
RNA Targeting in Inherited Neuromuscular Disorders: Novel Therapeutic Strategies to Counteract Mis-Splicing


Abstract
1. Introduction
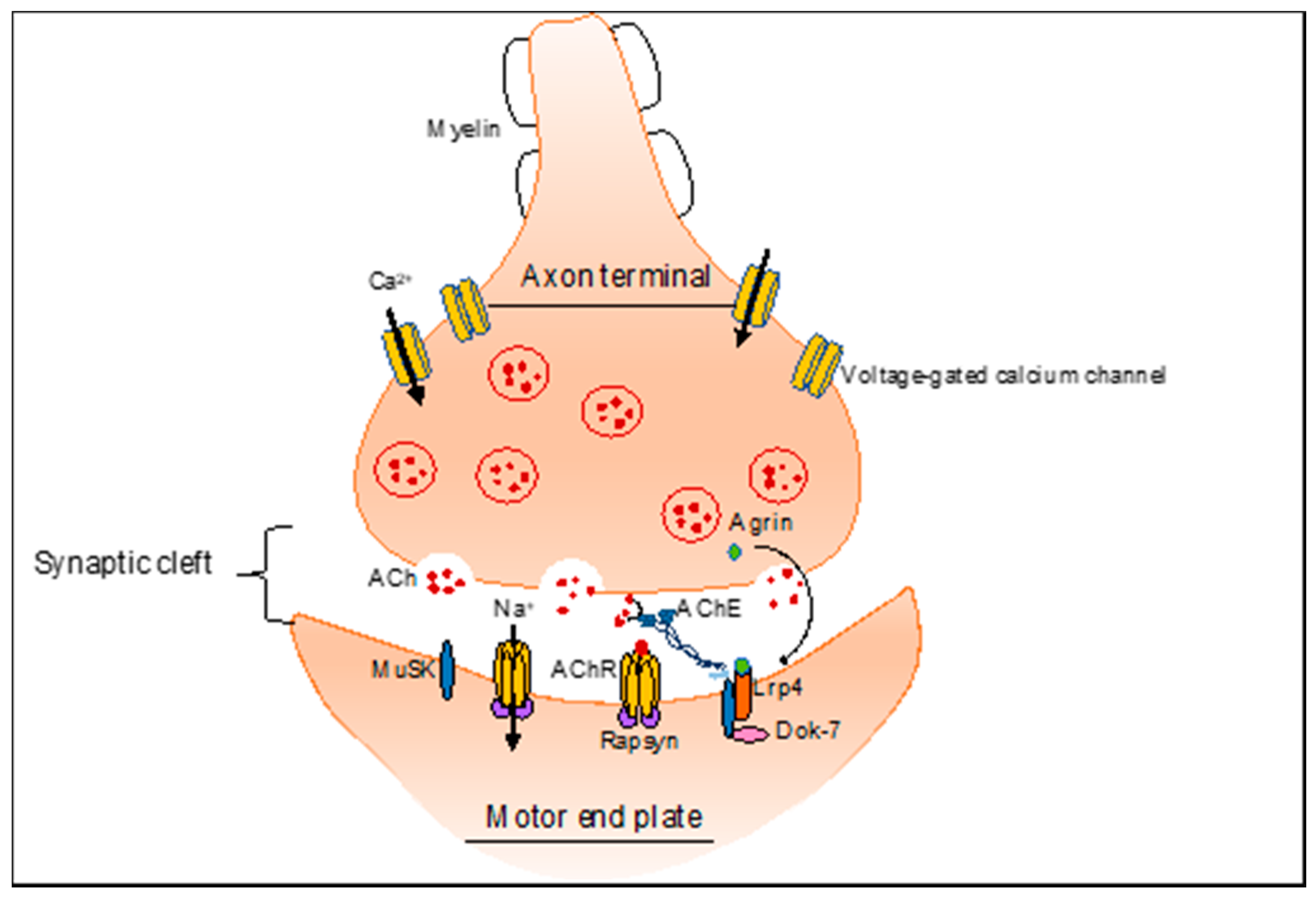
2. NMJ-Related Neuromuscular Disorders
3. Alternative Splicing in the Skeletal Muscle
4. Alternative Splicing and Neuromuscular Genetic Diseases
4.1. Alternative Splicing Defects in Congenital Myasthenic Syndrome (CMS)
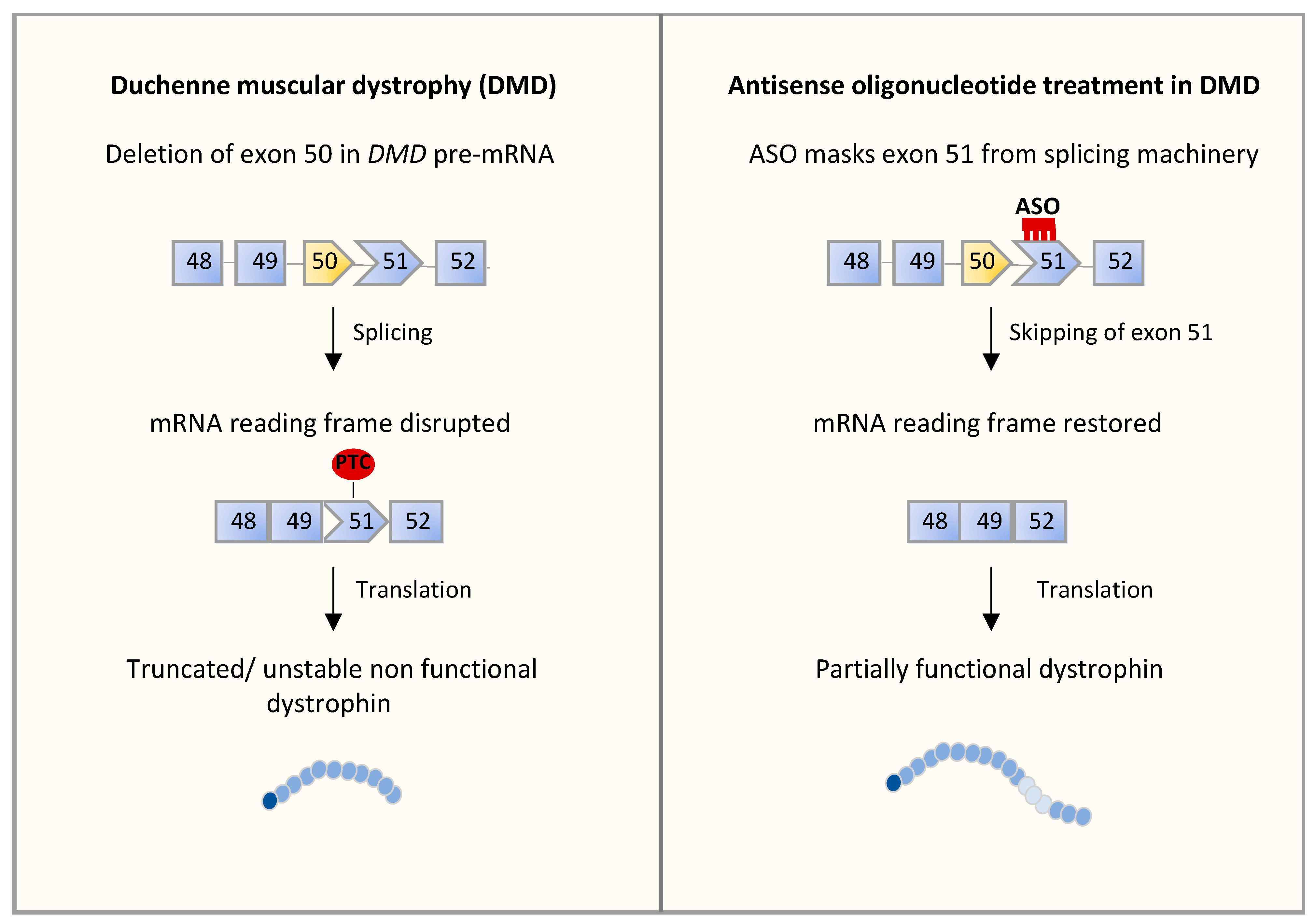
4.2. Alternative Splicing Defects in Duchenne Muscular Dystrophy (DMD)
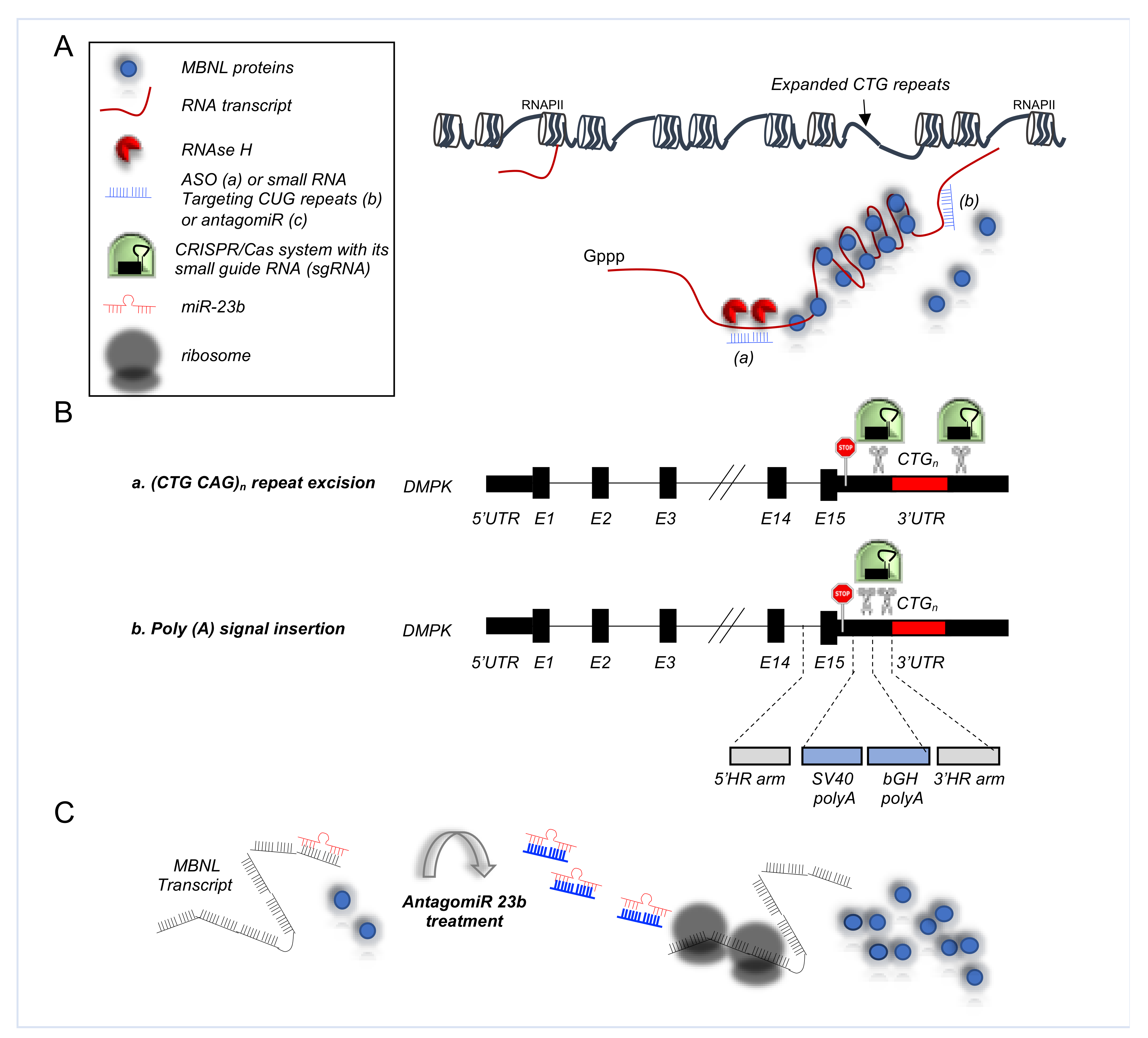
4.3. Alternative Splicing Defects in Myotonic Dystrophy Type 1 (DM1)
5. RNA-Based Approaches to Address Splicing Alterations
5.1. RNA-Based Approaches in CMS
5.2. RNA-Based Approaches in DMD
5.3. RNA-Based Approaches in DM1
5.3.1. Antisense Oligonucleotides in DM1
5.3.2. CRISPR/Cas9 Based Therapies for DM1
5.3.3. AntagomiRs in DM1
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tintignac, L.A.; Brenner, H.R.; Rüegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, M.F.; Saporta, M. Hereditary motor neuropathies. Curr. Opin. Neurol 2020, 33, 568–574. [Google Scholar] [CrossRef]
- Fox, M.A.; Sanes, J.R.; Borza, D.B.; Eswarakumar, V.P.; Fässler, R.; Hudson, B.G.; John, S.W.; Ninomiya, Y.; Pedchenko, V.; Pfaff, S.L.; et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 2007, 129, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Mudd, J.; Nichol, M.; Chu, G.C.; Sanes, J.R.; Merlie, J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 1995, 377, 232–236. [Google Scholar] [CrossRef]
- Gautam, M.; Noakes, P.G.; Moscoso, L.; Rupp, F.; Scheller, R.H.; Merlie, J.P.; Sanes, J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 1996, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Ruff, R.L. Neurophysiology of the neuromuscular junction: Overview. Ann. N. Y. Acad. Sci. 2003, 998, 1–10. [Google Scholar] [CrossRef]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin--glycoprotein complex. Neuron 2000, 25, 279–293. [Google Scholar] [CrossRef]
- Adams, M.E.; Kramarcy, N.; Krall, S.P.; Rossi, S.G.; Rotundo, R.L.; Sealock, R.; Froehner, S.C. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000, 150, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Belhasan, D.C.; Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 2020, 722, 134833. [Google Scholar] [CrossRef]
