
The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective

 and
and 
Abstract
:1. Introduction
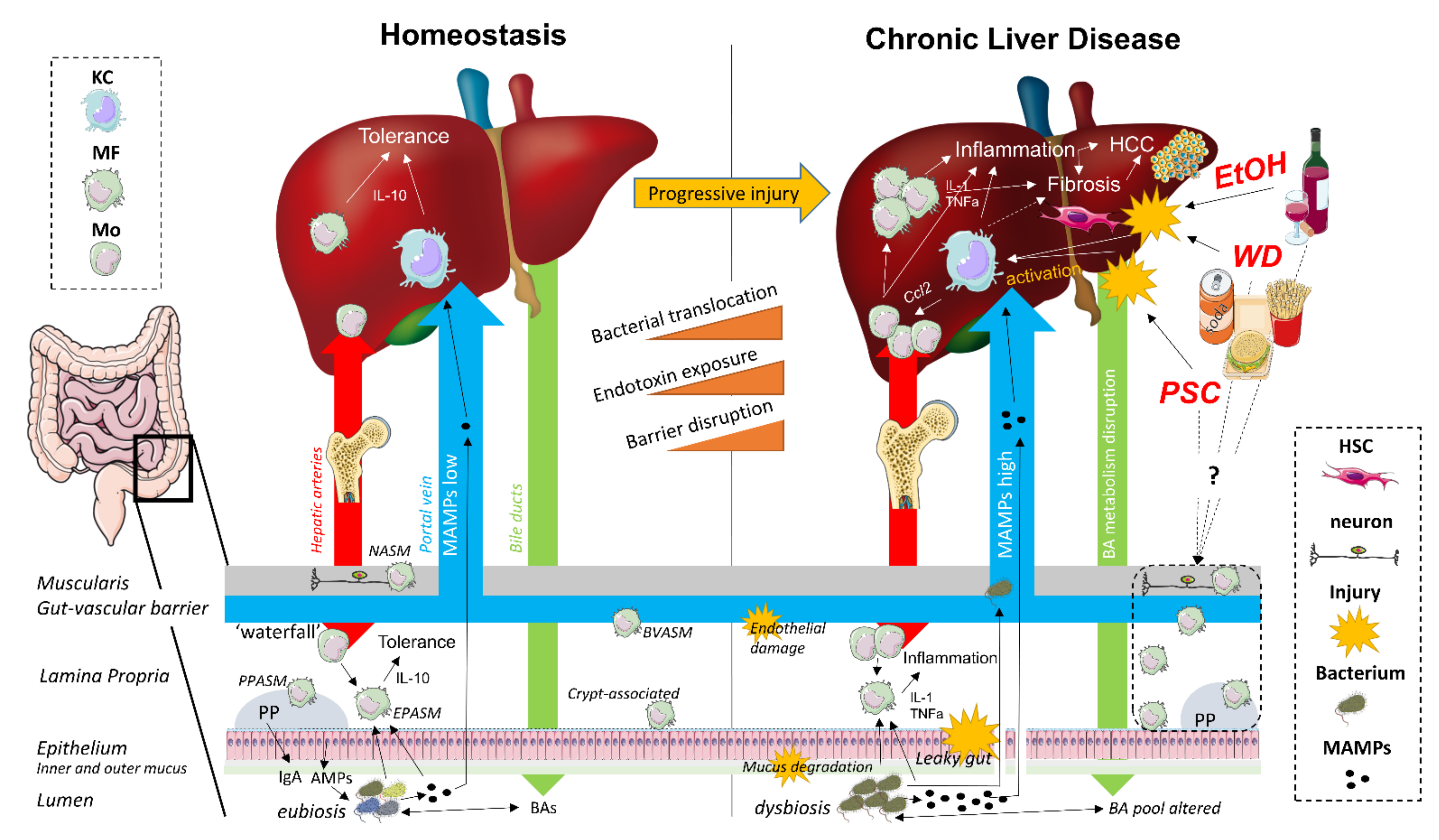
2. The Gut–Liver Axis
3. Macrophages during Gut–Liver Axis Homeostasis

{kind=link}
| Macrophage Subset (Niche/Development) | Specific Markers | Intestinal Segment | Intestinal Wall Substructure | Reported Function | Properties | Reference(s) |
|---|---|---|---|---|---|---|
| MOUSE | ||||||
| Epithelium-associated | CD11c, CD121b | Colon | Lamina propria | Unknown | Regulated by microbiota | [52] |
| Blood vessel-associated | ADAMDEC1, CD169 | Small intestine, Colon | Lamina propria | Blood vessel maintenance | Self-renewing | [44,45,46] |
| Neuron-associated | Fcrls | Small intestine | Muscularis layers | Facilitate enteric neuron survival | Self-renewing | [44] |
| Crypt-associated | (CD169) | Small intestine, Colon | Lamina propria | Unknown | Self-renewing | [44,46] |
| Peyer’s patch-associated | CD4, TIM4 | Small intestine | Lamina propria | TIM4+: phagocytic TIM4−: microbial defense | TIM4 expression can discriminate location | [48] |
| Unknown | CD4, TIM4 | Colon | Unknown | Unknown | Long-lived | [43] |
| HUMAN | ||||||
| Late intermediate (Clusters M4–6) | CD11cint C1QCint CD206+ CD11a+ CD1cint | Small intestine, Colon | Lamina propria, excl. ILF and GALT | M5: ↑ NF-κB, TNF-α, EGFR, and MAPK signaling | M4,6: ↑ JAK-STAT, Wnt signaling; (less inflammatory than M5, 7) M5: inflammatory + ↑ EGFR and MAPK signaling | [49] |
| Mature (Clusters M7–8) | CD209hi, C1QCint, CD206+ M7: CD14hi, LYVE1+ CD169+ FOLR2+ MAF+ | Small intestine, Colon | Lamina propria, excl. ILF and GALT | M7: ↑ NF-κB, TNF-α, TGF-β, EGFR, and MAPK signaling M8: ↑ JAK-STAT and Wnt signaling | M7: inflammatory + similar to BVAMs, cfr. Honda et al., 2020 | [49] |
| Mature-like (Clusters M9–10) | MHCIIhi C1QChi CD209lo CD163lo CD206− M9: CD11c+ CD4+ C2+ ADAMDEC1+ M10: CD11c− FTL+ RBP1+ PLP1+ PMEPA1+ GPM6B+ SPARC+ ADIRF+ SCD+ | Small intestine, Colon | Lamina propria, excl. ILF and GALT | M9: ↑ JAK-STAT, Wnt, TGF-β signaling M10: genes in iron metabolism, fibrosis, adipocyte-regulation; not involved in efferocytosis | M9: cfr. De Schepper et al., 2018 and Shaw et al., 2018 | [49] |
4. Macrophages during Gut-Liver Axis Disruption in Chronic Liver Disease
4.1. Non-Alcoholic Fatty Liver Disease
4.2. Alcoholic Liver Disease
4.3. Primary Sclerosing Cholangitis
4.4. Liver Fibrosis/Cirrhosis
4.5. Hepatocellular Carcinoma
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38 (Suppl. S1), 2–6. [Google Scholar] [CrossRef] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Larusso, N.F. Primary sclerosing cholangitis: The gut-liver axis. Clin. Gastroenterol. Hepatol. 2012, 10, 819–820. [Google Scholar] [CrossRef]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, H.; Wiest, R. Changes of intestinal functions in liver cirrhosis. Inflamm. Intest. Dis. 2016, 1, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Diet, microbiota, obesity, and nafld: A dangerous quartet. Int. J. Mol. Sci. 2016, 17, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piglionica, M.; Cariello, M.; Moschetta, A. The gut-liver axis in hepatocarcinoma: A focus on the nuclear receptor fxr and the enterokine FGF19. Curr. Opin. Pharmacol. 2018, 43, 93–98. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut⁻liver axis: How do gut bacteria influence the liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Bogdanos, D.P.; Gao, B.; Gershwin, M.E. Liver immunology. Compr. Physiol. 2013, 3, 567–598. [Google Scholar]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, B.S.; Lefrançois, L. Intraepithelial lymphocytes: To serve and protect. Curr. Gastroenterol. Rep. 2010, 12, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboziev, I.; Karlsson, F.; Grisham, M.B. Gut-associated lymphoid tissue, t cell trafficking, and chronic intestinal inflammation. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. S1), E86–E93. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef]
- Doherty, D.G. Antigen-specific immune tolerance in the liver. Nat. Biomed. Eng. 2019, 3, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-liver axis at the frontier of host-microbial interactions. Am. J. Physiol. Gastrointest. Liver. Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chiang, J.Y. Nuclear receptors in bile acid metabolism. Drug Metab Rev. 2013, 45, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Mowat, A.M.; Scott, C.L.; Bain, C.C. Barrier-tissue macrophages: Functional adaptation to environmental challenges. Nat. Med. 2017, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, L.; Shi, J.; Shultz, L.D.; Ren, G. Genetic models of macrophage depletion. Methods Mol. Biol. 2018, 1784, 243–258. [Google Scholar]
- Li, C.; Menoret, A.; Farragher, C.; Ouyang, Z.; Bonin, C.; Holvoet, P.; Vella, A.T.; Zhou, B. Single cell transcriptomics based-macspectrum reveals novel macrophage activation signatures in diseases. JCI Insight 2019, 5, e126453. [Google Scholar] [CrossRef]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; McCauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4, e124574. [Google Scholar] [CrossRef] [Green Version]
- Thornton, S.; Tan, R.; Sproles, A.; Do, T.; Schick, J.; Grom, A.A.; DeLay, M.; Schulert, G.S. A multiparameter flow cytometry analysis panel to assess CD163 mRNA and protein in monocyte and macrophage populations in hyperinflammatory diseases. J. Immunol. 2019, 202, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Allen, J.; Hankey-Giblin, P.A. Ontogeny and polarization of macrophages in inflammation: Blood monocytes versus tissue macrophages. Front. Immunol. 2014, 5, 683. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. New insights into tissue macrophages: From their origin to the development of memory. Immune Netw. 2015, 15, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS One 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.C.; Haschak, M.J.; Popovic, B.; Brown, B.N. Macrophages in the aging liver and age-related liver disease. Front. Immunol. 2018, 9, 2795. [Google Scholar] [CrossRef] [Green Version]
- Triantafyllou, E.; Woollard, K.J.; McPhail, M.J.W.; Antoniades, C.G.; Possamai, L.A. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front. Immunol. 2018, 9, 2948. [Google Scholar] [CrossRef]
- Bain, C.C.; Schridde, A. Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 2018, 9, 2733. [Google Scholar] [CrossRef]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of macrophages in the maintenance of intestinal homeostasis. J. Immunol. Res. 2019, 2019, 1512969. [Google Scholar] [CrossRef] [Green Version]
- Grainger, J.R.; Konkel, J.E.; Zangerle-Murray, T.; Shaw, T.N. Macrophages in gastrointestinal homeostasis and inflammation. Pflugers Arch. 2017, 469, 527–539. [Google Scholar] [CrossRef]
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.J.L.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by tim-4 and cd4 expression. J. Exp. Med. 2018, 215, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterlé, I.; et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell 2018, 175, 400–415.e413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, M.; Surewaard, B.G.J.; Watanabe, M.; Hedrick, C.C.; Lee, W.-Y.; Brown, K.; McCoy, K.D.; Kubes, P. Perivascular localization of macrophages in the intestinal mucosa is regulated by nr4a1 and the microbiome. Nat. Commun. 2020, 11, 1329. [Google Scholar] [CrossRef]
- Asano, K.; Kikuchi, K.; Tanaka, M. Cd169 macrophages regulate immune responses toward particulate materials in the circulating fluid. J. Biochem. 2018, 164, 77–85. [Google Scholar] [CrossRef]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Bonnardel, J.; Da Silva, C.; Henri, S.; Tamoutounour, S.; Chasson, L.; Montañana-Sanchis, F.; Gorvel, J.P.; Lelouard, H. Innate and adaptive immune functions of peyer’s patch monocyte-derived cells. Cell Rep. 2015, 11, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.M.; Wulff, L.; Jones, G.-R.; Vandamme, J.; Jørgensen, P.B.; Bain, C.C.; Lee, J.; Izarzugaza, J.M.G.; Belling, K.G.; Ho, G.-T.; et al. Single-cell characterisation of mononuclear phagocytes in the human intestinal mucosa. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bujko, A.; Atlasy, N.; Landsverk, O.J.B.; Richter, L.; Yaqub, S.; Horneland, R.; Øyen, O.; Aandahl, E.M.; Aabakken, L.; Stunnenberg, H.G.; et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J. Exp. Med. 2018, 215, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, D.; Marin, A.C.; Fernández-Tomé, S.; Montalban-Arques, A.; Carrasco, A.; Tristán, E.; Ortega-Moreno, L.; Mora-Gutiérrez, I.; Díaz-Guerra, A.; Caminero-Fernández, R.; et al. Human intestinal pro-inflammatory CD11C(high)CCR2(+)CX3CR1(+) macrophages, but not their tolerogenic CD11C(-)CCR2(-)CX3CR1(-) counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 2018, 11, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Zhang, Z.; Wang, J.; Wang, G.; Yan, Y.; Qian, H.; Zhang, X.; Xu, W.; Mao, F. Hucmscs attenuate IBD through releasing mir148b-5p to inhibit the expression of 15-lox-1 in macrophages. Mediators Inflamm. 2019, 2019, 6953963. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell rna sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016, 151, 1176–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef]
