
IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1

 ,
,  and
and
Abstract
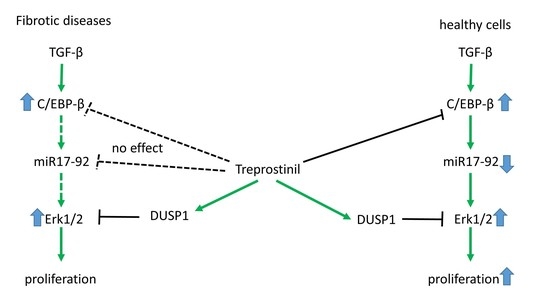
:
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Cell Generation
2.3. Western Blotting
2.4. Transfection
2.5. Luciferase Reporters
2.6. Proliferation
2.7. Statistics
3. Results
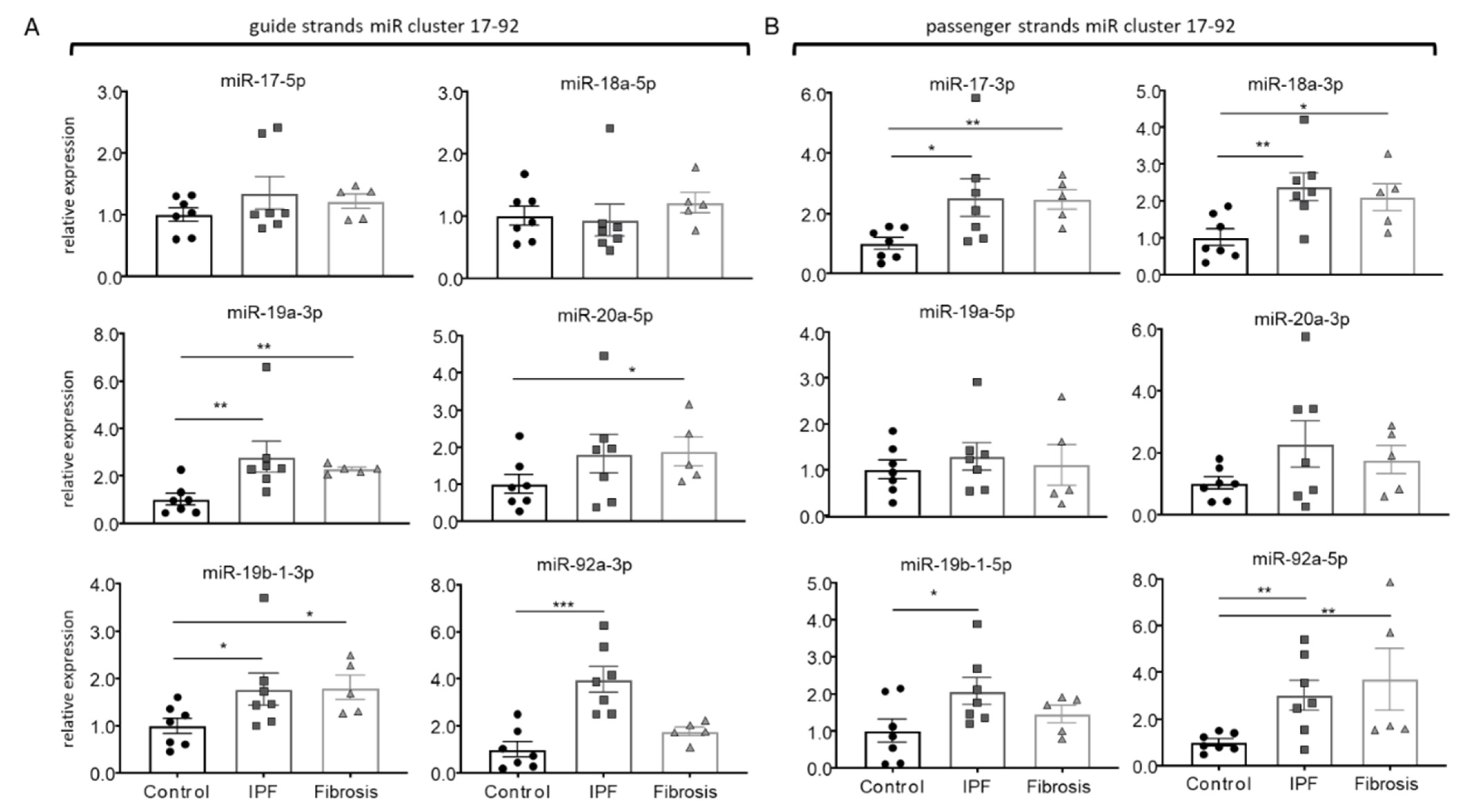