- Herbst, R.; Iskratsch, T.; Unger, E.; Bittner, R.E. Aberrant development of neuromuscular junctions in glycosylation-defective Large(myd) mice. Neuromuscul. Disord. 2009, 19, 366–378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pratt, S.J.P.; Shah, S.B.; Ward, C.W.; Kerr, J.P.; Stains, J.P.; Lovering, R.M. Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell Mol. Life Sci. 2015, 72, 153–164. [Google Scholar] [CrossRef]
- Hesser, B.A.; Henschel, O.; Witzemann, V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol. Cell Neurosci. 2006, 31, 470–480. [Google Scholar] [CrossRef]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet. J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- André, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768.e756. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Pediatr. 2016, 42, 78. [Google Scholar] [CrossRef]
- Thangarajh, M. The Dystrophinopathies. Continuum 2019, 25, 1619–1639. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol. 2020, 39, 179–186. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Howard, M.T.; Sampson, J.B.; Swoboda, K.J.; Bromberg, M.B.; Mendell, J.R.; Taylor, L.E.; et al. Nonsense mutation-associated Becker muscular dystrophy: Interplay between exon definition and splicing regulatory elements within the DMD gene. Hum. Mutat. 2011, 32, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.T.; Darmahkasih, A.J.; Horn, P.S.; Rybalsky, I.; Shellenbarger, K.C.; Tian, C.; Wong, B.L. Neurodevelopmental, behavioral, and emotional symptoms in Becker muscular dystrophy. Muscle Nerve 2020, 61, 156–162. [Google Scholar] [CrossRef]
- Yadava, R.S.; Mandal, M.; Giese, J.M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Modeling muscle regeneration in RNA toxicity mice. Hum. Mol. Genet. 2021, 30, 1111–1130. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Modrek, B.; Lee, C.J. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 2003, 34, 177–180. [Google Scholar] [CrossRef]
- Xu, Q.; Modrek, B.; Lee, C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002, 30, 3754–3766. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Seidman, S. Acetylcholinesterase--new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Chan, W.Y.; Soloviev, M.M.; Ciruela, F.; McIlhinney, R.A. Molecular determinants of metabotropic glutamate receptor 1B trafficking. Mol. Cell Neurosci. 2001, 17, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Dunker, A.K.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M.M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef]