- Blériot, C.; Barreby, E.; Dunsmore, G.; Ballaire, R.; Chakarov, S.; Ficht, X.; De Simone, G.; Andreata, F.; Fumagalli, V.; Guo, W.; et al. A subset of kupffer cells regulates metabolism through the expression of CD36. Immunity 2021, 54, 2101–2116.e6. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Devisscher, L.; Scott, C.L.; Lefere, S.; Raevens, S.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Geerts, A.; Guilliams, M.; Van Vlierberghe, H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol. 2017, 322, 74–83. [Google Scholar] [CrossRef]
- Lefere, S.; Degroote, H.; Van Vlierberghe, H.; Devisscher, L. Unveiling the depletion of kupffer cells in experimental hepatocarcinogenesis through liver macrophage subtype-specific markers. J. Hepatol. 2019, 71, 631–633. [Google Scholar] [CrossRef] [Green Version]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.J.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 2017, 47, 374–388.e6. [Google Scholar] [CrossRef]
- Wang, J.; Kubes, P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Aizarani, N.; Saviano, A.; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Matchett, K.P.; Dobie, R.; Wilson-Kanamori, J.R.; Henderson, N.C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.P.; Cheng, L.; Ju, C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008, 84, 1410–1421. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential ly-6c expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.Y.; Chen, J.B.; Tsai, T.F.; Tsai, Y.C.; Tsai, C.Y.; Liang, P.H.; Hsu, T.L.; Wu, C.Y.; Netea, M.G.; Wong, C.H.; et al. Clec4f is an inducible c-type lectin in f4/80-positive cells and is involved in alpha-galactosylceramide presentation in liver. PLoS ONE 2013, 8, e65070. [Google Scholar] [CrossRef] [Green Version]
- You, Q.; Holt, M.; Yin, H.; Li, G.; Hu, C.J.; Ju, C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem. Pharmacol. 2013, 86, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. Ccl2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, X.; Li, X.; Fan, Y.; Li, G.; Guo, C.; Zhu, F.; Zhang, L.; Shi, Y. Cd68(+)hla-dr(+) m1-like macrophages promote motility of hcc cells via nf-κb/fak pathway. Cancer Lett. 2014, 345, 91–99. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol 2014, 193, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blériot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef]
- Movita, D.; van de Garde, M.D.; Biesta, P.; Kreefft, K.; Haagmans, B.; Zuniga, E.; Herschke, F.; De Jonghe, S.; Janssen, H.L.; Gama, L.; et al. Inflammatory monocytes recruited to the liver within 24 h after virus-induced inflammation resemble kupffer cells but are functionally distinct. J. Virol. 2015, 89, 4809–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutchfield, B.M.; Antoine, D.J.; Mackinnon, A.C.; Gow, D.J.; Bain, C.C.; Hawley, C.A.; Hughes, M.J.; Francis, B.; Wojtacha, D.; Man, T.Y.; et al. Csf1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology 2015, 149, 1896–1909.e14. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chi, F.; Guo, T.; Punj, V.; Lee, W.N.; French, S.W.; Tsukamoto, H. Notch reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Invest. 2015, 125, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Beattie, L.; Sawtell, A.; Mann, J.; Frame, T.C.M.; Teal, B.; de Labastida Rivera, F.; Brown, N.; Walwyn-Brown, K.; Moore, J.W.J.; MacDonald, S.; et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J. Hepatol. 2016, 65, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (c-c motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Reid, D.T.; Reyes, J.L.; McDonald, B.A.; Vo, T.; Reimer, R.A.; Eksteen, B. Kupffer cells undergo fundamental changes during the development of experimental nash and are critical in initiating liver damage and inflammation. PLoS One 2016, 11, e0159524. [Google Scholar] [CrossRef] [Green Version]
- Graubardt, N.; Vugman, M.; Mouhadeb, O.; Caliari, G.; Pasmanik-Chor, M.; Reuveni, D.; Zigmond, E.; Brazowski, E.; David, E.; Chappell-Maor, L.; et al. Ly6c(hi) monocytes and their macrophage descendants regulate neutrophil function and clearance in acetaminophen-induced liver injury. Front. Immunol. 2017, 8, 626. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Suganami, T.; Kato, H.; Kanai, S.; Shirakawa, I.; Sakai, T.; Goto, T.; Asakawa, M.; Hidaka, I.; Sakugawa, H.; et al. Cd11c+ resident macrophages drive hepatocyte death-triggered liver fibrosis in a murine model of nonalcoholic steatohepatitis. JCI Insight 2017, 2, e92902. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Frasch, S.C.; Li, G.; Feng, D.; Gao, B.; Xu, L.; Ir, D.; Frank, D.N.; Bratton, D.L.; Ju, C. Role of gp91(phox) in hepatic macrophage programming and alcoholic liver disease. Hepatol. Commun. 2017, 1, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Campana, L.; Starkey Lewis, P.J.; Pellicoro, A.; Aucott, R.L.; Man, J.; O’Duibhir, E.; Mok, S.E.; Ferreira-Gonzalez, S.; Livingstone, E.; Greenhalgh, S.N.; et al. The stat3-il-10-il-6 pathway is a novel regulator of macrophage efferocytosis and phenotypic conversion in sterile liver injury. J. Immunol. 2018, 200, 1169–1187. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Trussoni, C.E.; Krishnan, A.; Bronk, S.F.; Lorenzo Pisarello, M.J.; O’Hara, S.P.; Splinter, P.L.; Gao, Y.; Vig, P.; Revzin, A.; et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol. 2018, 69, 676–686. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The transcription factor zeb2 is required to maintain the tissue-specific identities of macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. Mertk expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of intercellular crosstalk in healthy and nash liver revealed by single-cell secretome gene analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage mertk promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef]
- Jin, K.; Liu, Y.; Shi, Y.; Zhang, H.; Sun, Y.; Zhangyuan, G.; Wang, F.; Yu, W.; Wang, J.; Tao, X.; et al. Ptprot aggravates inflammation by enhancing nf-κb activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics 2020, 10, 5290–5304. [Google Scholar] [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020, 69, 551–563. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity 2020, 53, 641–657.e14. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef] [PubMed]
- She, S.; Wu, X.; Zheng, D.; Pei, X.; Ma, J.; Sun, Y.; Zhou, J.; Nong, L.; Guo, C.; Lv, P.; et al. Psmp/msmp promotes hepatic fibrosis through ccr2 and represents a novel therapeutic target. J. Hepatol. 2020, 72, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity 2020, 53, 627–640.e5. [Google Scholar] [CrossRef]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in nash. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef]
- Devisscher, L.; Van Campenhout, S.; Lefere, S.; Raevens, S.; Tilleman, L.; Van Nieuwerburgh, F.; Van Eeckhoutte, H.P.; Hoorens, A.; Lynes, M.A.; Geerts, A.; et al. Metallothioneins alter macrophage phenotype and represent novel therapeutic targets for acetaminophen-induced liver injury. J. Leukoc. Biol. 2021. [Google Scholar] [CrossRef]
- Raevens, S.; Van Campenhout, S.; Debacker, P.J.; Lefere, S.; Verhelst, X.; Geerts, A.; Van Vlierberghe, H.; Colle, I.; Devisscher, L. Combination of sivelestat and n-acetylcysteine alleviates the inflammatory response and exceeds standard treatment for acetaminophen-induced liver injury. J. Leukoc. Biol. 2020, 107, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Van de Velde, F.; Hoorens, A.; Raevens, S.; Van Campenhout, S.; Vandierendonck, A.; Neyt, S.; Vandeghinste, B.; Vanhove, C.; Debbaut, C.; et al. Angiopoietin-2 promotes pathological angiogenesis and is a therapeutic target in murine nonalcoholic fatty liver disease. Hepatology 2019, 69, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ros to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, M.A.; McMullen, M.R.; Roychowdhury, S.; Madhun, N.Z.; Niese, K.; Olman, M.A.; Stavitsky, A.B.; Bucala, R.; Nagy, L.E. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J. Leukoc. Biol. 2015, 97, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, J.; Zhang, S.; Weinman, S.A. Foxo3-dependent apoptosis limits alcohol-induced liver inflammation by promoting infiltrating macrophage differentiation. Cell Death Discov. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen-Lefebvre, A.T.; Ajith, A.; Portik-Dobos, V.; Horuzsko, D.D.; Arbab, A.S.; Dzutsev, A.; Sadek, R.; Trinchieri, G.; Horuzsko, A. The innate immune receptor trem-1 promotes liver injury and fibrosis. J. Clin. Invest. 2018, 128, 4870–4883. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Bala, S.; Hosseini, N.; Kodys, K.; Szabo, G. Krüppel-like factor 4 is a transcriptional regulator of m1/m2 macrophage polarization in alcoholic liver disease. J. Leukoc. Biol. 2015, 97, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of mir-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via ccl2/ccr2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The ccr2(+) macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M.; et al. M2 macrophages mediate sorafenib resistance by secreting hgf in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Ikarashi, M.; Nakashima, H.; Kinoshita, M.; Sato, A.; Nakashima, M.; Miyazaki, H.; Nishiyama, K.; Yamamoto, J.; Seki, S. Distinct development and functions of resident and recruited liver kupffer cells/macrophages. J. Leukoc. Biol. 2013, 94, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Zannetti, C.; Roblot, G.; Charrier, E.; Ainouze, M.; Tout, I.; Briat, F.; Isorce, N.; Faure-Dupuy, S.; Michelet, M.; Marotel, M.; et al. Characterization of the inflammasome in human kupffer cells in response to synthetic agonists and pathogens. J. Immunol. 2016, 197, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Hollingshead, N.; Roberto, J.; Knupp, A.; Kenerson, H.; Chen, A.; Strickland, I.; Horton, H.; Yeung, R.; Soysa, R.; et al. Human liver macrophage subsets defined by cd32. Front. Immunol. 2020, 11, 2108. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.G.; Quaglia, A.; Taams, L.S.; Mitry, R.R.; Hussain, M.; Abeles, R.; Possamai, L.A.; Bruce, M.; McPhail, M.; Starling, C.; et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012, 56, 735–746. [Google Scholar] [CrossRef]