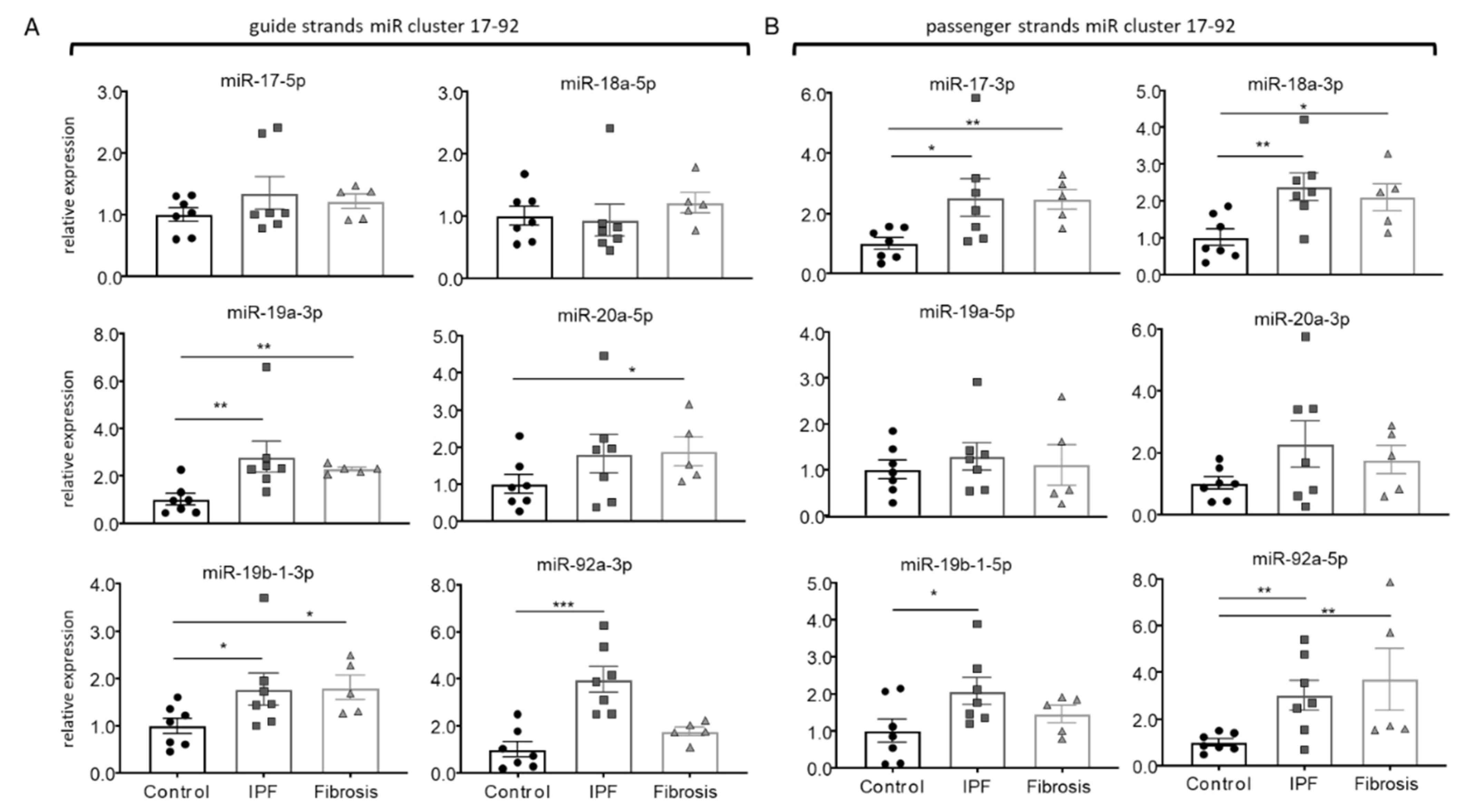
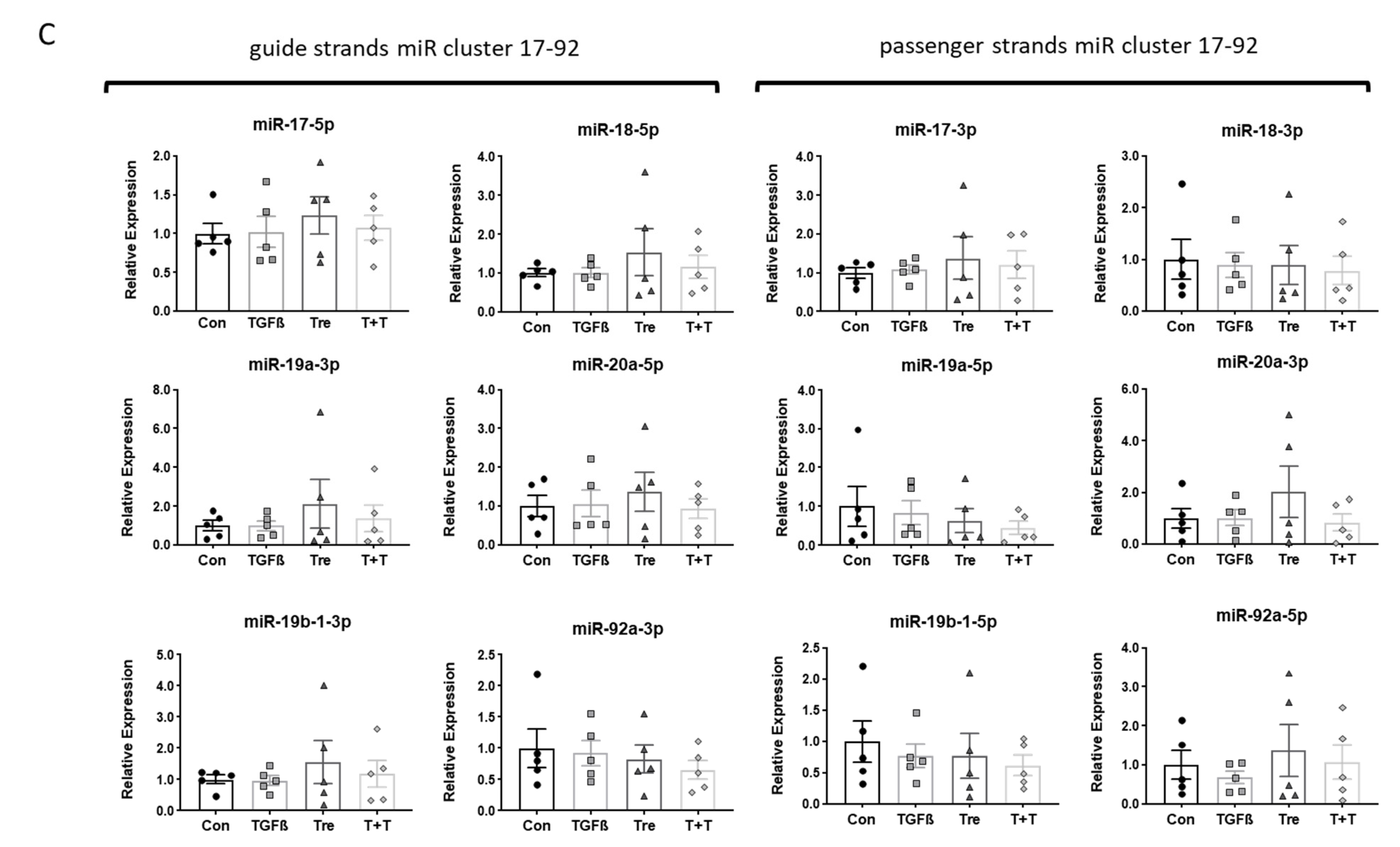
3.1. Disease-Specific