- Black, D.L.; Grabowski, P.J. Alternative pre-mRNA splicing and neuronal function. Prog Mol. Subcell. Biol. 2003, 31, 187–216. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Söllner, J.F.; Leparc, G.; Hildebrandt, T.; Klein, H.; Thomas, L.; Stupka, E.; Simon, E. An RNA-Seq atlas of gene expression in mouse and rat normal tissues. Sci. Data 2017, 4, 170185. [Google Scholar] [CrossRef]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef]
- Bland, C.S.; Wang, E.T.; Vu, A.; David, M.P.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010, 38, 7651–7664. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Xia, Z.; Loehr, J.A.; Li, W.; Rodney, G.G.; Cooper, T.A. Extensive alternative splicing transitions during postnatal skeletal muscle development are required for calcium handling functions. eLife 2017, 6, e27192. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef]
- Tapial, J.; Ha, K.C.H.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallières, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017, 27, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Nakka, K.; Ghigna, C.; Gabellini, D.; Dilworth, F.J. Diversification of the muscle proteome through alternative splicing. Skelet Muscle 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Helfman, D.M.; Ricci, W.M.; Finn, L.A. Alternative splicing of tropomyosin pre-mRNAs in vitro and in vivo. Genes Dev. 1988, 2, 1627–1638. [Google Scholar] [CrossRef]
- Mullen, M.P.; Smith, C.W.; Patton, J.G.; Nadal-Ginard, B. Alpha-tropomyosin mutually exclusive exon selection: Competition between branchpoint/polypyrimidine tracts determines default exon choice. Genes Dev. 1991, 5, 642–655. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gateva, G.; Kremneva, E.; Reindl, T.; Kotila, T.; Kogan, K.; Gressin, L.; Gunning, P.W.; Manstein, D.J.; Michelot, A.; Lappalainen, P. Tropomyosin Isoforms Specify Functionally Distinct Actin Filament Populations In Vitro. Curr. Biol. 2017, 27, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Zot, A.S.; Potter, J.D. Structural aspects of troponin-tropomyosin regulation of skeletal muscle contraction. Annu Rev. Biophys Biophys. Chem. 1987, 16, 535–559. [Google Scholar] [CrossRef]
- Giudice, J.; Loehr, J.A.; Rodney, G.G.; Cooper, T.A. Alternative Splicing of Four Trafficking Genes Regulates Myofiber Structure and Skeletal Muscle Physiology. Cell Rep. 2016, 17, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef]
- Palace, J.; Beeson, D. The congenital myasthenic syndromes. J. Neuroimmunol. 2008, 201–202, 2–5. [Google Scholar] [CrossRef]