- Gadd, V.L.; Melino, M.; Roy, S.; Horsfall, L.; O’Rourke, P.; Williams, M.R.; Irvine, K.M.; Sweet, M.J.; Jonsson, J.R.; Clouston, A.D.; et al. Portal, but not lobular, macrophages express matrix metalloproteinase-9: Association with the ductular reaction and fibrosis in chronic hepatitis c. Liver Int. 2013, 33, 569–579. [Google Scholar] [CrossRef]
- Tan-Garcia, A.; Wai, L.E.; Zheng, D.; Ceccarello, E.; Jo, J.; Banu, N.; Khakpoor, A.; Chia, A.; Tham, C.Y.L.; Tan, A.T.; et al. Intrahepatic cd206(+) macrophages contribute to inflammation in advanced viral-related liver disease. J. Hepatol. 2017, 67, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 2019, 179, 829–845. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, P.; Ma, L.; Liu, L.; Zhao, G.; Zhang, S.; Dong, L.; Xue, R.; Chen, S. Cd86⁺/cd206⁺, diametrically polarized tumor-associated macrophages, predict hepatocellular carcinoma patient prognosis. Int. J. Mol. Sci. 2016, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Yu, X.J.; Wang, Y.C.; Huang, L.Y.; Liu, C.Q.; Zheng, L.; Fang, Y.J.; Xu, J. Distinct patterns and prognostic values of tumor-infiltrating macrophages in hepatocellular carcinoma and gastric cancer. J. Transl. Med. 2017, 15, 37. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.X.; Leng, R.X.; Fan, Y.G.; Pan, H.F.; Li, B.Z.; Wu, C.H.; Wu, Q.; Wang, N.N.; Xiong, Q.R.; Geng, X.P.; et al. Intratumoral and peritumoral expression of cd68 and cd206 in hepatocellular carcinoma and their prognostic value. Oncol. Rep. 2017, 38, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Gao, W.; Tang, Q.; Yu, Y.; You, W.; Wu, Z.; Fan, Y.; Zhang, L.; Wu, C.; Han, G.; et al. M2 macrophage-derived exosomes facilitate hcc metastasis by transferring α(m) β(2) integrin to tumor cells. Hepatology 2021, 73, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Mandrekar, P. Macrophages and alcohol-related liver inflammation. Alcohol Res. 2015, 37, 251–262. [Google Scholar]
- Dong, X.; Liu, J.; Xu, Y.; Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 2019, 17, 3835–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillot, A.; Tacke, F. Liver macrophages: Old dogmas and new insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Degroote, H.; Lefere, S.; Vandierendonck, A.; Vanderborght, B.; Meese, T.; Van Nieuwerburgh, F.; Verhelst, X.; Geerts, A.; Van Vlierberghe, H.; Devisscher, L. Characterization of the inflammatory microenvironment and hepatic macrophage subsets in experimental hepatocellular carcinoma models. Oncotarget 2021, 12, 562–577. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut microbiome, liver immunology, and liver diseases. Cell Mol. Immunol. 2021, 18, 4–17. [Google Scholar] [CrossRef]
- Won, S.M.; Park, E.; Jeong, J.J.; Ganesan, R.; Gupta, H.; Gebru, Y.A.; Sharma, S.; Kim, D.J.; Suk, K.T. The gut microbiota-derived immune response in chronic liver disease. Int. J. Mol. Sci. 2021, 22, 8309. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Vespasiani-Gentilucci, U.; Carotti, S.; Perrone, G.; Mazzarelli, C.; Galati, G.; Onetti-Muda, A.; Picardi, A.; Morini, S. Hepatic toll-like receptor 4 expression is associated with portal inflammation and fibrosis in patients with nafld. Liver Int. 2015, 35, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Singanayagam, A.; Triantafyllou, E. Macrophages in chronic liver failure: Diversity, plasticity and therapeutic targeting. Front. Immunol. 2021, 12, 661182. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Bao, T.; He, F.; Zhang, X.; Zhu, L.; Wang, Z.; Lu, H.; Wang, T.; Li, Y.; Yang, S.; Wang, H. Inulin exerts beneficial effects on non-alcoholic fatty liver disease via modulating gut microbiome and suppressing the lipopolysaccharide-toll-like receptor 4-mψ-nuclear factor-κb-nod-like receptor protein 3 pathway via gut-liver axis in mice. Front. Pharmacol. 2020, 11, 558525. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, A.; Hundertmark, J.; Guillot, A.; Tacke, F. Molecular and cellular mediators of the gut-liver axis in the progression of liver diseases. Front. Med. 2021, 8, 725390. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Zwicker, C.; Bujko, A.; Scott, C.L. Hepatic macrophage responses in inflammation, a function of plasticity, heterogeneity or both? Front. Immunol. 2021, 12, 690813. [Google Scholar] [CrossRef]
- Jones, G.R.; Bain, C.C.; Fenton, T.M.; Kelly, A.; Brown, S.L.; Ivens, A.C.; Travis, M.A.; Cook, P.C.; MacDonald, A.S. Dynamics of colon monocyte and macrophage activation during colitis. Front. Immunol. 2018, 9, 2764. [Google Scholar] [CrossRef]
- Hine, A.M.; Loke, P.n. Intestinal macrophages in resolving inflammation. J. Immunol. 2019, 203, 593. [Google Scholar] [CrossRef]
- Salvador, P.; Macías-Ceja, D.C.; Gisbert-Ferrándiz, L.; Hernández, C.; Bernardo, D.; Alós, R.; Navarro-Vicente, F.; Esplugues, J.V.; Ortiz-Masiá, D.; Barrachina, M.D.; et al. Cd16+ macrophages mediate fibrosis in inflammatory bowel disease. J. Crohn’s Colitis 2018, 12, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Du Plessis, J.; Vanheel, H.; Janssen, C.E.; Roos, L.; Slavik, T.; Stivaktas, P.I.; Nieuwoudt, M.; van Wyk, S.G.; Vieira, W.; Pretorius, E.; et al. Activated intestinal macrophages in patients with cirrhosis release no and il-6 that may disrupt intestinal barrier function. J. Hepatol. 2013, 58, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of nafld and nash: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.J.; Singal, A.K. Trends in liver disease etiology among adults awaiting liver transplantation in the United States, 2014-2019. JAMA Netw. Open. 2020, 3, e1920294. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla Bertot, L.; Adams, L.A. The natural course of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A distinct entity shaped by differential metabolic adaptation. Hepatology 2019, 71, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. No need for a large belly to have nash. J. Hepatol. 2011, 54, 1090–1093. [Google Scholar] [CrossRef]
- Devisscher, L.; Verhelst, X.; Colle, I.; Van Vlierberghe, H.; Geerts, A. The role of macrophages in obesity-driven chronic liver disease. J. Leukoc. Biol. 2016, 99, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, H.; Boesch, M.; Meelberghs, L.; van der Merwe, S. Macrophages as key players during adipose tissue-liver crosstalk in nonalcoholic fatty liver disease. Semin. Liver Dis. 2019, 39, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caslin, H.L.; Bhanot, M.; Bolus, W.R.; Hasty, A.H. Adipose tissue macrophages: Unique polarization and bioenergetics in obesity. Immunol. Rev. 2020, 295, 101–113. [Google Scholar] [CrossRef]
- Hill, D.A.; Lim, H.-W.; Kim, Y.H.; Ho, W.Y.; Foong, Y.H.; Nelson, V.L.; Nguyen, H.C.B.; Chegireddy, K.; Kim, J.; Habertheuer, A.; et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Nat. Acad. Sci. USA 2018, 115, E5096–E5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazankov, K.; Barrera, F.; Moller, H.J.; Rosso, C.; Bugianesi, E.; David, E.; Younes, R.; Esmaili, S.; Eslam, M.; McLeod, D.; et al. The macrophage activation marker scd163 is associated with morphological disease stages in patients with non-alcoholic fatty liver disease. Liver Int. 2016, 36, 1549–1557. [Google Scholar] [CrossRef]
- Rodgaard-Hansen, S.; St George, A.; Kazankov, K.; Bauman, A.; George, J.; Gronbaek, H.; Jon Moller, H. Effects of lifestyle intervention on soluble cd163, a macrophage activation marker, in patients with non-alcoholic fatty liver disease. Scand. J. Clin. Lab. Invest. 2017, 77, 498–504. [Google Scholar] [CrossRef]
