
Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
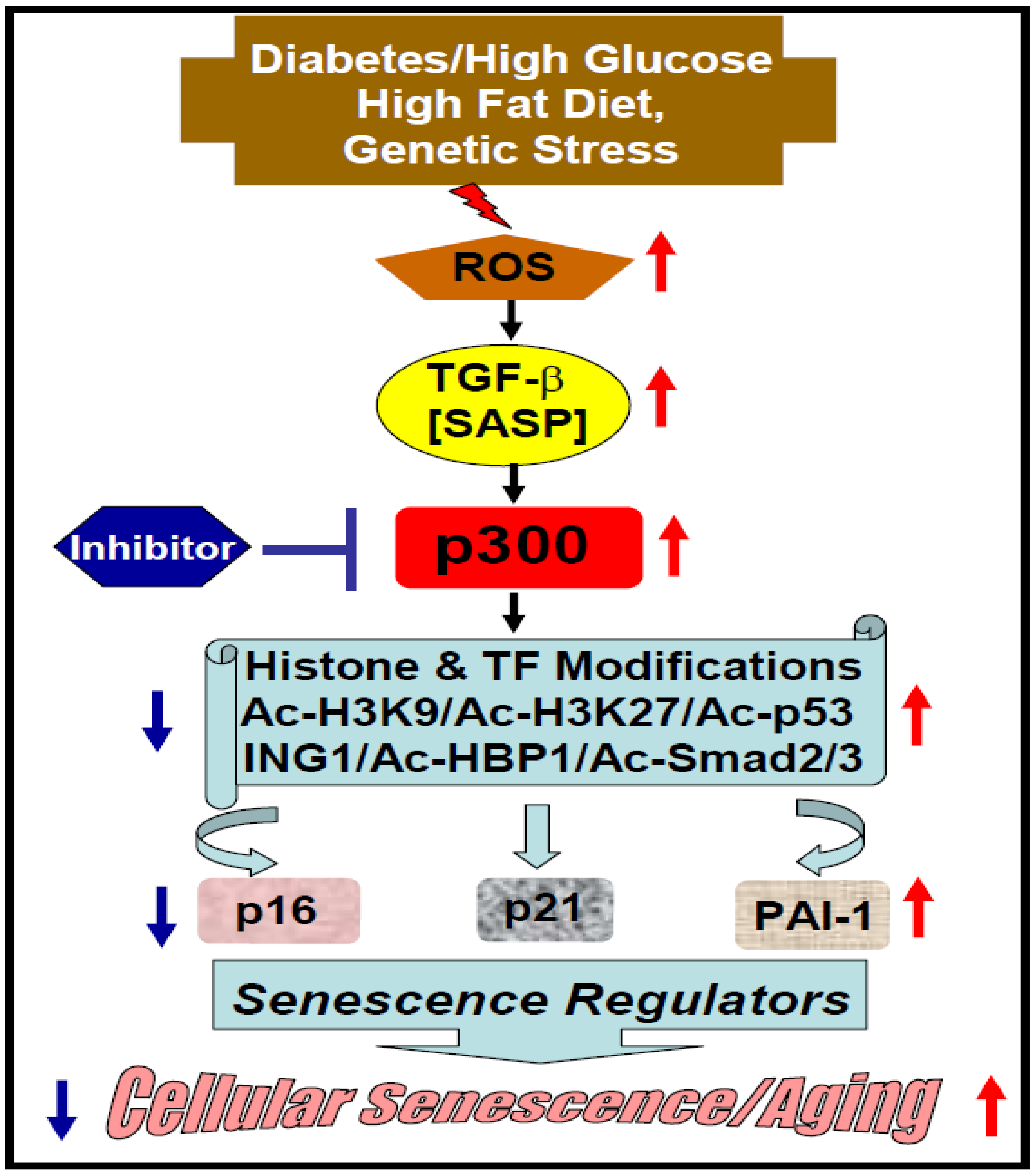
2. Acetyltransferase p300 in Cellular Senescence and Aging
2.1. Cardiac Senescence
2.2. Diabetes-Induced Vascular Senescence and Dysfunction
2.3. Vascular Senescence in Atherosclerosis
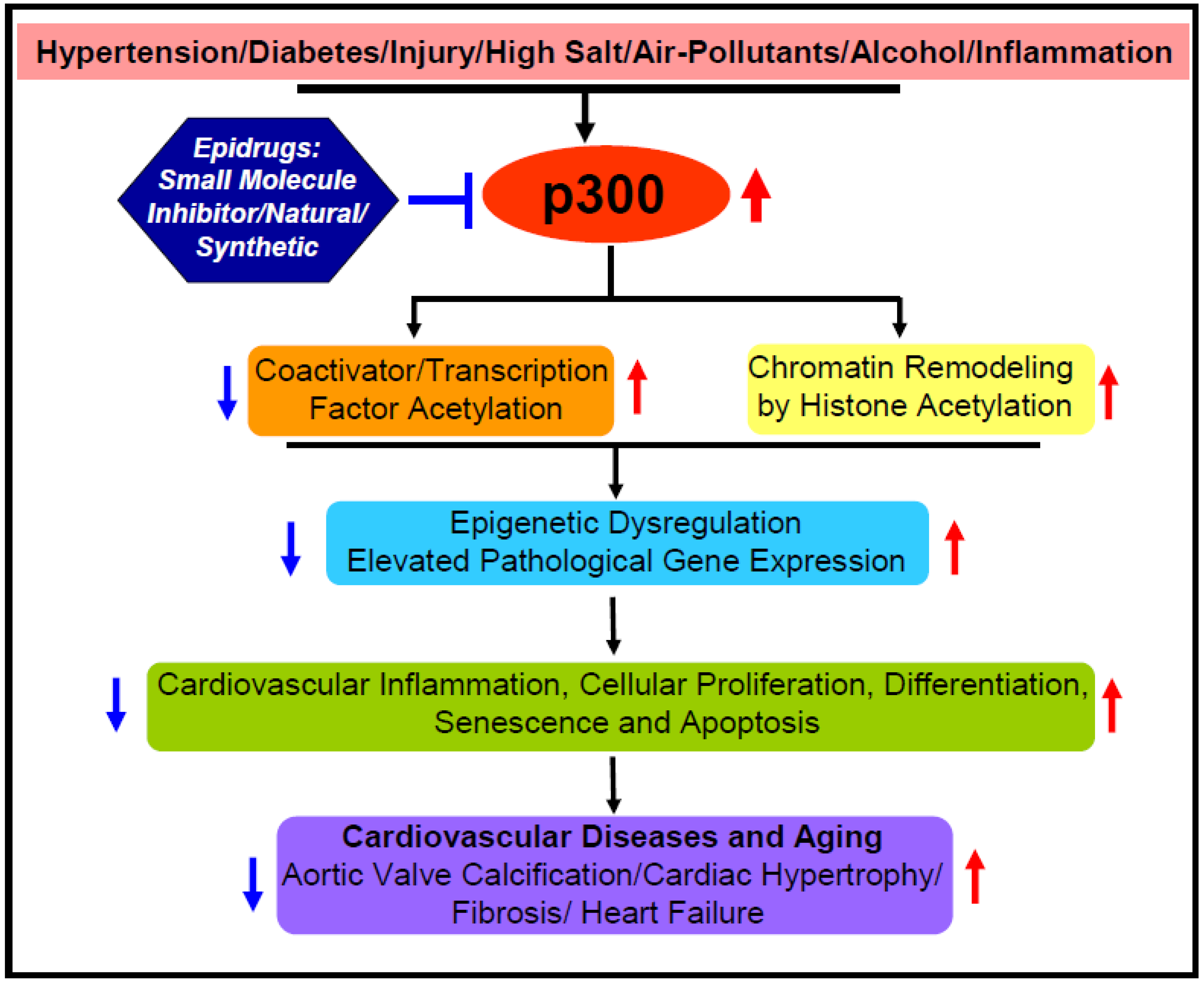
2.4. Possible Underlying Mechanisms and Epidrug Development
3. Acetyltransferase p300 Boosts Cardiac Hypertrophy, Myocardial Fibrosis, and Heart Failure
3.1. α-Adrenergic Receptor Agonist Phenylephrine-Induced Cardiac Hypertrophy and Fibrosis: Epigenetic Regulation by Acetyltransferase p300
3.2. Peptide Hormone Angiotensin II-Induced Cardiac Hypertrophy and Fibrosis: Pivotal Role of Epigenetic Regulator Acetyltransferase p300
3.3. Acetyltransferase p300 Contributes to Transverse Aortic Constriction-Induced Cardiac Hypertrophy and Fibrosis
3.4. Genetic Models of Cardiac Hypertrophy, Fibrosis and Heart Failure: Contribution of Acetyltransferase p300
3.5. Involvement of p300 in Environmental Air Pollution and Alcohol-Induced Cardiovascular Disease
3.6. Myocardial Infarction Associated Cardiovascular Pathologies Are Epigenetically Modulated by Acetyltransferase p300
4. Epigenetic Regulation of Aortic Valve Calcification by Acetyltransferase p300
5. Perspective and Future Direction
Funding
Acknowledgments
Conflicts of Interest
References
- Safi-Stibler, S.; Gabory, A. Epigenetics and the developmental origins of health and disease: Parental environment signalling to the epigenome, critical time windows and sculpting the adult phenotype. Semin. Cell Dev. Biol. 2020, 97, 172–180. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic modifications in cardiovascular aging and diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Eliezer Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell. 2013, 49, 825–837. [Google Scholar] [CrossRef]
- Eckner, R.; Arany, Z.; Ewen, M.; Sellers, W.; Livingston, D.M. The adenovirus E1A-associated 300-kD protein exhibits properties of a transcriptional coactivator and belongs to an evolutionarily conserved family. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 85–95. [Google Scholar] [CrossRef]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Yuan, W.; Mori, Y.; Varga, J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene 2000, 19, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Bhattacharyya, S.; Varga, J. The tumor suppressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. J. Biol. Chem. 2004, 279, 47455–47463. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, S.; Wei, J.; Kim, S.; Barak, Y.; Mori, Y.; Varga, J. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Verma, S.K.; Kim, D.; Ramirez, V.; Lux, E.; Li, C.; Sahoo, S.; Wilsbacher, L.D.; Vaughan, D.E.; Quaggin, S.E.; et al. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 2017, 12, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chen, C.C. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell Biol. 2005, 25, 6592–6602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Meng, J.; Liu, X.; Zhang, X.; Peng, X.; Cheng, Z.; Zhang, F. ING5 differentially regulates protein lysine acetylation and promotes p300 autoacetylation. Oncotarget 2017, 9, 1617–1629. [Google Scholar] [CrossRef]