Expression and Response of miR17-92Members in IPF-Fibroblasts
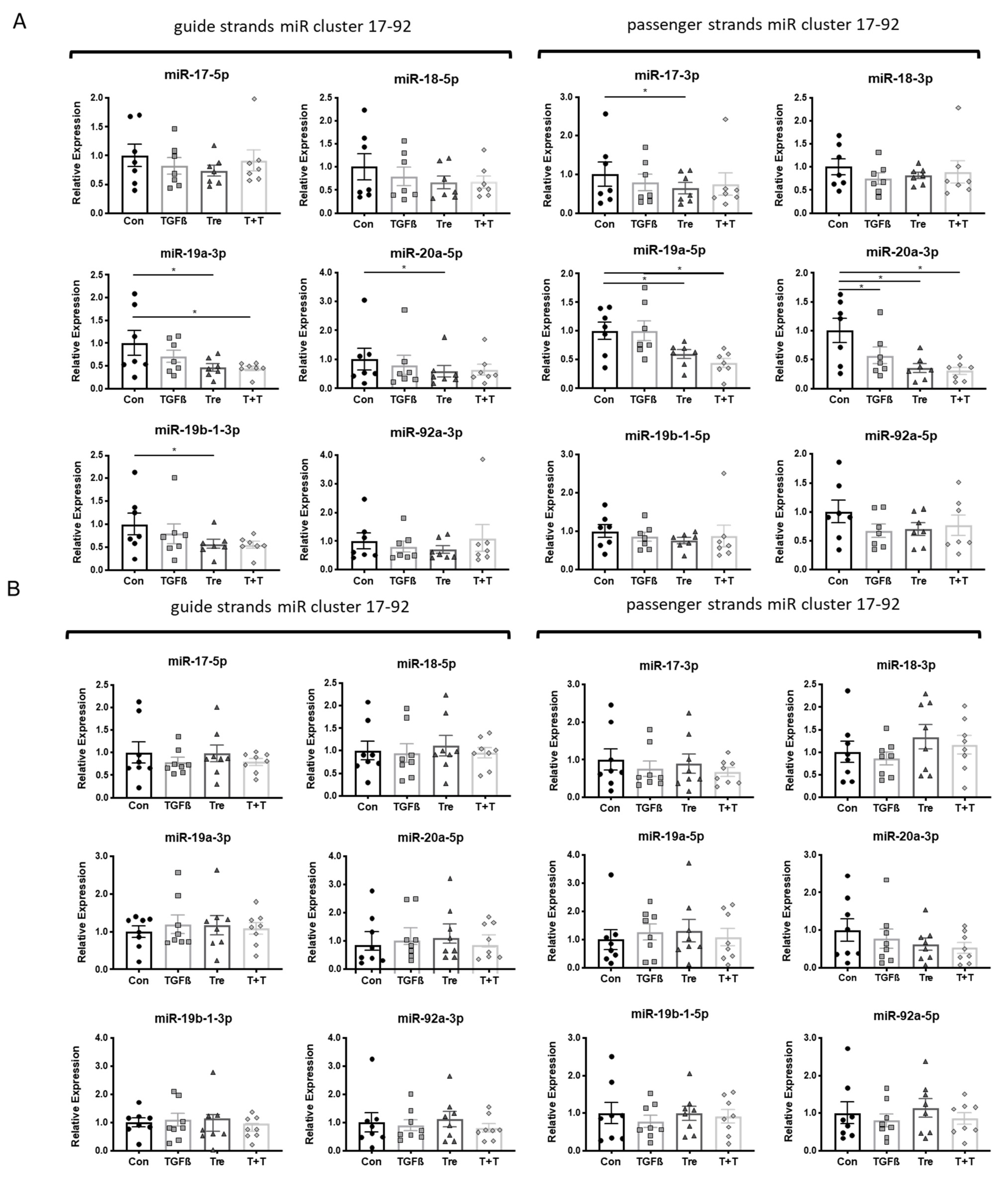