- Du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.Y.; Lee, S.Y.; Shih, C.K.; Chou, M.J.; Wu, M.C.; Teng, I.C.; Bai, C.H.; Sabrina, N.; Tinkov, A.A.; Skalny, A.V.; et al. Soluble cd163-associated dietary patterns and the risk of metabolic syndrome. Nutrients 2019, 11, 940. [Google Scholar] [CrossRef] [Green Version]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Wieland, A.; Frank, D.N.; Harnke, B.; Bambha, K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2015, 42, 1051–1063. [Google Scholar] [CrossRef]
- Dai, X.; Wang, B. Role of gut barrier function in the pathogenesis of nonalcoholic fatty liver disease. Gastroenterol. Res. Pract. 2015, 2015, 287348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human nafld: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, I.C.; de Lima, J.G. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease? - a mechanistic hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Canbay, A. Why bile acids are so important in non-alcoholic fatty liver disease (NAFLD) progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef] [PubMed]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Stahlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte myd88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Asgari-Taee, F.; Zerafati-Shoae, N.; Dehghani, M.; Sadeghi, M.; Baradaran, H.R.; Jazayeri, S. Association of sugar sweetened beverages consumption with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Nutr. 2019, 58, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome p450-2e1-mediated oxidative and nitrative stress. Hepatology 2019, 73, 2180–2195. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Dallio, M.; Godos, J.; Loguercio, C.; Salomone, F. Targeting gut-liver axis for the treatment of nonalcoholic steatohepatitis: Translational and clinical evidence. Transl. Res. 2016, 167, 116–124. [Google Scholar] [CrossRef]
- Schneider, K.M.; Bieghs, V.; Heymann, F.; Hu, W.; Dreymueller, D.; Liao, L.; Frissen, M.; Ludwig, A.; Gassler, N.; Pabst, O.; et al. Cx3cr1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology 2015, 62, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Cai, C.; Chen, Q.; Jin, S.; Yang, B.; Li, N. High-fat diet promotes dss-induced ulcerative colitis by downregulated fxr expression through the tgfb pathway. Biomed. Res. Int. 2020, 2020, 3516128. [Google Scholar] [CrossRef]
- Gabele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Scholmerich, J.; Hellerbrand, C.; Obermeier, F. Dss induced colitis increases portal lps levels and enhances hepatic inflammation and fibrogenesis in experimental nash. J. Hepatol 2011, 55, 1391–1399. [Google Scholar] [CrossRef]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced nafld mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Jeong, G.; Kim, S.J.; Kim, M.K.; Park, S.M. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: Comprehensive study of clinical and immunohistochemical findings in younger asian patients. J. Gastroenterol. Hepatol. 2007, 22, 491–497. [Google Scholar] [CrossRef]
- Govaere, O.; Kragh Petersen, S.; Martinez-Lopez, N.; Wouters, J.; Van Haele, M.; Mancina, R.M.; Jamialahmadi, O.; Bilkei-Gorzo, O.; Bel Lassen, P.; Darlay, R.; et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. bioRxiv 2021. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Yoshio, S.; Doi, H.; Kawai, H.; Shimagaki, T.; Mori, T.; Matsuda, M.; Aoki, Y.; Osawa, Y.; Yoshida, Y.; et al. Serum soluble sialic acid-binding immunoglobulin-like lectin-7 concentration as an indicator of liver macrophage activation and advanced fibrosis in patients with non-alcoholic fatty liver disease. Hepatol. Res. 2020, 50, 466–477. [Google Scholar] [CrossRef]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Neyrinck, A.M.; Cani, P.D.; Dewulf, E.M.; De Backer, F.; Bindels, L.B.; Delzenne, N.M. Critical role of kupffer cells in the management of diet-induced diabetes and obesity. Biochem. Biophys. Res. Commun. 2009, 385, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrene, E.; et al. Differential effects of selective- and pan-ppar agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Kiki, I.; Altunkaynak, B.Z.; Altunkaynak, M.E.; Vuraler, O.; Unal, D.; Kaplan, S. Effect of high fat diet on the volume of liver and quantitative feature of kupffer cells in the female rat: A stereological and ultrastructural study. Obes. Surg. 2007, 17, 1381–1388. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in hfd/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Clementi, A.H.; Gaudy, A.M.; van Rooijen, N.; Pierce, R.H.; Mooney, R.A. Loss of kupffer cells in diet-induced obesity is associated with increased hepatic steatosis, stat3 signaling, and further decreases in insulin signaling. Biochim. Biophys. Acta 2009, 1792, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.B.; Liu, Y.; Liu, C.; Xiang, X.; Wang, J.; Cheng, Z.; Shah, S.V.; Zhang, S.; Zhang, L.; Zhuang, X.; et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009, 50, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine c-c motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing ly-6c(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Sugimoto, M.; Murayama, T.; Ueda, Y.; Kanamori, H.; Ono, K.; Ariyasu, H.; Akamizu, T.; Kita, T.; Yokode, M.; et al. Inhibition of ccr2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2195–2201. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.; Abdelmalek, M.F.; Younossi, Z.M.; Yuan, J.; Pecoraro, M.L.; Seyedkazemi, S.; Fischer, L.; Bedossa, P.; et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: Aurora phase 3 study design. Contemp. Clin. Trials 2020, 89, 105922. [Google Scholar] [CrossRef] [Green Version]
- Ægidius, H.M.; Veidal, S.S.; Feigh, M.; Hallenborg, P.; Puglia, M.; Pers, T.H.; Vrang, N.; Jelsing, J.; Kornum, B.R.; Blagoev, B.; et al. Multi-omics characterization of a diet-induced obese model of non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Davanso, M.R.; Crisma, A.R.; Murata, G.; Newsholme, P.; Curi, R. Impact of dietary fatty acids on macrophage lipid metabolism, signaling and function. Immunometabolism 2020, 2, e200008. [Google Scholar]
- Gwag, T.; Mooli, R.G.R.; Li, D.; Lee, S.; Lee, E.Y.; Wang, S.X. Macrophage-derived thrombospondin 1 promotes obesity-associated non-alcoholic fatty liver disease. J. Hep. Rep. 2021, 3, 100193. [Google Scholar]
- Robert, O.; Boujedidi, H.; Bigorgne, A.; Ferrere, G.; Voican, C.S.; Vettorazzi, S.; Tuckermann, J.P.; Bouchet-Delbos, L.; Tran, T.; Hemon, P.; et al. Decreased expression of the glucocorticoid receptor-gilz pathway in kupffer cells promotes liver inflammation in obese mice. J. Hepatol. 2016, 64, 916–924. [Google Scholar] [CrossRef]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of hif-1α in tumor-associated macrophages enhances m2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; de Carvalho Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-specific hypoxia-inducible factor-1α contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Van Steenkiste, C.; Verhelst, X.; Van Vlierberghe, H.; Devisscher, L.; Geerts, A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2016, 73, 3419–3431. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, Y.; Wang, W.; Scott, J.; Kim, K.; Sun, Z.; Guo, Q.; Lu, Y.; Gonzales, N.M.; Wu, H.; et al. Vitamin D receptor activation in liver macrophages ameliorates hepatic inflammation, steatosis, and insulin resistance in mice. Hepatology 2020, 71, 1559–1574. [Google Scholar] [CrossRef] [PubMed]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 kupffer cells promote m1 kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Wang, S.W.; Lan, T.; Sheng, H.; Zheng, F.; Lei, M.K.; Wang, L.X.; Chen, H.F.; Xu, C.Y.; Zhang, F. Nobiletin alleviates non-alcoholic steatohepatitis in mcd-induced mice by regulating macrophage polarization. Front. Physiol. 2021, 12, 687744. [Google Scholar] [CrossRef] [PubMed]
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the native study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Xiao, P.; Xu, H.-Q.; Niu, J.-Q.; Gao, Y.-H. Growing burden of alcoholic liver disease in China. World J. Gastroenterol. 2019, 25, 1445–1456. [Google Scholar] [CrossRef]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teli, M.R.; Day, C.P.; Burt, A.D.; Bennett, M.K.; James, O.F. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Moos, R.H.; Moos, B.S. Rates and predictors of relapse after natural and treated remission from alcohol use disorders. Addiction 2006, 101, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.K.; Mathurin, P. Diagnosis and treatment of alcohol-associated liver disease: A review. Jama 2021, 326, 165–176. [Google Scholar] [CrossRef]
- Lieber, C.S. The metabolic basis of alcohol’s toxicity. Hosp. Pract. 1977, 12, 73–80. [Google Scholar] [CrossRef]
- Magdaleno, F.; Blajszczak, C.C.; Nieto, N. Key events participating in the pathogenesis of alcoholic liver disease. Biomolecules 2017, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, B.; Tornai, D.; Kodys, K.; Adejumo, A.; Lowe, P.; McClain, C.; Mitchell, M.; McCullough, A.; Dasarathy, S.; Kroll-Desrosiers, A.; et al. Biomarkers of macrophage activation and immune danger signals predict clinical outcomes in alcoholic hepatitis. Hepatology 2019, 70, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Petrasek, J. Gut-liver axis and sterile signals in the development of alcoholic liver disease. Alcohol Alcohol. 2017, 52, 414–424. [Google Scholar] [CrossRef]