- Xu, W.; Chen, H.; Du, K.; Asahara, H.; Tini, M.; Emerson, B.M.; Montminy, M.; Evans, R.M. A transcriptional switch mediated by cofactor methylation. Science 2001, 294, 2507–2511. [Google Scholar] [CrossRef]
- Giles, R.H.; Peters, D.J.M.; Breuning, M.H. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998, 14, 178–183. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arter. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone acetyltransferase p300 induces de novo super-enhancers to drive cellular senescence. Mol. Cell. 2019, 73, 684–698. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell–cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2020, 598, 2923–2939. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Tam, B.T.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Rudd, J.A.; Siu, P.M. Resveratrol protects against doxorubicin-induced cardiotoxicity in aged hearts through the SIRT1-USP7 axis. J. Physiol. 2015, 593, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Adachi, S.; Ito, H.; Hirao, K.; Isobe, M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell 2008, 7, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, 380–391. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rai, R.; Park, K.E.; Eren, M.; Miyata, T.; Wilsbacher, L.D.; Vaughan, D.E. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget 2016, 7, 72443–72457. [Google Scholar] [CrossRef]
- Di Tomo, P.; Alessio, N.; Falone, S.; Pietrangelo, L.; Lanuti, P.; Cordone, V.; Santini, S.J.; Di Pietrantonio, N.; Marchisio, M.; Protasi, F.; et al. Endothelial cells from umbilical cord of women affected by gestational diabetes: A suitable in vitro model to study mechanisms of early vascular senescence in diabetes. FASEB J. 2021, 35, e21662. [Google Scholar] [CrossRef] [PubMed]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Guo, Q.; Gao, H.; Xu, R.; Teng, S.; Wu, Y. Metformin and Resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/ p53/p21 Pathway. PLoS ONE 2015, 10, e0143814. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Vlad, M.L.; Manea, S.A.; Lazar, A.G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Histone acetyltransferase-dependent pathways mediate upregulation of NADPH oxidase 5 in human macrophages under inflammatory conditions: A potential mechanism of reactive oxygen species overproduction in atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3201062. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Q.; Cao, X.; Zhao, G.; Xue, L.; Tong, T. The tumor suppressor p33ING1b upregulates p16INK4a expression and induces cellular senescence. FEBS Lett. 2011, 585, 3106–3112. [Google Scholar] [CrossRef]