- Rahman, M.A.; Azuma, Y.; Nasrin, F.; Takeda, J.; Nazim, M.; Bin Ahsan, K.; Masuda, A.; Engel, A.G.; Ohno, K. SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci. Rep. 2015, 5, 13208. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Engel, A.G. Congenital myasthenic syndromes:gene mutations. Neuromuscul. Disord. 2003, 13, 854–857. [Google Scholar] [CrossRef]
- Cossins, J.; Liu, W.W.; Belaya, K.; Maxwell, S.; Oldridge, M.; Lester, T.; Robb, S.; Beeson, D. The spectrum of mutations that underlie the neuromuscular junction synaptopathy in DOK7 congenital myasthenic syndrome. Hum. Mol. Genet. 2012, 21, 3765–3775. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.S.; Baumeister, S.K.; Rasic, V.M.; Krause, S.; Todorovic, S.; Kugler, K.; Müller-Felber, W.; Abicht, A.; Lochmüller, H. Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations. Neurology 2006, 67, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Krull, M.; Petrusma, M.; Makalowski, W.; Brosius, J.; Schmitz, J. Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs). Genome Res. 2007, 17, 1139–1145. [Google Scholar] [CrossRef]
- Masuda, A.; Shen, X.M.; Ito, M.; Matsuura, T.; Engel, A.G.; Ohno, K. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum. Mol. Genet. 2008, 17, 4022–4035. [Google Scholar] [CrossRef]
- Nasrin, F.; Rahman, M.A.; Masuda, A.; Ohe, K.; Takeda, J.; Ohno, K. HnRNP C, YB-1 and hnRNP L coordinately enhance skipping of human MUSK exon 10 to generate a Wnt-insensitive MuSK isoform. Sci. Rep. 2014, 4, 6841. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Grootscholten, P.M.; Bakker, E.; Blonden, L.A.; Ginjaar, H.B.; Wapenaar, M.C.; van Paassen, H.M.; van Broeckhoven, C.; Pearson, P.L.; van Ommen, G.J. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am. J. Hum. Genet. 1989, 45, 835–847. [Google Scholar]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Malhotra, S.B.; Hart, K.A.; Klamut, H.J.; Thomas, N.S.; Bodrug, S.E.; Burghes, A.H.; Bobrow, M.; Harper, P.S.; Thompson, M.W.; Ray, P.N. Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science 1988, 242, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Martone, J.; Briganti, F.; Legnini, I.; Morlando, M.; Picillo, E.; Sthandier, O.; Politano, L.; Bozzoni, I. The lack of the Celf2a splicing factor converts a Duchenne genotype into a Becker phenotype. Nat. Commun 2016, 7, 10488. [Google Scholar] [CrossRef]