- Zhong, W.; McClain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z.X. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalny, A.V.; Skalnaya, M.G.; Grabeklis, A.R.; Skalnaya, A.A.; Tinkov, A.A. Zinc deficiency as a mediator of toxic effects of alcohol abuse. Eur. J. Nutr. 2018, 57, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef]
- Grewal, R.K.; Mahmood, A. The effects of ethanol administration on brush border membrane glycolipids in rat intestine. Alcohol 2010, 44, 515–522. [Google Scholar] [CrossRef]
- Kaur, J. Chronic ethanol feeding affects intestinal mucus lipid composition and glycosylation in rats. Ann. Nutr. Metabolism 2002, 46, 38–44. [Google Scholar] [CrossRef]
- Zhong, W.; Li, Q.; Sun, Q.; Zhang, W.; Zhang, J.; Sun, X.; Yin, X.; Zhang, X.; Zhou, Z. Preventing gut leakiness and endotoxemia contributes to the protective effect of zinc on alcohol-induced steatohepatitis in rats. J. Nutr. 2015, 145, 2690–2698. [Google Scholar] [CrossRef] [Green Version]
- Jung, F.; Burger, K.; Staltner, R.; Brandt, A.; Mueller, S.; Bergheim, I. Markers of intestinal permeability are rapidly improved by alcohol withdrawal in patients with alcohol-related liver disease. Nutrients 2021, 13, 1659. [Google Scholar] [CrossRef]
- Smirnova, E.; Puri, P.; Muthiah, M.D.; Daitya, K.; Brown, R.; Chalasani, N.; Liangpunsakul, S.; Shah, V.H.; Gelow, K.; Siddiqui, M.S.; et al. Fecal microbiome distinguishes alcohol consumption from alcoholic hepatitis but does not discriminate disease severity. Hepatology 2020, 72, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, V.; Horvath, A.; Komarova, I.; Schmerboeck, B.; Feldbacher, N.; Wurm, S.; Klymiuk, I.; Durdevic, M.; Rainer, F.; Blesl, A.; et al. A single alcohol binge impacts on neutrophil function without changes in gut barrier function and gut microbiome composition in healthy volunteers. PLoS One 2019, 14, e0211703. [Google Scholar] [CrossRef] [Green Version]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar] [PubMed]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor i and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Canesso, M.C.C.; Lacerda, N.L.; Ferreira, C.M.; Gonçalves, J.L.; Almeida, D.; Gamba, C.; Cassali, G.; Pedroso, S.H.; Moreira, C.; Martins, F.S.; et al. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: The role of the gut microbiota. BMC Microbiol. 2014, 14, 240. [Google Scholar]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota protects mice against acute alcohol-induced liver injury. Alcohol Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Xu, M.-J.; Bertola, A.; Wang, H.; Zhou, Z.; Liangpunsakul, S. Animal models of alcoholic liver disease: Pathogenesis and clinical relevance. Gene Expr. 2017, 17, 173–186. [Google Scholar] [CrossRef]
- Souza, H.S.P.; Elia, C.C.S.; Braulio, V.B.; Cortes, M.Q.; Furtado, V.C.S.; Garrofe, H.C.; Martinusso, C.A. Effects of ethanol on gut-associated lymphoid tissues in a model of bacterial translocation: A possible role of apoptosis. Alcohol 2003, 30, 183–191. [Google Scholar] [CrossRef]
- Glueck, B.; Han, Y.; Cresci, G.A.M. Tributyrin supplementation protects immune responses and vasculature and reduces oxidative stress in the proximal colon of mice exposed to chronic-binge ethanol feeding. J. Immunol. Res. 2018, 2018, 9671919. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Song, B.J. Pomegranate prevents binge alcohol-induced gut leakiness and hepatic inflammation by suppressing oxidative and nitrative stress. Redox Biol. 2018, 18, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.S.; Hong, S.H.; Suk, K.T.; Kim, J.B.; Han, S.H.; Sung, H.; Kim, E.J.; Kim, M.J.; Kim, M.Y.; Baik, S.K.; et al. Effects of korean red ginseng (Panax ginseng), urushiol (Rhus vernicifera stokes), and probiotics (Lactobacillus rhamnosus r0011 and Lactobacillus acidophilus r0052) on the gut-liver axis of alcoholic liver disease. J. Ginseng Res. 2014, 38, 167–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Lu, Y.; Leung, T.M.; Sorensen, E.S.; Nieto, N. Milk osteopontin, a nutritional approach to prevent alcohol-induced liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G929–G939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.M.; Forsyth, C.B.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Oats supplementation prevents alcohol-induced gut leakiness in rats by preventing alcohol-induced oxidative tissue damage. J. Pharmacol. Exp. Ther. 2009, 329, 952–958. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, J.P. Diet, food components and the intestinal barrier. Nutr Bull. 2017, 42, 123–131. [Google Scholar] [CrossRef]
- Yang, X.; He, F.; Zhang, Y.; Xue, J.; Li, K.; Zhang, X.; Zhu, L.; Wang, Z.; Wang, H.; Yang, S. Inulin ameliorates alcoholic liver disease via suppressing lps-tlr4-mpsi axis and modulating gut microbiota in mice. Alcohol Clin. Exp. Res. 2019, 43, 411–424. [Google Scholar] [CrossRef]
- Han, Y.; Glueck, B.; Shapiro, D.; Miller, A.; Roychowdhury, S.; Cresci, G.A.M. Dietary synbiotic supplementation protects barrier integrity of hepatocytes and liver sinusoidal endothelium in a mouse model of chronic-binge ethanol exposure. Nutrients 2020, 12, 373. [Google Scholar] [CrossRef] [Green Version]
- Ballway, J.W.; Song, B.J. Translational approaches with antioxidant phytochemicals against alcohol-mediated oxidative stress, gut dysbiosis, intestinal barrier dysfunction, and fatty liver disease. Antioxidants 2021, 10, 384. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.X.; Zhu, L.L.; Yang, X.L.; He, F.; Wang, T.; Bao, T.; Lu, H.X.; Wang, H.; Yang, S.Q. Inulin alleviates inflammation of alcoholic liver disease via scfas-inducing suppression of m1 and facilitation of m2 macrophages in mice. Int. Immunopharmacol. 2020, 78, 12. [Google Scholar] [CrossRef]
- Zheng, T.-x.; Pu, S.-l.; Tan, P.; Du, Y.-c.; Qian, B.-l.; Chen, H.; Fu, W.-g.; Huang, M.-z. Liver metabolomics reveals the effect of lactobacillus reuteri on alcoholic liver disease. Front. Physiol. 2020, 11, 595382. [Google Scholar] [CrossRef]
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665. [Google Scholar] [CrossRef]
- Marentette, J.O.; Wang, M.; Michel, C.R.; Powell, R.; Zhang, X.; Reisdorph, N.; Fritz, K.S.; Ju, C. Multi-omics analysis of liver infiltrating macrophages following ethanol consumption. Sci. Rep. 2019, 9, 7776. [Google Scholar] [CrossRef] [PubMed]
- Alharshawi, K.; Fey, H.; Vogle, A.; Klenk, T.; Kim, M.; Aloman, C. Alcohol consumption accumulation of monocyte derived macrophages in female mice liver is interferon alpha receptor dependent. Front. Immunol. 2021, 12, 663548. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C.; et al. Pharmacological inhibition of ccr2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef]
- Tornai, D.; Furi, I.; Shen, Z.T.; Sigalov, A.B.; Coban, S.; Szabo, G. Inhibition of triggering receptor expressed on myeloid cells 1 ameliorates inflammation and macrophage and neutrophil activation in alcoholic liver disease in mice. Hepatol. Commun. 2019, 3, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.D.; Chen, X.; Xu, J.J.; Du, X.S.; Yang, Y.; Li, J.J.; Yang, X.J.; Huang, H.M.; Li, X.F.; Wu, M.F.; et al. Dnmt3b-mediated methylation of zswim3 enhances inflammation in alcohol-induced liver injury via regulating traf2-mediated nf-kappa b pathway. Clin. Sci. 2020, 134, 1935–1956. [Google Scholar] [CrossRef]
- Denaes, T.; Lodder, J.; Chobert, M.N.; Ruiz, I.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. The cannabinoid receptor 2 protects against alcoholic liver disease via a macrophage autophagy-dependent pathway. Sci. Rep. 2016, 6, 28806. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Zhong, Z.; Kim, S.Y.; Uchiyama, R.; Roh, Y.S.; Matsushita, H.; Gottlieb, R.A.; Seki, E. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (irf1) and removing damaged mitochondria. J. Biol. Chem. 2019, 294, 12359–12369. [Google Scholar] [CrossRef]
- Ilyas, G.; Cingolani, F.; Zhao, E.; Tanaka, K.; Czaja, M.J. Decreased macrophage autophagy promotes liver injury and inflammation from alcohol. Alcoholism 2019, 43, 1403–1413. [Google Scholar] [CrossRef]
- Babuta, M.; Furi, I.; Bala, S.; Bukong, T.N.; Lowe, P.; Catalano, D.; Calenda, C.; Kodys, K.; Szabo, G. Dysregulated autophagy and lysosome function are linked to exosome production by micro-RNA 155 in alcoholic liver disease. Hepatology 2019, 70, 2123–2141. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Bala, S.; Kodys, K.; Szabo, G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific mirna-122 and sensitize monocytes to lps. Sci. Rep. 2015; 5, 9991. [Google Scholar]