- Dantas, A.; Al Shueili, B.; Yang, Y.; Nabbi, A.; Fink, D.; Riabowol, K. Biological functions of the ING proteins. Cancers 2019, 11, 1817. [Google Scholar] [CrossRef]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef]
- Yuan, H.; Reddy, M.A.; Sun, G.; Lanting, L.; Wang, M.; Kato, M.; Natarajan, R. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol. Ren. Physiol. 2013, 304, 601–613. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021, 11, 1118–1137. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.C.; van Oort, R.J.; Wehrens, X.H. Transverse aortic constriction in mice. J. Vis. Exp. 2010, 38, 1729. [Google Scholar]
- Tannu, S.; Allocco, J.; Yarde, M.; Wong, P.; Ma, X. Experimental model of congestive heart failure induced by transverse aortic constriction in BALB/c mice. J. Pharmacol. Toxicol. Methods 2020, 106, 106935. [Google Scholar] [CrossRef] [PubMed]
- Yanazume, T.; Hasegawa, K.; Morimoto, T.; Kawamura, T.; Wada, H.; Matsumori, A.; Kawase, Y.; Hirai, M.; Kita, T. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell Biol. 2003, 23, 3593–3606. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, Y.; Morimoto, T.; Takaya, T.; Kaichi, S.; Wada, H.; Kawamura, T.; Fujita, M.; Shimatsu, A.; Kita, T.; Hasegawa, K. Cyclin-dependent kinase-9 is a component of the p300/GATA4 complex required for phenylephrine-induced hypertrophy in cardiomyocytes. J. Biol. Chem. 2010, 285, 9556–9568. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Katanasaka, Y.; Sunagawa, Y.; Miyazaki, Y.; Funamoto, M.; Wada, H.; Hasegawa, K.; Morimoto, T. Tyrosine phosphorylation of RACK1 triggers cardiomyocyte hypertrophy by regulating the interaction between p300 and GATA4. Biochim. Biophys. Acta 2016, 1862, 1544–1557. [Google Scholar] [CrossRef]
- Funamoto, M.; Sunagawa, Y.; Katanasaka, Y.; Shimizu, K.; Miyazaki, Y.; Sari, N.; Shimizu, S.; Mori, K.; Wada, H.; Hasegawa, K.; et al. Histone acetylation domains are differentially induced during development of heart failure in dahl salt-sensitive rats. Int. J. Mol. Sci. 2021, 22, 1771. [Google Scholar] [CrossRef]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef]
- Shen, P.; Feng, X.; Zhang, X.; Huang, X.; Liu, S.; Lu, X.; Li, J.; You, J.; Lu, J.; Li, Z.; et al. SIRT6 suppresses phenylephrine-induced cardiomyocyte hypertrophy though inhibiting p300. J. Pharmacol. Sci. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Peng, C.; Luo, X.; Li, S.; Sun, H. Phenylephrine-induced cardiac hypertrophy is attenuated by a histone acetylase inhibitor anacardic acid in mice. Mol. Biosyst. 2017, 13, 714–724. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef]
