. [Google Scholar] [CrossRef]
- Li, X.; Ruan, X.; Gu, M.; Mueck, A.O. PGRMC1 can trigger estrogen-dependent proliferation of breast cancer cells: Estradiol vs. equilin vs. ethinylestradiol. Climacteric 2019, 22, 483–488. [Google Scholar] [CrossRef]
- Neubauer, H.; Yang, Y.; Seeger, H.; Fehm, T.; Cahill, M.A.; Tong, X.; Ruan, X.; Mueck, A.O. The presence of a membrane-bound progesterone receptor sensitizes the estradiol-induced effect on the proliferation of human breast cancer cells. Menopause 2011, 18, 845–850. [Google Scholar] [CrossRef]
- Izquierdo, C.; Martín-Martínez, M.; Gómez-Monterrey, I.; González-Muñiz, R. TRPM8 Channels: Advances in Structural Studies and Pharmacological Modulation. Int. J. Mol. Sci. 2021, 22, 8502. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. (−)-Epicatechin acts as a potent agonist of the membrane androgen receptor, ZIP9 (SLC39A9), to promote apoptosis of breast and prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2021, 211, 105906. [Google Scholar] [CrossRef]
- Bulldan, A.; Malviya, V.N.; Upmanyu, N.; Konrad, L.; Scheiner-Bobis, G. Testosterone/bicalutamide antagonism at the predicted extracellular androgen binding site of ZIP9. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 2402–2414. [Google Scholar] [CrossRef]
- Stepniewski, T.M.; Torrens-Fontanals, M.; Rodríguez-Espigares, I.; Giorgino, T.; Primdahl, K.G.; Vik, A.; Stenstrøm, Y.; Selent, J.; Hansen, T.V. Synthesis, molecular modelling studies and biological evaluation of new oxoeicosanoid receptor 1 agonists. Bioorg. Med. Chem. 2018, 26, 3580–3587. [Google Scholar] [CrossRef]
- Chourey, S.; Ye, Q.; Reddy, C.N.; Cossette, C.; Gravel, S.; Zeller, M.; Slobodchikova, I.; Vuckovic, D.; Rokach, J.; Powell, W.S. In vivo α-hydroxylation of a 2-alkylindole antagonist of the OXE receptor for the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid in monkeys. Biochem. Pharmacol. 2017, 138, 107–118. [Google Scholar] [CrossRef]
- Cossette, C.; Chourey, S.; Ye, Q.; Reddy, C.N.; Gore, V.; Gravel, S.; Slobodchikova, I.; Vuckovic, D.; Rokach, J.; Powell, W.S. Pharmacokinetics and Metabolism of Selective Oxoeicosanoid (OXE) Receptor Antagonists and Their Effects on 5-Oxo-6,8,11,14-eicosatetraenoic Acid (5-Oxo-ETE)-Induced Granulocyte Activation in Monkeys. J. Med. Chem. 2016, 59, 10127–10146. [Google Scholar] [CrossRef]
- Ye, Q.; Chourey, S.; Reddy, C.N.; Wang, R.; Cossette, C.; Gravel, S.; Slobodchikova, I.; Vuckovic, D.; Rokach, J.; Powell, W.S. Novel highly potent OXE receptor antagonists with prolonged plasma lifetimes that are converted to active metabolites in vivo in monkeys. Br. J. Pharmacol. 2020, 177, 388–401. [Google Scholar] [CrossRef]
- Chourey, S.; Ye, Q.; Reddy, C.N.; Wang, R.; Cossette, C.; Gravel, S.; Slobodchikova, I.; Vuckovic, D.; Rokach, J.; Powell, W.S. Novel Highly Potent and Metabolically Resistant Oxoeicosanoid (OXE) Receptor Antagonists That Block the Actions of the Granulocyte Chemoattractant 5-Oxo-6,8,11,14-Eicosatetraenoic Acid (5-oxo-ETE). J. Med. Chem. 2018, 61, 5934–5948. [Google Scholar] [CrossRef]
- Reddy, C.N.; Alhamza, H.; Chourey, S.; Ye, Q.; Gore, V.; Cossette, C.; Gravel, S.; Slobodchikova, I.; Vuckovic, D.; Rokach, J.; et al. Metabolism and pharmacokinetics of a potent N-acylindole antagonist of the OXE receptor for the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid (5-oxo-ETE) in rats and monkeys. Eur. J. Pharm. Sci. 2018, 115, 88–99. [Google Scholar] [CrossRef]
- Pi, M.; Kapoor, K.; Ye, R.; Hwang, D.-J.; Miller, D.D.; Smith, J.C.; Baudry, J.; Quarles, L.D. Computationally identified novel agonists for GPRC6A. PLoS ONE 2018, 13, e0195980. [Google Scholar] [CrossRef]
- Ishida, Y.; Kitayama, K.; Hanada, K.; Shibutani, S.; Nishizaki, K.; Kinjo, T.; Endo, T.; Suzuki, A.; Tateyama, S.; Nishizaki, F.; et al. Diltiazem Inhibits Coronary Spasm via Inhibition of Cav1.2Phosphorylation and Protein Kinase C Activation in a Mouse Model of Coronary Spastic Angina. Int. Hearth J. 2021, 62, 910–918. [Google Scholar] [CrossRef]