- Verma, V.K.; Li, H.Y.; Wang, R.S.; Hirsova, P.; Cao, S.; Malhi, H.; Contreras, P.C.; Kamath, P.S.; Gores, G.J.; Shah, V. Alcohol stimulates macrophage activation through caspase dependent hepatocyte derived release of CD40l containing extracellular vesicles. J. Hepatol. 2015, 64, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Momen-Heravi, F.; Furi, I.; Kodys, K.; Catalano, D.; Gangopadhyay, A.; Haraszti, R.; Satishchandran, A.; Iracheta-Vellve, A.; Adejumo, A.; et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology 2018, 67, 1986–2000. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Momen-Heravi, F.; Kodys, K.; Szabo, G. Microrna cargo of extracellular vesicles from alcohol-exposed monocytes signals naive monocytes to differentiate into m2 macrophages. J. Biol. Chem. 2016, 291, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Bruneau, J.C.; Kodys, K.; Szabo, G. Alcohol-induced mir-27a regulates differentiation and m2 macrophage polarization of normal human monocytes. J. Immunol. 2015, 194, 3079–3087. [Google Scholar] [CrossRef] [Green Version]
- Momen-Heravi, F.; Saha, B.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J. Transl. Med. 2015, 13, 261. [Google Scholar] [CrossRef] [Green Version]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis - a comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Mehta, T.I.; Weissman, S.; Fung, B.M.; Sotiriadis, J.; Lindor, K.D.; Tabibian, J.H. Global incidence, prevalence, and features of primary sclerosing cholangitis: A systematic review and meta-analysis. Liver Int. 2021, 41, 2418–2426. [Google Scholar] [CrossRef]
- Vesterhus, M.; Karlsen, T.H. Emerging therapies in primary sclerosing cholangitis: Pathophysiological basis and clinical opportunities. J. Gastroenterol. 2020, 55, 588–614. [Google Scholar] [CrossRef] [Green Version]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary sclerosing cholangitis, part 1: Epidemiology, etiopathogenesis, clinical features, and treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar]
- Eksteen, B. The gut-liver axis in primary sclerosing cholangitis. Clin. Liver Dis. 2016, 20, 1–14. [Google Scholar] [CrossRef]
- Tanaka, A.; Mertens, J.C. Ulcerative colitis with and without primary sclerosing cholangitis: Two different diseases? Inflamm. Intest. Dis. 2016, 1, 9–14. [Google Scholar] [CrossRef]
- De Krijger, M.; Wildenberg, M.E.; de Jonge, W.J.; Ponsioen, C.Y. Return to sender: Lymphocyte trafficking mechanisms as contributors to primary sclerosing cholangitis. J. Hepatol. 2019, 71, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Milkiewicz, M.; Klak, M.; Kempinska-Podhorodecka, A.; Wiechowska-Kozlowska, A.; Urasinska, E.; Blatkiewicz, M.; Wunsch, E.; Elias, E.; Milkiewicz, P. Impaired hepatic adaptation to chronic cholestasis induced by primary sclerosing cholangitis. Sci. Rep. 2016, 6, 39573. [Google Scholar] [CrossRef] [Green Version]
- Mousa, O.Y.; Juran, B.D.; McCauley, B.M.; Vesterhus, M.N.; Folseraas, T.; Turgeon, C.T.; Ali, A.H.; Schlicht, E.M.; Atkinson, E.J.; Hu, C.; et al. Bile acid profiles in primary sclerosing cholangitis and their ability to predict hepatic decompensation. Hepatology 2021, 74, 281–295. [Google Scholar] [CrossRef]
- Dhillon, A.K.; Kummen, M.; Trøseid, M.; Åkra, S.; Liaskou, E.; Moum, B.; Vesterhus, M.; Karlsen, T.H.; Seljeflot, I.; Hov, J.R. Circulating markers of gut barrier function associated with disease severity in primary sclerosing cholangitis. Liver Int. 2019, 39, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Tornai, T.; Palyu, E.; Vitalis, Z.; Tornai, I.; Tornai, D.; Antal-Szalmas, P.; Norman, G.L.; Shums, Z.; Veres, G.; Dezsofi, A.; et al. Gut barrier failure biomarkers are associated with poor disease outcome in patients with primary sclerosing cholangitis. World J. Gastroenterol. 2017, 23, 5412–5421. [Google Scholar] [CrossRef] [Green Version]
- Hov, J.R.; Karlsen, T.H. The microbiome in primary sclerosing cholangitis: Current evidence and potential concepts. Semin. Liver Dis. 2017, 37, 314–331. [Google Scholar] [CrossRef] [Green Version]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered gut microbial metabolism of essential nutrients in primary sclerosing cholangitis. Gastroenterology 2021, 160, 1784–1798.e0. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutus, G.; Ghosh, S.; Rossiter, A.; Loman, N.; van Schaik, W.; et al. A pilot integrative analysis of colonic gene expression, gut microbiota and immune infiltration in primary sclerosing cholangitis-inflammatory bowel disease: Association of disease with bile acid pathways. J. Crohn’s Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iravani, S.; Dooghaie-Moghadam, A.; Razavi-Khorasani, N.; Moazzami, B.; Dowlati Beirami, A.; Mansour-Ghanaei, A.; Majidzadeh, A.K.; Mehrvar, A.; Khoshdel, A.; Nasiri Toosi, M.; et al. An update on treatment options for primary sclerosing cholangitis. Gastroenterol. Hepatol. Bed. Bench 2020, 13, 115–124. [Google Scholar] [PubMed]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of antibiotic therapy in primary sclerosing cholangitis with and without inflammatory bowel disease: A systematic review and meta-analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal microbiota transplantation in patients with primary sclerosing cholangitis: A pilot clinical trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rubio, R.; Patterson, A.M.; Cotter, P.D.; Beraza, N. Cholestasis induced by bile duct ligation promotes changes in the intestinal microbiome in mice. Sci. Rep. 2019, 9, 12324. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ma, C.; Duan, Y.; Heinrich, B.; Rosato, U.; Diggs, L.P.; Ma, L.; Roy, S.; Fu, Q.; Brown, Z.J.; et al. Gut microbiome directs hepatocytes to recruit mdscs and promote cholangiocarcinoma. Cancer Discov. 2021, 11, 1248–1267. [Google Scholar] [CrossRef]
- Isaacs-Ten, A.; Echeandia, M.; Moreno-Gonzalez, M.; Brion, A.; Goldson, A.; Philo, M.; Patterson, A.M.; Parker, A.; Galduroz, M.; Baker, D.; et al. Intestinal microbiome-macrophage crosstalk contributes to cholestatic liver disease by promoting intestinal permeability. Hepatology 2020, 72, 2090–2108. [Google Scholar] [CrossRef] [Green Version]
- Liao, L.; Schneider, K.M.; Galvez, E.J.C.; Frissen, M.; Marschall, H.U.; Su, H.; Hatting, M.; Wahlstrom, A.; Haybaeck, J.; Puchas, P.; et al. Intestinal dysbiosis augments liver disease progression via NLRP3 in a murine model of primary sclerosing cholangitis. Gut 2019, 68, 1477–1492. [Google Scholar] [CrossRef]
- Schrumpf, E.; Kummen, M.; Valestrand, L.; Greiner, T.U.; Holm, K.; Arulampalam, V.; Reims, H.M.; Baines, J.; Bäckhed, F.; Karlsen, T.H.; et al. The gut microbiota contributes to a mouse model of spontaneous bile duct inflammation. J. Hepatol. 2017, 66, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Juanola, O.; Hassan, M.; Kumar, P.; Yilmaz, B.; Keller, I.; Simillion, C.; Engelmann, C.; Tacke, F.; Dufour, J.-F.; De Gottardi, A.; et al. Intestinal microbiota drives cholestasis-induced specific hepatic gene expression patterns. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, N.; Sasaki, N.; Aoki, R.; Miyamoto, K.; Suda, W.; Teratani, T.; Suzuki, T.; Koda, Y.; Chu, P.S.; Taniki, N.; et al. Gut pathobionts underlie intestinal barrier dysfunction and liver t helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 2019, 4, 492–503. [Google Scholar] [CrossRef]
- Peng, Z.-W.; Rothweiler, S.; Wei, G.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Longhi, M.S.; Kuang, M.; Robson, S.C.; et al. The ectonucleotidase entpd1/cd39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatol. Commun. 2017, 1, 957–972. [Google Scholar] [CrossRef]
- Jahnel, J.; Fickert, P.; Langner, C.; Högenauer, C.; Silbert, D.; Gumhold, J.; Fuchsbichler, A.; Trauner, M. Impact of experimental colitis on hepatobiliary transporter expression and bile duct injury in mice. Liver Int. 2009, 29, 1316–1325. [Google Scholar] [CrossRef]
- Gao, R.Y.; Shearn, C.T.; Orlicky, D.J.; Battista, K.D.; Alexeev, E.E.; Cartwright, I.M.; Lanis, J.M.; Kostelecky, R.E.; Ju, C.; Colgan, S.P.; et al. Bile acids modulate colonic madcam-1 expression in a murine model of combined cholestasis and colitis. Mucosal Immunol. 2021, 14, 479–490. [Google Scholar] [CrossRef]
- Isidro, R.A.; Appleyard, C.B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G59–G73. [Google Scholar] [CrossRef] [PubMed]
- Gijbels, E.; Pieters, A.; De Muynck, K.; Vinken, M.; Devisscher, L. Rodent models of cholestatic liver disease: A practical guide for translational research. Liver Int. 2021, 41, 656–682. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Trauner, M.; Fickert, P. Will we ever model psc? - "it’s hard to be a psc model!". Clin. Res. Hepatol. Gastroenterol. 2011, 35, 792–804. [Google Scholar] [CrossRef]
- Cameron, R.G.; Blendis, L.M.; Neuman, M.G. Accumulation of macrophages in primary sclerosing cholangitis. Clin. Biochem. 2001, 34, 195–201. [Google Scholar] [CrossRef]
- Shearn, C.T.; Orlicky, D.J.; Petersen, D.R. Dysregulation of antioxidant responses in patients diagnosed with concomitant primary sclerosing cholangitis/inflammatory bowel disease. Exp. Mol. Pathol. 2018, 104, 1–8. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Arndtz, K.; Webb, G.; Corrigan, M.; Akiror, S.; Liaskou, E.; Woodward, P.; Adams, D.H.; Weston, C.J.; Hirschfield, G.M. Intrahepatic macrophage populations in the pathophysiology of primary sclerosing cholangitis. JHEP Rep. 2019, 1, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Bossen, L.; Vesterhus, M.; Hov, J.R.; Färkkilä, M.; Rosenberg, W.M.; Møller, H.J.; Boberg, K.M.; Karlsen, T.H.; Grønbæk, H. Circulating macrophage activation markers predict transplant-free survival in patients with primary sclerosing cholangitis. Clin. Transl. Gastroenter. 2021, 12, e00315. [Google Scholar] [CrossRef]