- Xing, W.; Zhang, T.C.; Cao, D.; Wang, Z.; Antos, C.L.; Li, S.; Wang, Y.; Olson, E.N.; Wang, D.Z. Myocardin induces cardiomyocyte hypertrophy. Circ. Res. 2006, 98, 1089–1097. [Google Scholar] [CrossRef]
- Cao, D.; Wang, C.; Tang, R.; Chen, H.; Zhang, Z.; Tatsuguchi, M.; Wang, D.Z. Acetylation of myocardin is required for the activation of cardiac and smooth muscle genes. J. Biol. Chem. 2012, 287, 38495–38504. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.S.; Luo, Y.X.; Zhang, R.; Zhang, X.D.; Chen, H.Z.; Zhang, Y.; Chen, K.; Zhang, S.M.; Fan, G.C.; Liu, P.P.; et al. Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension 2014, 63, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Peng, B.; Luo, X.; Sun, H.; Peng, C. Anacardic acid attenuates pressure-overload cardiac hypertrophy through inhibiting histone acetylases. J. Cell Mol. Med. 2019, 23, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Sunagawa, Y.; Funamoto, M.; Wakabayashi, H.; Genpei, M.; Miyazaki, Y.; Katanasaka, Y.; Sari, N.; Shimizu, S.; Katayama, A.; et al. The Synthetic Curcumin Analogue GO-Y030 Effectively suppresses the development of pressure overload-induced heart failure in mice. Sci. Rep. 2020, 10, 7172. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jéhanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zähringer, U.; et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef]
- Lin, H.B.; Naito, K.; Oh, Y.; Farber, G.; Kanaan, G.; Valaperti, A.; Dawood, F.; Zhang, L.; Li, G.H.; Smyth, D.; et al. Innate immune Nod1/RIP2 signaling is essential for cardiac hypertrophy but requires mitochondrial antiviral signaling protein for signal transductions and energy balance. Circulation 2020, 142, 2240–2258. [Google Scholar] [CrossRef] [PubMed]
- Doris, P.A. Genetics of hypertension: An assessment of progress in the spontaneously hypertensive rat. Physiol. Genom. 2017, 49, 601–617. [Google Scholar] [CrossRef]
- Jin, L.; Piao, Z.H.; Liu, C.P.; Sun, S.; Liu, B.; Kim, G.R.; Choi, S.Y.; Ryu, Y.; Kee, H.J.; Jeong, M.H. Gallic acid attenuates calcium calmodulin-dependent kinase II-induced apoptosis in spontaneously hypertensive rats. J. Cell Mol. Med. 2018, 22, 1517–1526. [Google Scholar] [CrossRef]
- Kim, M.J.; Seong, A.R.; Yoo, J.Y.; Jin, C.H.; Lee, Y.H.; Kim, Y.J.; Lee, J.; Jun, W.J.; Yoon, H.G. Gallic acid, a histone acetyltransferase inhibitor, suppresses β-amyloid neurotoxicity by inhibiting microglial-mediated neuroinflammation. Mol. Nutr. Food Res. 2011, 55, 1798–1808. [Google Scholar] [CrossRef]
- Lee, W.; Lee, S.Y.; Son, Y.J.; Yun, J.M. Gallic acid decreases inflammatory cytokine secretion through histone acetyltransferase/histone deacetylase regulation in high glucose-induced human monocytes. J. Med. Food 2015, 18, 793–801. [Google Scholar] [CrossRef]