- Dwianingsih, E.K.; Malueka, R.G.; Nishida, A.; Itoh, K.; Lee, T.; Yagi, M.; Iijima, K.; Takeshima, Y.; Matsuo, M. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J. Hum. Genet. 2014, 59, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Shiga, N.; Takeshima, Y.; Sakamoto, H.; Inoue, K.; Yokota, Y.; Yokoyama, M.; Matsuo, M. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J. Clin. Invest. 1997, 100, 2204–2210. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne muscular dystrophy: Meeting the therapeutic challenge. Lancet Neurol. 2016, 15, 785–787. [Google Scholar] [CrossRef]
- Deburgrave, N.; Daoud, F.; Llense, S.; Barbot, J.C.; Récan, D.; Peccate, C.; Burghes, A.H.; Béroud, C.; Garcia, L.; Kaplan, J.C.; et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum. Mutat. 2007, 28, 183–195. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Ferlini, A.; Galié, N.; Merlini, L.; Sewry, C.; Branzi, A.; Muntoni, F. A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1998, 63, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ishmukhametova, A.; Khau Van Kien, P.; Méchin, D.; Thorel, D.; Vincent, M.C.; Rivier, F.; Coubes, C.; Humbertclaude, V.; Claustres, M.; Tuffery-Giraud, S. Comprehensive oligonucleotide array-comparative genomic hybridization analysis: New insights into the molecular pathology of the DMD gene. Eur. J. Hum. Genet. 2012, 20, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Greer, K.; Mizzi, K.; Rice, E.; Kuster, L.; Barrero, R.A.; Bellgard, M.I.; Lynch, B.J.; Foley, A.R.; Rathallaigh, O.E.; Wilton, S.D.; et al. Pseudoexon activation increases phenotype severity in a Becker muscular dystrophy patient. Mol. Genet. Genomic. Med. 2015, 3, 320–326. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Dansithong, W.; Paul, S.; Comai, L.; Reddy, S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J. Biol. Chem. 2005, 280, 5773–5780. [Google Scholar] [CrossRef]
- Fardaei, M.; Larkin, K.; Brook, J.D.; Hamshere, M.G. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001, 29, 2766–2771. [Google Scholar] [CrossRef]
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.; Carrascosa, J.M.; Ermel, B.; Ullrich, A.; Häring, H.U. The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem. Biophys. Res. Commun. 1991, 177, 1013–1018. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Rau, F.; Lainé, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun 2015, 6, 7205. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Newey, S.E.; Benson, M.A.; Ponting, C.P.; Davies, K.E.; Blake, D.J. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr. Biol. 2000, 10, 1295–1298. [Google Scholar] [CrossRef]