- Rothweiler, S.; Feldbrugge, L.; Jiang, Z.G.; Csizmadia, E.; Longhi, M.S.; Vaid, K.; Enjyoji, K.; Popov, Y.V.; Robson, S.C. Selective deletion of entpd1/cd39 in macrophages exacerbates biliary fibrosis in a mouse model of sclerosing cholangitis. Purinergic Signal. 2019, 15, 375–385. [Google Scholar] [CrossRef]
- Best, J.; Verhulst, S.; Syn, W.K.; Lagaisse, K.; van Hul, N.; Heindryckx, F.; Sowa, J.P.; Peeters, L.; Van Vlierberghe, H.; Leclercq, I.A.; et al. Macrophage depletion attenuates extracellular matrix deposition and ductular reaction in a mouse model of chronic cholangiopathies. PLoS ONE 2016, 11, e0162286. [Google Scholar]
- Abshagen, K.; König, M.; Hoppe, A.; Müller, I.; Ebert, M.; Weng, H.; Holzhütter, H.-G.; Zanger, U.M.; Bode, J.; Vollmar, B.; et al. Pathobiochemical signatures of cholestatic liver disease in bile duct ligated mice. BMC Syst. Biol. 2015, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duwaerts, C.C.; Gehring, S.; Cheng, C.-W.; van Rooijen, N.; Gregory, S.H. Contrasting responses of kupffer cells and inflammatory mononuclear phagocytes to biliary obstruction in a mouse model of cholestatic liver injury. Liver Int. 2013, 33, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Trussoni, C.E.; LaRusso, N.F.; Gores, G.J. The spectrum of reactive cholangiocytes in primary sclerosing cholangitis. Hepatology 2020, 71, 741–748. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fiorotto, R.; Cadamuro, M.; Spirli, C.; Mariotti, V.; Kaffe, E.; Scirpo, R.; Fabris, L. Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1374–1379. [Google Scholar] [CrossRef]
- Guillot, A.; Guerri, L.; Feng, D.; Kim, S.J.; Ahmed, Y.A.; Paloczi, J.; He, Y.; Schuebel, K.; Dai, S.; Liu, F.; et al. Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin αvβ6 upregulation following liver injury. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Eksteen, B.; Bowlus, C.L.; Montano-Loza, A.J.; Lefebvre, E.; Fischer, L.; Vig, P.; Martins, E.B.; Ahmad, J.; Yimam, K.K.; Pockros, P.J.; et al. Efficacy and safety of cenicriviroc in patients with primary sclerosing cholangitis: Perseus study. Hepatol. Commun. 2021, 5, 478–490. [Google Scholar] [CrossRef]
- Yu, D.K.; Cai, S.Y.; Mennone, A.; Vig, P.; Boyer, J.L. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int. 2018, 38, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of kupffer cells in cholestatic liver injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemail, L.; Miyao, M.; Kotani, H.; Kawai, C.; Minami, H.; Abiru, H.; Tamaki, K. Pivotal roles of kupffer cells in the progression and regression of ddc-induced chronic cholangiopathy. Sci. Rep. 2018, 8, 6415. [Google Scholar] [CrossRef]
- Locatelli, L.; Cadamuro, M.; Spirlì, C.; Fiorotto, R.; Lecchi, S.; Morell, C.M.; Popov, Y.; Scirpo, R.; De Matteis, M.; Amenduni, M.; et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 2016, 63, 965–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, J.M.; Faiola, B.; Melich, D.H.; Peterson, R.A.; Jordan, H.L.; Kimbrough, C.L.; Prescott, J.S.; Miller, R.T. Effects of kupffer cell depletion on acute alpha-naphthylisothiocyanate-induced liver toxicity in male mice. Toxicol. Pathol. 2013, 41, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Katsumi, T.; Guicciardi, M.E.; Azad, A.; Bronk, S.F.; Krishnan, A.; Gores, G.J. Activated cholangiocytes release macrophage-polarizing extracellular vesicles bearing the damp s100a11. Am. J. Physiol. Cell Physiol. 2019, 317, C788–c799. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Wang, Y.; Zhu, W.; Zhao, D.; Wang, X.; Yang, H.; Gurley, E.C.; Chen, W.; Hylemon, P.B.; et al. Cholangiocyte-derived exosomal lncrna h19 promotes macrophage activation and hepatic inflammation under cholestatic conditions. Cells 2020, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Pope, C.; Mishra, S.; Russell, J.; Zhou, Q.; Zhong, X.-B. Targeting h19, an imprinted long non-coding rna, in hepatic functions and liver diseases. Diseases 2017, 5, 11. [Google Scholar] [CrossRef]
- Thompson, M.D.; Monga, S.P.S. Wnt/β-catenin signaling in liver health and disease. Hepatology 2007, 45, 1298–1305. [Google Scholar] [CrossRef]
- Jiang, A.; Okabe, H.; Popovic, B.; Preziosi, M.E.; Pradhan-Sundd, T.; Poddar, M.; Singh, S.; Bell, A.; England, S.G.; Nagarajan, S.; et al. Loss of wnt secretion by macrophages promotes hepatobiliary injury after administration of 3,5-diethoxycarbonyl-1, 4-dihydrocollidine diet. Am. J. Pathol. 2019, 189, 590–603. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Suk, K.T.; Kim, D.J. Staging of liver fibrosis or cirrhosis: The role of hepatic venous pressure gradient measurement. World J. Hepatol. 2015, 7, 607–615. [Google Scholar] [CrossRef]
- Raevens, S.; Geerts, A.; Van Steenkiste, C.; Verhelst, X.; Van Vlierberghe, H.; Colle, I. Hepatopulmonary syndrome and portopulmonary hypertension: Recent knowledge in pathogenesis and overview of clinical assessment. Liver Int. 2015, 35, 1646–1660. [Google Scholar] [CrossRef]
- Kalaitzakis, E. Gastrointestinal dysfunction in liver cirrhosis. World J. Gastroenterol. 2014, 20, 14686–14695. [Google Scholar] [CrossRef]
- Arab, J.P.; Martin-Mateos, R.M.; Shah, V.H. Gut-liver axis, cirrhosis and portal hypertension: The chicken and the egg. Hepatol. Int. 2018, 12, 24–33. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H. Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J. Hepatol. 2015, 7, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Aguirre Valadez, J.M.; Rivera-Espinosa, L.; Mendez-Guerrero, O.; Chavez-Pacheco, J.L.; Garcia Juarez, I.; Torre, A. Intestinal permeability in a patient with liver cirrhosis. Ther. Clin. Risk Manag. 2016, 12, 1729–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.J.; Zimmermann, H.W.; Adams, D.H. The role of myeloid-derived cells in the progression of liver disease. Front. Immunol. 2019, 10, 893. [Google Scholar] [CrossRef]
- Ritz, T.; Krenkel, O.; Tacke, F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol. 2018, 330, 175–182. [Google Scholar] [CrossRef]
- Berres, M.L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Invest. 2010, 120, 4129–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichs, D.; Berres, M.L.; Nellen, A.; Fischer, P.; Scholten, D.; Trautwein, C.; Wasmuth, H.E.; Sahin, H. The chemokine ccl3 promotes experimental liver fibrosis in mice. PLoS ONE 2013, 8, e66106. [Google Scholar]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H.W.; Seidler, S.; Nattermann, J.; Gassler, N.; Hellerbrand, C.; Zernecke, A.; Tischendorf, J.J.; Luedde, T.; Weiskirchen, R.; Trautwein, C.; et al. Functional contribution of elevated circulating and hepatic non-classical cd14cd16 monocytes to inflammation and human liver fibrosis. PLoS ONE 2010, 5, e11049. [Google Scholar] [CrossRef]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Tan-Garcia, A.; Lai, F.; Sheng Yeong, J.P.; Irac, S.E.; Ng, P.Y.; Msallam, R.; Tatt Lim, J.C.; Wai, L.E.; Tham, C.Y.L.; Choo, S.P.; et al. Liver fibrosis and cd206(+) macrophage accumulation are suppressed by anti-gm-csf therapy. JHEP Rep. 2020, 2, 100062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Resolution of liver fibrosis: Basic mechanisms and clinical relevance. Semin. Liver Dis. 2015, 35, 119–131. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic effects of the dual ccr2/ccr5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: Final analysis of the phase 2b centaur study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Devisscher, L.; Tacke, F. Targeting ccr2/5 in the treatment of nonalcoholic steatohepatitis (nash) and fibrosis: Opportunities and challenges. Expert Opin. Investig. Drugs 2020, 29, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, N.; Kawada, N. Role of the gut-liver axis in liver inflammation, fibrosis, and cancer: A special focus on the gut microbiota relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Trautwein, C. Mechanisms of liver fibrosis resolution. J. Hepatol. 2015, 63, 1038–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.; Couton, D.; Couty, J.P.; Anson, M.; Crain, A.M.; Bizet, V.; Renia, L.; Pol, S.; Mallet, V.; Gilgenkrantz, H. Dual role of ccr2 in the constitution and the resolution of liver fibrosis in mice. Am. J. Pathol. 2009, 174, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Zimmermann, H.W.; Roderburg, C.; Gassler, N.; Wasmuth, H.E.; Luedde, T.; Trautwein, C.; Tacke, F. The fractalkine receptor cx(3)cr1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 2010, 52, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.F.; Gao, C.C.; Yi, J.; Zhao, J.L.; Liang, S.Q.; Zhao, Y.; Ye, Y.C.; Bai, J.; Zheng, Q.J.; Dou, K.F.; et al. Cytotherapy with m1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J. Hepatol. 2017, 67, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5, 879. [Google Scholar] [CrossRef] [PubMed]