- Lu, Z.; Cui, Y.; Wei, X.; Gao, P.; Zhang, H.; Wei, X.; Li, Q.; Sun, F.; Yan, Z.; Zheng, H.; et al. Deficiency of PKD2L1 (TRPP3) exacerbates pathological cardiac hypertrophy by augmenting NCX1-mediated mitochondrial calcium overload. Cell Rep. 2018, 24, 1639–1652. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.W.; Tse, M.Y.; O’Tierney-Ginn, P.F.; Wong, P.G.; Ventura, N.M.; Janzen-Pang, J.J.; Matangi, M.F.; Johri, A.M.; Croy, B.A.; Adams, M.A.; et al. Gestational hypertension in atrial natriuretic peptide knockout mice and the developmental origins of salt-sensitivity and cardiac hypertrophy. Regul. Pept. 2013, 186, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.M.; Lu, Y.; Jeyaraj, D.; Kawanami, D.; Cui, Y.; Eapen, S.J.; Hao, C.; Li, Y.; Doughman, Y.Q.; Watanabe, M.; et al. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci. Transl. Med. 2010, 2, 26ra26. [Google Scholar] [CrossRef]
- Su, H.; Zeng, H.; He, X.; Zhu, S.H.; Chen, J.X. Histone acetyltransferase p300 inhibitor improves coronary flow reserve in sirt3 (Sirtuin 3) knockout mice. J. Am. Heart Assoc. 2020, 9, e017176. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Ghosh, S. Environmental exposures, epigenetics and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 323–329. [Google Scholar] [CrossRef]
- Micheu, M.M.; Birsan, M.V.; Szép, R.; Keresztesi, A.; Nita, I.A. From air pollution to cardiovascular diseases: The emerging role of epigenetics. Mol. Biol. Rep. 2020, 47, 5559–5567. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dasgupta, S.; Mishra, P.K.; Chaudhury, K. Air pollution-induced epigenetic changes: Disease development and a possible link with hypersensitivity pneumonitis. Environ. Sci. Pollut. Res. 2021, 28, 55981–56002. [Google Scholar] [CrossRef]
- Wu, X.; Pan, B.; Liu, L.; Zhao, W.; Zhu, J.; Huang, X.; Tian, J. In utero exposure to PM2.5 during gestation caused adult cardiac hypertrophy through histone acetylation modification. J. Cell. Biochem. 2019, 120, 4375–4384. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, Y.; Shi, J.; Zhao, C.; Xie, P.; Huang, W.; Yong, T.; Cai, Z. Effects of PM2.5 exposure in utero on heart injury, histone acetylation and GATA4 expression in offspring mice. Chemosphere 2020, 256, 127133. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, J.; Sun, H.-C.; Huang, X.-P.; Zhao, W.-A.; Zheng, M.; Liu, L.-J.; Tian, J. Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLoS ONE 2014, 9, e104135. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhang, W.; Zhao, W.; Zhu, J.; Huang, X.; Tian, J. Alcohol-induced histone H3K9 hyperacetylation and cardiac hypertrophy are reversed by a histone acetylases inhibitor anacardic acid in developing murine hearts. Biochimie 2015, 113, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kawamura, T.; Morimoto, T.; Ono, K.; Wada, H.; Kawase, Y.; Matsumori, A.; Nishio, R.; Kita, T.; Hasegawa, K. Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation 2006, 113, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Sunagawa, Y.; Fujita, M.; Hasegawa, K. Novel heart failure therapy targeting transcriptional pathway in cardiomyocytes by a natural compound, curcumin. Circ. J. 2010, 74, 1059–1066. [Google Scholar] [CrossRef]