- Nakamori, M.; Kimura, T.; Kubota, T.; Matsumura, T.; Sumi, H.; Fujimura, H.; Takahashi, M.P.; Sakoda, S. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology 2008, 70, 677–685. [Google Scholar] [CrossRef]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum. Mol. Genet. 2012, 21, 1312–1324. [Google Scholar] [CrossRef]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef]
- Sultana, N.; Dienes, B.; Benedetti, A.; Tuluc, P.; Szentesi, P.; Sztretye, M.; Rainer, J.; Hess, M.W.; Schwarzer, C.; Obermair, G.J.; et al. Restricting calcium currents is required for correct fiber type specification in skeletal muscle. Development 2016, 143, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Piacentini, R.; Masciullo, M.; Bianchi, M.L.; Modoni, A.; Podda, M.V.; Ricci, E.; Silvestri, G.; Grassi, C. Alternative splicing alterations of Ca2+ handling genes are associated with Ca2+ signal dysregulation in myotonic dystrophy type 1 (DM1) and type 2 (DM2) myotubes. Neuropathol. Appl. Neurobiol. 2014, 40, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Overby, S.J.; Cerro-Herreros, E.; Llamusi, B.; Artero, R. RNA-mediated therapies in myotonic dystrophy. Drug Discov. Today 2018, 23, 2013–2022. [Google Scholar] [CrossRef]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef]
- Raaijmakers, R.H.L.; Ripken, L.; Ausems, C.R.M.; Wansink, D.G. CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. Int. J. Mol. Sci. 2019, 20, 3689. [Google Scholar] [CrossRef] [PubMed]
- Cerro-Herreros, E.; Sabater-Arcis, M.; Fernandez-Costa, J.M.; Moreno, N.; Perez-Alonso, M.; Llamusi, B.; Artero, R. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9, 2482. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Network, E.S.G.a.T.F.D.I. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Ottesen, E.W. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Tei, S.; Ishii, H.T.; Mitsuhashi, H.; Ishiura, S. Antisense oligonucleotide-mediated exon skipping of CHRNA1 pre-mRNA as potential therapy for Congenital Myasthenic Syndromes. Biochem. Biophys. Res. Commun. 2015, 461, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Exon-skipping therapy for Duchenne muscular dystrophy. Lancet 2011, 378, 546–547. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Hodgkinson, L.; Sorbera, L.; Graul, A.I. Duchenne muscular dystrophy drugs face tough path to approval. Drugs Today 2016, 52, 199–202. [Google Scholar] [CrossRef]
- Sarepta Therapeutics Announces Positive Expression Results from the Casimersen (SRP-4045) Arm of the ESSENCE Study. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeuticsannounces-positive-expression-results (accessed on 28 August 2020).