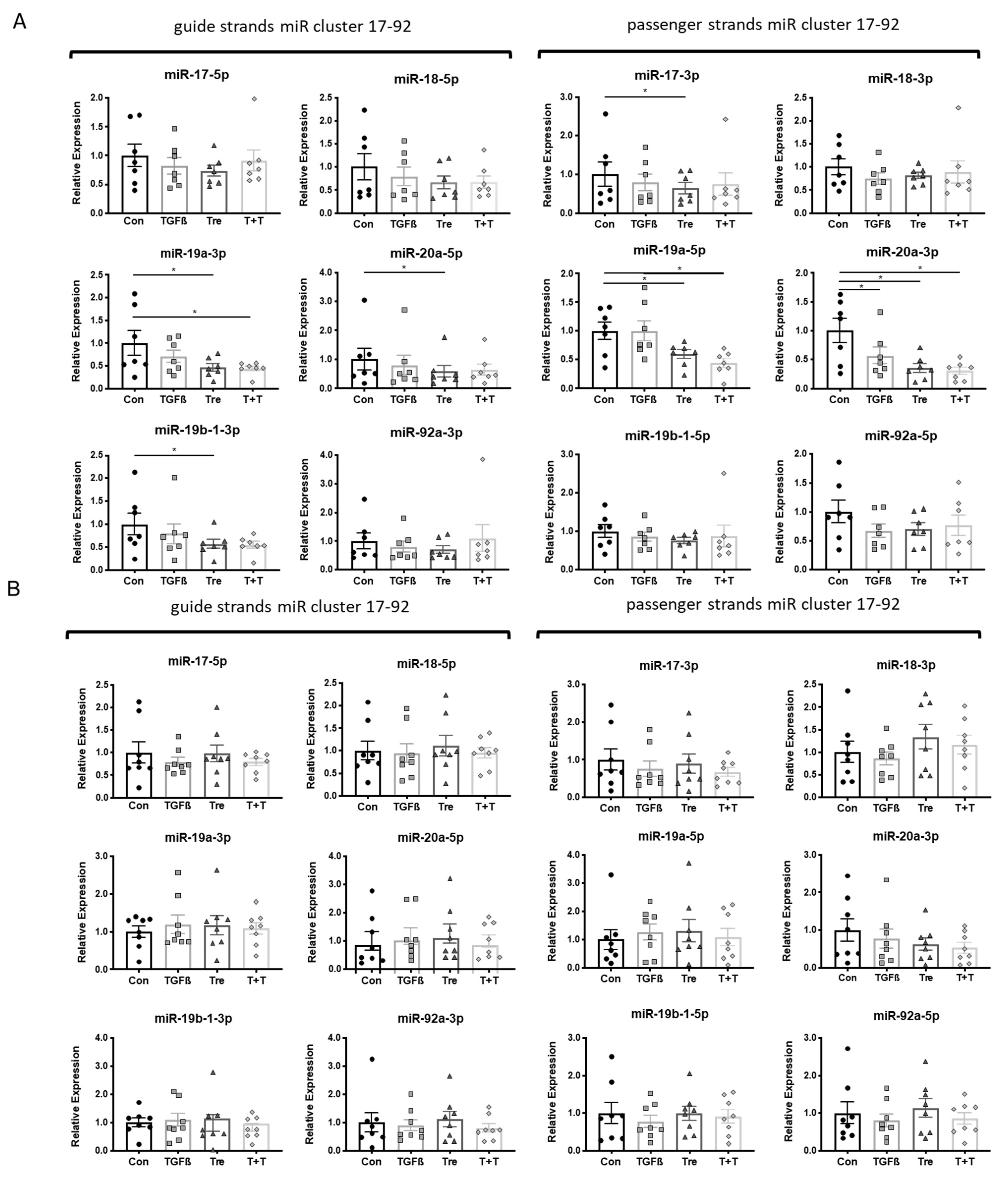
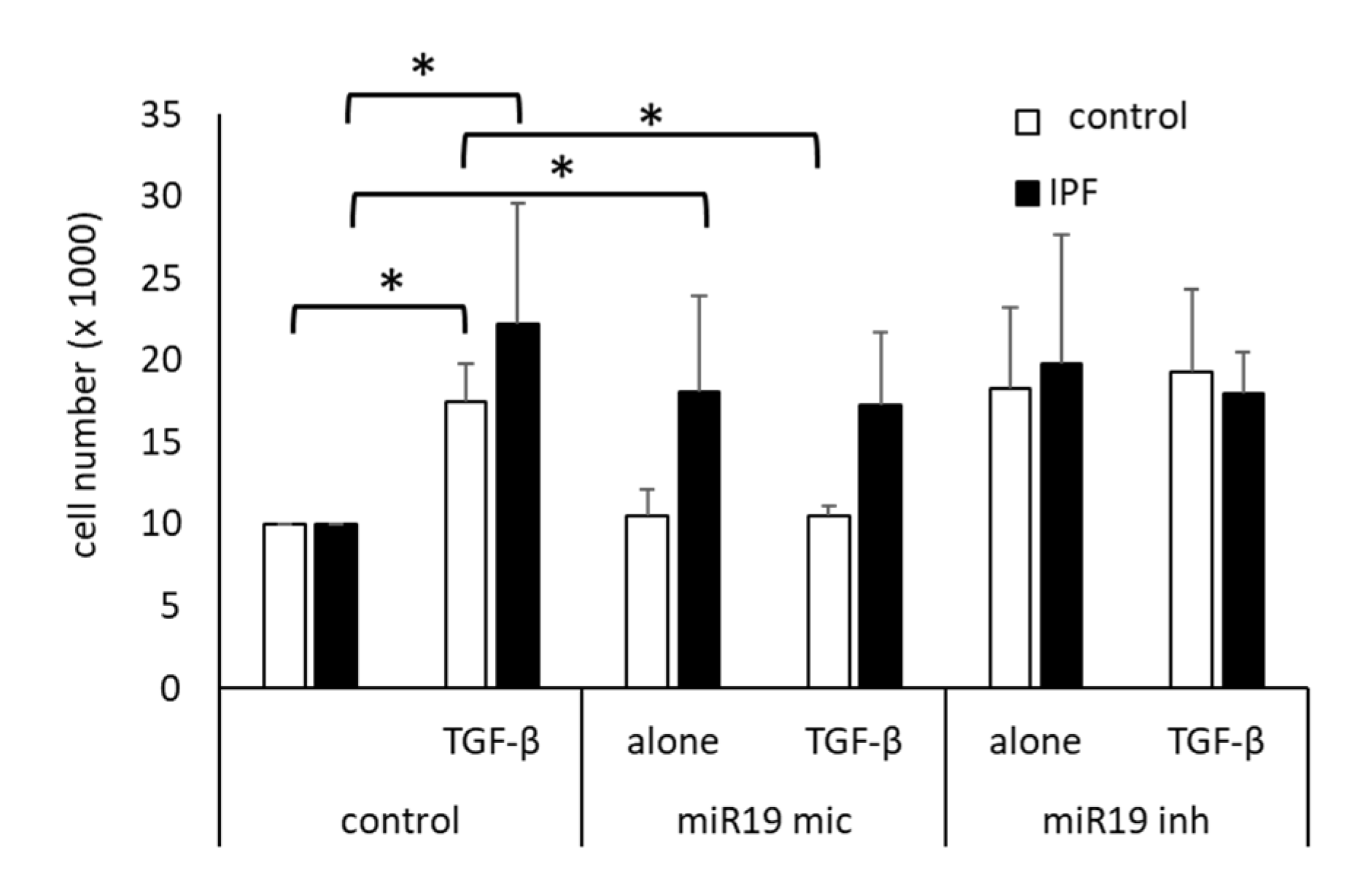
3.2. MiR Inhibition