- Sunagawa, Y.; Wada, H.; Suzuki, H.; Sasaki, H.; Imaizumi, A.; Fukuda, H.; Hashimoto, T.; Katanasaka, Y.; Shimatsu, A.; Kimura, T.; et al. A novel drug delivery system of oral curcumin markedly improves efficacy of treatment for heart failure after myocardial infarction in rats. Biol. Pharm. Bull. 2012, 35, 139–144. [Google Scholar] [CrossRef][Green Version]
- Zhang, C.L.; Chen, Z.J.; Feng, H.; Zhao, Q.; Cao, Y.P.; Li, L.; Wang, J.Y.; Zhang, Y.; Wu, L.L. C1q/tumor necrosis factor-related protein-3 enhances the contractility of cardiomyocyte by increasing calcium sensitivity. Cell Calcium. 2017, 66, 90–97. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, L.; Feng, P.; Tan, Y.; Zhang, B.; Gao, E.; Wang, X.; Fan, C.; Wang, X.; Yi, W.; et al. C1q/tumor necrosis factor-related protein-3-engineered mesenchymal stromal cells attenuate cardiac impairment in mice with myocardial infarction. Cell Death Dis. 2019, 10, 530. [Google Scholar] [CrossRef]
- Wu, D.; Lei, H.; Wang, J.Y.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Li, L.; Wu, L.L. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J. Mol. Med. 2015, 93, 1311–1325. [Google Scholar] [CrossRef]
- Gu, J.; Lu, Y.; Deng, M.; Qiu, M.; Tian, Y.; Ji, Y.; Zong, P.; Shao, Y.; Zheng, R.; Zhou, B.; et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Messika-Zeitoun, D.; Bielak, L.F.; Peyser, P.A.; Sheedy, P.F.; Turner, S.T.; Nkomo, V.T.; Breen, J.F.; Maalouf, J.; Scott, C.; Tajik, A.J.; et al. Aortic valve calcification: Determinants and progression in the population. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Li, S.J.; Kao, Y.H.; Chung, C.C.; Chen, W.Y.; Cheng, W.L.; Chen, Y.J. Activated p300 acetyltransferase activity modulates aortic valvular calcification with osteogenic transdifferentiation and downregulation of Klotho. Int. J. Cardiol. 2017, 232, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Fauquier, L.; Azzag, K.; Parra, M.A.M.; Quillien, A.; Boulet, M.; Diouf, S.; Carnac, G.; Waltzer, L.; Gronemeyer, H.; Vandel, L. CBP and p300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci. Rep. 2018, 8, 12629. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, A.K. Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease. Cells 2021, 10, 2839. https://doi.org/10.3390/cells10112839
Ghosh AK. Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease. Cells. 2021; 10(11):2839. https://doi.org/10.3390/cells10112839
Chicago/Turabian StyleGhosh, Asish K. 2021. "Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease" Cells 10, no. 11: 2839. https://doi.org/10.3390/cells10112839
APA StyleGhosh, A. K. (2021). Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease. Cells, 10(11), 2839. https://doi.org/10.3390/cells10112839