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Vulin, A.; Fougerousse, F.; Leturcq, F.; Kaplan, J.C.; Garcia, L.; Danos, O. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 2004, 306, 1796–1799. [Google Scholar] [CrossRef]
- Hammond, S.M.; Wood, M.J. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef] [PubMed]
- AAV9 U7snRNA Gene Therapy to Treat Boys with DMD Exon 2 Duplications. Available online: https://clinicaltrials.gov/ct2/show/NCT04240314 (accessed on 10 October 2021).
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Kim, E.; Antoury, L.; Li, J.; González-Pérez, P.; Rutkove, S.B.; Wheeler, T.M. Antisense oligonucleotide and adjuvant exercise therapy reverse fatigue in old mice with myotonic dystrophy. Mol. Ther. Nucleic Acids 2021, 23, 393–405. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef]
- Gudde, A.E.E.G.; van Heeringen, S.J.; de Oude, A.I.; van Kessel, I.D.G.; Estabrook, J.; Wang, E.T.; Wieringa, B.; Wansink, D.G. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. 2017, 14, 1374–1388. [Google Scholar] [CrossRef]
- Lo Scrudato, M.; Poulard, K.; Sourd, C.; Tomé, S.; Klein, A.F.; Corre, G.; Huguet, A.; Furling, D.; Gourdon, G.; Buj-Bello, A. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Mol. Ther. 2019, 27, 1372–1388. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Nelles, D.A.; Roth, D.M.; Krach, F.; Nutter, C.A.; Tadokoro, T.; Thomas, J.D.; Sznajder, Ł.; Blue, S.M.; Gutierrez, H.L.; et al. The sustained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat. Biomed. Eng. 2021, 5, 157–168. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753. [Google Scholar] [CrossRef]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; González-Martínez, I.; Moreno-Cervera, N.; Overby, S.; Pérez-Alonso, M.; Llamusí, B.; Artero, R. Therapeutic Potential of AntagomiR-23b for Treating Myotonic Dystrophy. Mol. Ther. Nucleic Acids 2020, 21, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Gilabert, M.; López-Castel, A.; Artero, R. Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discov. Today 2021, 26, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Marwick, C. First “antisense” drug will treat CMV retinitis. JAMA 1998, 280, 871. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Arechavala-Gomeza, V. Why dystrophin quantification is key in the eteplirsen saga. Nat. Rev. Neurol. 2018, 14, 454–456. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid. Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Wurster, C.D.; Winter, B.; Wollinsky, K.; Ludolph, A.C.; Uzelac, Z.; Witzel, S.; Schocke, M.; Schneider, R.; Kocak, T. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J. Neurol. 2019, 266, 183–194. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Epidemiology | Age at Onset | Symptoms and Life Expectancy |
|---|---|---|---|
| Congenital myasthenic syndrome (CMS) | Estimated 2.5 to 12 per 1,000,000 individuals | Intrauterine and congenital onset to rarely adolescence | Severe generalized muscle weakness causes bulbar, ocular, limbs and respiratory muscles failure. Heart block leads early death. |
| Duchenne muscular dystrophy (DMD) | <10 per 100,000 in male <1 per million in female | 2 to 6 years | Muscle weakness and wasting affect pelvis, upper arms, and upper legs. Most patients need wheelchair and assisted ventilation before the age of 20. Rarely optimal treatments for cardiopulmonary dysfunction extend life expectancy to late thirties. |