- Borella, G.; Da Ros, A.; Borile, G.; Porcù, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting mesenchymal stromal cells plasticity to reroute acute myeloid leukemia course. Blood 2021, 138, 7. [Google Scholar] [CrossRef]
- Chen, H.; Vandorpe, D.H.; Xie, X.; Alper, S.L.; Zeidel, M.L.; Yu, W. Disruption of Cav1.2-mediated signaling is a pathway for ketamine-induced pathology. Nat. Commun. 2020, 11, 4328. [Google Scholar] [CrossRef]
- Fusi, F.; Trezza, A.; Sgaragli, G.; Spiga, O.; Saponara, S.; Bova, S. Ritanserin blocks CaV1.2 channels in rat artery smooth muscles: Electrophysiological, functional, and computational studies. Acta Pharmacol. Sin. 2020, 41, 1158–1166. [Google Scholar] [CrossRef]
- Yarotskyy, V.; Gao, G.; Du, L.; Ganapathi, S.B.; Peterson, B.Z.; Elmslie, K. Roscovitine Binds to Novel L-channel (CaV1.2) Sites That Separately Affect Activation and Inactivation. J. Biol. Chem. 2010, 285, 43–53. [Google Scholar] [CrossRef]
- Lee, J.-H.; Liu, J.; Shin, M.; Hong, M.; Nah, S.-Y.; Bae, H. Metergoline inhibits the neuronal Nav1.2 voltage-dependent Na+ channels expressed in Xenopus oocytes. Acta Pharmacol. Sin. 2014, 35, 862–868. [Google Scholar] [CrossRef]
- Peters, C.; Sokolov, S.; Rajamani, S.; Ruben, P. Effects of the antianginal drug, ranolazine, on the brain sodium channel NaV1.2 and its modulation by extracellular protons. Br. J. Pharmacol. 2013, 169, 704–716. [Google Scholar] [CrossRef]
- Rivara, M.; Baheti, A.R.; Fantini, M.; Cocconcelli, G.; Ghiron, C.; Kalmar, C.L.; Singh, N.; Merrick, E.C.; Patel, M.K.; Zuliani, V. 2,4(5)-Diarylimidazoles: Synthesis and biological evaluation of a new class of sodium channel blockers against hNav1.2. Bioorg. Med. Chem. Lett. 2008, 18, 5460–5462. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, X.; Lu, X.; Xie, M.; He, B.; He, S.; You, S.; Chen, Q. Progesterone/Org inhibits lung adenocarcinoma cell growth via membrane progesterone receptor alpha. Thorac. Cancer 2020, 11, 2209–2223. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y. Anti-apoptotic Actions of Allopregnanolone and Ganaxolone Mediated Through Membrane Progesterone Receptors (PAQRs) in Neuronal Cells. Front. Endocrinol. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Wang-Eckhardt, L.; Eckhardt, M. A progesterone receptor membrane component 1 antagonist induces large vesicles independent of progesterone receptor membrane component 1 expression. Biol. Chem. 2020, 401, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Teakel, S.L.; Ludescher, M.; Thejer, B.M.; Poschmann, G.; Forwood, J.K.; Neubauer, H.; Cahill, M.A. Protein complexes including PGRMC1 and actin-associated proteins are disrupted by AG-205. Biochem. Biophys. Res. Commun. 2020, 524, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Will, E.A.; Liu, X.; Peluso, J.J. AG 205, a progesterone receptor membrane component 1 antagonist, ablates progesterone’s ability to block oxidative stress-induced apoptosis of human granulosa/luteal cells. Biol. Reprod. 2017, 96, 843–854. [Google Scholar] [CrossRef]
- D’Arrigo, G.; Gianquinto, E.; Rossetti, G.; Cruciani, G.; Lorenzetti, S.; Spyrakis, F. Binding of Androgen- and Estrogen-Like Flavonoids to Their Cognate (Non)Nuclear Receptors: A Comparison by Computational Prediction. Molecules 2021, 26, 1613. [Google Scholar] [CrossRef]
- Kowalska, M.; Nowaczyk, J.; Nowaczyk, A. KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099. [Google Scholar] [CrossRef]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1.2 Ca2+ channel exacerbates the symptoms in a Parkinson’s disease model. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Jørgensen, C.V.; Bräuner-Osborne, H. Pharmacology and physiological function of the orphan GPRC6A receptor. Basic Clin. Pharmacol. Toxicol. 2020, 126, 77–87. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Receptor | Profile | Cell Line/Model | Pathway | Effect | Ref. |
|---|---|---|---|---|---|---|
| (−)-Epicatechin | ZIP9 | agonist | PC3 (PC) MDA-MB-468 (BC) | ERK1/2, JNK and Bax | Proapoptotic action (increased Caspase-3 levels), increased cAMP and intracellular Zn2+ levels | [205] |