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular carcinoma: Risk factors, diagnosis and treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [Green Version]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.X.; Qiu, X.Y.; Hu, D.X.; Chen, X.Q. Advances in hypoxia-mediated mechanisms in hepatocellular carcinoma. Mol. Pharmacol. 2017, 92, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Wang, N.; Qin, W. Gut microbiota and hepatocellular carcinoma. Gastrointest. Tumors 2015, 2, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in hcc - mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achiwa, K.; Ishigami, M.; Ishizu, Y.; Kuzuya, T.; Honda, T.; Hayashi, K.; Hirooka, Y.; Katano, Y.; Goto, H. Dss colitis promotes tumorigenesis and fibrogenesis in a choline-deficient high-fat diet-induced nash mouse model. Biochem. Biophys. Res. Commun. 2016, 470, 15–21. [Google Scholar] [CrossRef]
- Zhang, H.L.; Yu, L.X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and tlr4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.W.; Chen, X.H.; Ren, Z.; Zheng, S.S. Gut microbial dysbiosis associates hepatocellular carcinoma via the gut-liver axis. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 19–27. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Salcedo, R.; Mivechi, N.F.; Trinchieri, G.; Horuzsko, A. The proinflammatory myeloid cell receptor trem-1 controls kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012, 72, 3977–3986. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ma, L.; Shen, S.; Guo, Y.; Cao, Q.; Cai, X.; Feng, J.; Yan, Y.; Hu, T.; Luo, S.; et al. Intestinal dysbacteriosis-induced il-25 promotes development of hcc via alternative activation of macrophages in tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 303. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [Green Version]
- Van Campenhout, S.; Tilleman, L.; Lefere, S.; Vandierendonck, A.; Raevens, S.; Verhelst, X.; Geerts, A.; Van Nieuwerburgh, F.; Van Vlierberghe, H.; Devisscher, L. Myeloid-specific ire1alpha deletion reduces tumour development in a diabetic, non-alcoholic steatohepatitis-induced hepatocellular carcinoma mouse model. Metabolism 2020, 107, 154220. [Google Scholar] [CrossRef]
- Vanderborght, B.; De Muynck, K.; Lefere, S.; Geerts, A.; Degroote, H.; Verhelst, X.; Van Vlierberghe, H.; Devisscher, L. Effect of isoform-specific hif-1α and hif-2α antisense oligonucleotides on tumorigenesis, inflammation and fibrosis in a hepatocellular carcinoma mouse model. Oncotarget 2020, 11, 4504–4520. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.C.; Zhao, J.L.; Lu, Y.T.; Gao, C.C.; Yang, Y.; Liang, S.Q.; Lu, Y.Y.; Wang, L.; Yue, S.Q.; Dou, K.F.; et al. Notch signaling via wnt regulates the proliferation of alternative, ccr2-independent tumor-associated macrophages in hepatocellular carcinoma. Cancer Res. 2019, 79, 4160–4172. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Zhang, Z.; Zheng, B.H.; Shi, Y.; Duan, M.; Ma, L.J.; Wang, Z.C.; Dong, L.Q.; Dong, P.P.; Shi, J.Y.; et al. Ccl15 recruits suppressive monocytes to facilitate immune escape and disease progression in hepatocellular carcinoma. Hepatology 2019, 69, 143–159. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.M.; Patten, D.A.; Shetty, S. Tumour-associated macrophages in hepatocellular carcinoma: Pressing the metabolic switch to prevent t cell responses. J. Hepatol. 2019, 71, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of cd8+ t cells in human hepatocellular carcinoma is mediated by b7-h1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 macrophages induce pd-l1 expression in hepatocellular carcinoma cells through il-1beta signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Wree, A.; Demir, M.; Schmelzle, M.; Tacke, F. The role of the innate immune system in the development and treatment of hepatocellular carcinoma. Hepat. Oncol. 2020, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degroote, H.; Van Dierendonck, A.; Geerts, A.; Van Vlierberghe, H.; Devisscher, L. Preclinical and clinical therapeutic strategies affecting tumor-associated macrophages in hepatocellular carcinoma. J. Immunol. Res. 2018, 2018, 7819520. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.H.; Ge, Y.S.; Ma, H.X.; Gao, X.Q.; Pan, J.J.; Liu, D.; Xu, G.L.; Ma, J.L.; Jia, W.D. Prognostic value of polarized macrophages in patients with hepatocellular carcinoma after curative resection. J. Cell. Mol. Med. 2016, 20, 1024–1035. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via wnt/beta-catenin signaling promotes m2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters hcc development by enhancing tgf-beta-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Tian, Z.; Hou, X.; Liu, W.; Han, Z.; Wei, L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019, 9, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.H.; Ng, H.H.M.; Lim, C.J.; Sim, X.N.; Malavasi, F.; Li, H.; Loh, J.J.H.; Sabai, K.; Kim, J.K.; Ong, C.C.H.; et al. Expression of cd38 on macrophages predicts improved prognosis in hepatocellular carcinoma. Front. Immunol. 2019, 10, 2093. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Ge, W.; Zhou, J.; Gao, B.; Qian, X.; Wang, W. The role of tumor associated macrophages in hepatocellular carcinoma. J. Cancer 2021, 12, 1284–1294. [Google Scholar] [CrossRef]
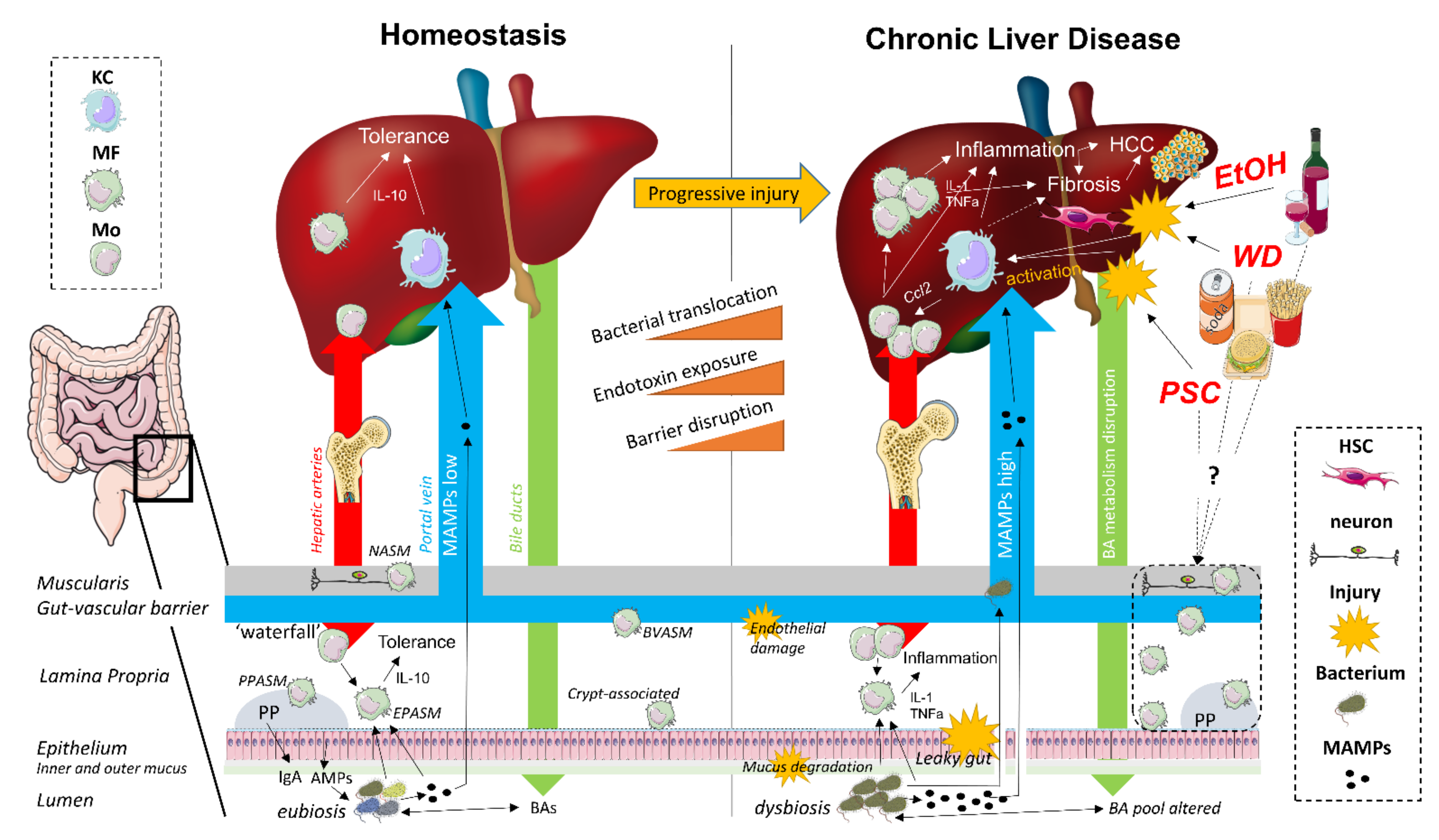
| Macrophage Subset | Markers | Reference(s) |
|---|---|---|
| MOUSE | ||
| Resident Kupffer cell (KC) | F4/80hi CD11bint Ly6Clo CX3CR1lo Clec4F+ TIM4+ Clec2hi VSIG4+ CD207+ CD163+ MARCO+ | [54,57,58,59,60,64,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101] |
| M1-like KC | CD11c+ | [54] |
| M2-like KC | CD206+ CD317+ CD1d+ | [54] |
| Metabolic KC | CD206hi ESAM+ | [56] |
| AILI-associated restorative KC | F4/80+ CD11b+ MHCIIhi Ly6Clo MerTK+ | [88] |
| Infiltrating monocyte-derived macrophage (MoMF) | F4/80int CD11bhi CX3CR1hi CCR2+ | [66,67,69,70,71,72,73,74,76,77,79,80,81,82,83,84,85,86,88,90,91,95,96,98,99,100,102] |
| Proinflammatory/profibrotic MoMF | Ly6Chi | [67,71,79,80,83,84,85,86,95,102,103,104,105] |
| Anti-inflammatory/restorative MoMF | Ly6Clo | [67,71,79,80,83,84,86,95,102,103] |
| Transitioning macrophage | CX3CR1hi CCR2hi CD11c+ Clec4F− TIM4− Clec2+ VSIG4− | [94,97,98] |
| Transitioning LAM | TREM2hi SPP1+ CD9+ CD63+ GPNMB+ | [98] |
| Monocyte-derived Kupffer cell (MoKC) | F4/80hi CD11blo/int CX3CR1lo CCR2lo Clec4F+ TIM4− Clec2hi VSIG4+ | [57,59,78,87,89,94,95,97,98] |
| NASH-associated macrophage (NAM) | F4/80hi Clec4Fhi TREM2hi CD9hi GPNMBhi | [106,107] |
| Lipid-associated macrophage (LAM) | CX3CR1lo CCR2lo TREM2hi CD9hi CD63hi SPP1hi GPNMBhi | [90,95] |
| ALD-associated M2-like macrophage | F4/80+ CD163+ CD206+ | [94,98,108] |
| Scar-associated macrophage (SAM) | F4/80+ CD11b+ TIM4− CD9+ TREM2+ | [64] |
| M1-like tumor-associated macrophage (TAM) | F4/80+ CD11b+ MHCIIhi CD206− | [109,110,111] |
| M2-like tumor-associated macrophage (TAM) | F4/80+ CD11b+ MHCIIlo CD206+ | [109,110,111] |
| Liver capsular macrophage | F4/80+ CD11blo CD11clo MHCIIhi CD64+ CX3CR1hi Ly6Clo TIM4− | [61] |
| Peritoneal macrophage | CD11b+ F4/80hi GATA6+ Ly6C− MHCII− CD206+ CD64+ CD68+ CD11c+ | [62] |
| HUMAN | ||
| Kupffer cell (KC) | CD68+ CD11b− CD14int CD32hi CD163+ VSIG4+ MARCO+ TIMD4+ CCR2− | [53,63,64,79,85,86,112,113,114] |
| Tolerogenic KC | CD163+ VSIG4+ LILRB5+ CD5L+ MARCO+ HMOX1hi | [53,63] |
| Inflammatory KC | CD163+ VSIG4+ CD1C+ FCER1A+ | [63] |
| Infiltrating monocyte-derived macrophage (MoMF) | CD68+ CD11b+ CD14hi CD32int CCR2+ S100A9+ MAC387+ MARCO− | [53,79,85,86,112,113,114,115,116] |
| HCV-associated pro-inflammatory macrophage | CD14+ HLA-DRhi CD206+ | [117] |
| Monocyte-derived Kupffer cell (MoKC) | CD163+ MARCO+ TIMD4− | [64] |
| ALF-associated restorative macrophage | MerTK+ HLA-DRhi CD163hi Tie-2hi | [79,88] |
| Fibrosis-associated macrophage | CD68+ CD163+ MMP9+ | [116,118] |
| Scar-associated macrophage (SAM) | TREM2+ CD9+ MNDA+ | [64] |
| Tumor-associated MDSC-like macrophage | THBS1+ | [119] |
| Tumor-associated macrophage (TAM) | CD68+ CD14+ C1QA+ | [119,120] |
| M1-like TAM | CD86+ CD169+ HLA-DR+ CCR2+ | [71,110,121,122,123,124] |
| M2-like TAM | CD163+ CD204+ CD206+ HLA-DR− CCR2− Arg1+ MR+ | [71,110,121,122,123,124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Muynck, K.; Vanderborght, B.; Van Vlierberghe, H.; Devisscher, L. The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective. Cells 2021, 10, 2959. https://doi.org/10.3390/cells10112959
De Muynck K, Vanderborght B, Van Vlierberghe H, Devisscher L. The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective. Cells. 2021; 10(11):2959. https://doi.org/10.3390/cells10112959
Chicago/Turabian StyleDe Muynck, Kevin, Bart Vanderborght, Hans Van Vlierberghe, and Lindsey Devisscher. 2021. "The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective" Cells 10, no. 11: 2959. https://doi.org/10.3390/cells10112959
APA StyleDe Muynck, K., Vanderborght, B., Van Vlierberghe, H., & Devisscher, L. (2021). The Gut–Liver Axis in Chronic Liver Disease: A Macrophage Perspective. Cells, 10(11), 2959. https://doi.org/10.3390/cells10112959