| Becker muscular dystrophy (BMD) | <8 per 100,000 in male <1 per million in female | Adolescence to early adulthood | Symptoms are almost identical to Duchenne, but less severe and progress slowly. Life quality and expectation is similar to healthy individuals. |
| Myotonic dystrophy type 1 (DM1) | 0.5 to 18 per 100,000 individuals | Birth to adulthood | Progressive weakness, atrophy of distal muscles and myotonia. Affected neonate shows hypotonia (floppy infant syndrome) and need intubation immediately after birth. |
| Disease | Gene | Alteration Observed | Tissue Expression | Therapeutic Approach |
|---|---|---|---|---|
| Congenital myasthenic syndrome (CMS) | CHRNA1 | Inclusion of exon P3A | NMJ | ASOs covering the 5′ splice site promotes the rescue of P3A skipping [102] |
| CHRNE | Exclusion of exon 6 | NMJ | ||
| COLQ | Exclusion of exon 16 | NMJ | ||
| RAPSN | Exclusion of exon 1 | NMJ | ||
| Duchenne muscular dystrophy (DMD) | DMD | Deletion of exon 50 (leads to an out-of-frame mRNA generating a premature stop codon) | Skeletal muscle | ASOs promoting the skipping of exon 51 are in clinical trials [104] |
| Deletion of a single exon (44) or multiple exons (44–45) (induces out-of-frame mutations) | Casimersen increases exon 45-skipping | |||
| Deletion of multiple exons (45–55) (leads to in-frame mutation) | Golodirsen increases the skipping of exon 53 [109] | |||
| Myotonic dystrophy type 1 (DM1) | INSR | Exclusion of exon 11 | Skeletal muscle | ASOs directed against the CUGexp RNA promote release sequestered MBNL1 protein, improving its splicing regulatory activity [116] Adeno-associated virus vectors encoding nuclease-dead Cas9 and a single-guide RNA targeting CUG repeats enhance redistribution of MBNL1 and the rescue of splicing defects [125] AntagomiR 23b upregulates MBNL proteins rescuing the molecular, cellular, and functional defects of DM1 muscle [128] |
| CLCN1 | Inclusion of intron 2 and inclusion of exon 7a | Skeletal muscle | ||
| DMD | Exclusion of exon 78 | Skeletal muscle | ||
| BIN1 | Exclusion of exon 11 | Skeletal muscle | ||
| SERCA1 | Exclusion of exon 22 | Skeletal muscle | ||
| RYR1 | Exclusion of exon 70 | Skeletal muscle | ||
| CACNA1S | Exclusion of exon 29 | Skeletal muscle | ||
| α-DB1 | Inclusion of exon 11A and exon 12 | Skeletal muscle |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdile, V.; Guizzo, G.; Ferrante, G.; Paronetto, M.P. RNA Targeting in Inherited Neuromuscular Disorders: Novel Therapeutic Strategies to Counteract Mis-Splicing. Cells 2021, 10, 2850. https://doi.org/10.3390/cells10112850
Verdile V, Guizzo G, Ferrante G, Paronetto MP. RNA Targeting in Inherited Neuromuscular Disorders: Novel Therapeutic Strategies to Counteract Mis-Splicing. Cells. 2021; 10(11):2850. https://doi.org/10.3390/cells10112850
Chicago/Turabian StyleVerdile, Veronica, Gloria Guizzo, Gabriele Ferrante, and Maria Paola Paronetto. 2021. "RNA Targeting in Inherited Neuromuscular Disorders: Novel Therapeutic Strategies to Counteract Mis-Splicing" Cells 10, no. 11: 2850. https://doi.org/10.3390/cells10112850
APA StyleVerdile, V., Guizzo, G., Ferrante, G., & Paronetto, M. P. (2021). RNA Targeting in Inherited Neuromuscular Disorders: Novel Therapeutic Strategies to Counteract Mis-Splicing. Cells, 10(11), 2850. https://doi.org/10.3390/cells10112850