| (+)-Catechin | ZIP9 | antagonist | PC3 MDA-MB-468 | ERK 1/2, JNK and Bax | [205] | |
| Bicalutamide | ZIP9 | antagonist | 93RS2 (non -cancerous testicular cell line) | ERK1/2, CREB and ATF-1 | Reduced claudin-5 and zonula occludens-1 (ZO-1) expression | [206] |
| Nandrolone | OXER1 | antagonist | MCF-7, MDA-MB-231 (BC) | PI3K/Akt/NF-κB and RACK1 | Reduced proliferation and migration | [35] |
| 5-oxo-EPE | OXER1 | agonist | In vitro assay | Increased β-Arrestin recruitment | [207] | |
| S-230 | OXER1 | antagonist | In vivo (monkeys), human neutrophils | Gβγ-mediated signaling | Reduced Gβγ-mediated Ca2+ mobilization | [208,209] |
| S-Y048 | OXER1 | antagonist | In vivo (monkeys), human neutrophils and human eosinophils | Gβγ-mediated signaling | Reduced Gβγ-mediated Ca2+ mobilization, actin polymerization and eosinophil infiltration | [210] |
| S-C025 | OXER1 | antagonist | In vivo (monkeys), human neutrophils | Gβγ-mediated signaling | Reduced Gβγ-mediated Ca2+ mobilization and eosinophil activation | [211] |
| 264 | OXER1 | antagonist | In vivo (monkeys, rats), monkey eosinophils and monkey neutrophils | Gβγ-mediated signaling | Reduced Gβγ-mediated Ca2+ mobilization, actin polymerization and chemotaxis in granulocytes | [212] |
| DJ-V-159 | GPRC6A | agonist | HEK-293 (human embryonic kidney), MIN-6 (mouse pancreatic β-cell) and in vivo (mice) | Gαs-dependent signaling, ERK1/2 | Increased cAMP levels, insulin secretion and decreased serum glucose (in vivo, mouse) | [213] |
| Diltiazem | CaV1.2 | antagonist | In vivo (mice) | CaV1.2-PKC | Inhibition of Ca2+ influx, PLCδ1 | [214] |
| Lercanidipine | CaV1.2 | antagonist | Healthy and pediatric acute myeloid leukemia (AML) mesenchymal stromal cells (MSCs) | Inhibition of Ca2+ influx | [215] | |
| Ketamine | CaV1.2 | antagonist | In vivo (mice), Xenopus laevis oocytes (ex vivo) | Inhibition of CaV1.2 expression; Ca2+ influx; and vascular smooth muscle contraction | [216] | |
| Ritanserin | CaV1.2 | antagonist | Rat vascular myocytes (ex vivo) | Inhibition of Ca2+ influx; in vitro vasodilation; and vascular smooth muscle relaxation | [217] | |
| (R)-Roscovitine | CaV1.2 | antagonist | HEK-293 | Slows activation and enhances inactivation | [218] | |
| Metergoline | NaV1.2 | antagonist | Xenopus laevis oocytes (ex vivo) | Inhibition of Na+ influx | [219] | |
| Ranolazine | NaV1.2 | antagonist | CHO | Inhibition of Na+ influx | [220] | |
| 2,4(5)-diarylimidazoles | NaV1.2 | antagonist | In vitro assay | Inhibition of Na+ influx | [221] | |
| Org OD 02-0 | mPRα | agonist | A549, PC-9 (human lung adenocarcinoma), HBE (human bronchial epithelial) and MCF-7 | PKA/CREB and PKA/β-catenin | Inhibition of cell growth and tumor growth (in vivo) | [222] |
| Ganaxolone | mPRδ | agonist | GT1-7 (rat hypothalamic cells), H19-7 (rat hippocampal neuronal cells) | Gαs-dependent signaling | Reduction of apoptosis and cell death | [223] |
| AG-205 | PGRMC1 | antagonist | CHO-K1, HeLa, COS-7, and H4 glioma Cells | Increased endosome formation | [224] | |
| PaCa-2 cells (pancreatic cancer) | RACK1, alpha-Actinin-1 | Reduced PGRMC1 interactions with the actin cytoskeleton | [225] | |||
| Human granulosa/luteal cell | B-cell lymphoma 2 (BCL2) pathway | Increased PGRMC1 monomeric form, increased proapoptotic Harakiri (Hrk) expression | [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masi, M.; Racchi, M.; Travelli, C.; Corsini, E.; Buoso, E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells 2021, 10, 2999. https://doi.org/10.3390/cells10112999
Masi M, Racchi M, Travelli C, Corsini E, Buoso E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells. 2021; 10(11):2999. https://doi.org/10.3390/cells10112999
Chicago/Turabian StyleMasi, Mirco, Marco Racchi, Cristina Travelli, Emanuela Corsini, and Erica Buoso. 2021. "Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers" Cells 10, no. 11: 2999. https://doi.org/10.3390/cells10112999
APA StyleMasi, M., Racchi, M., Travelli, C., Corsini, E., & Buoso, E. (2021). Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells, 10(11), 2999. https://doi.org/10.3390/cells10112999