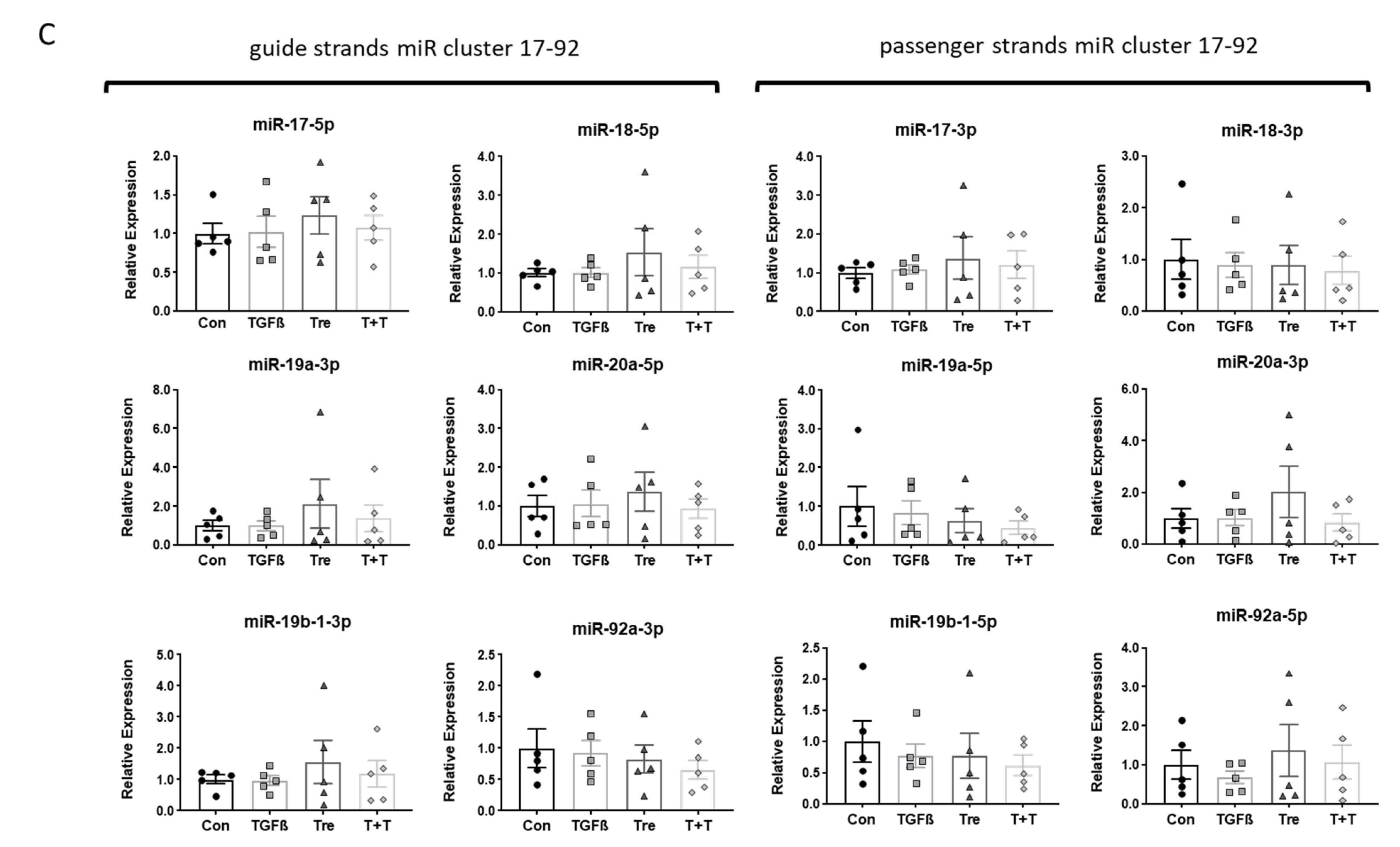
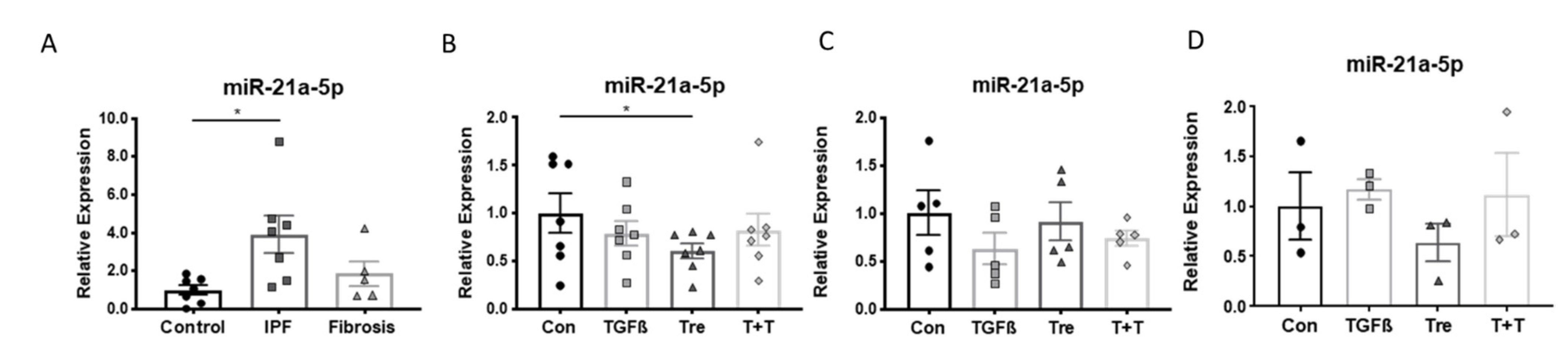
3.3. Disease-Specific Loss of miR-21a-5p Response
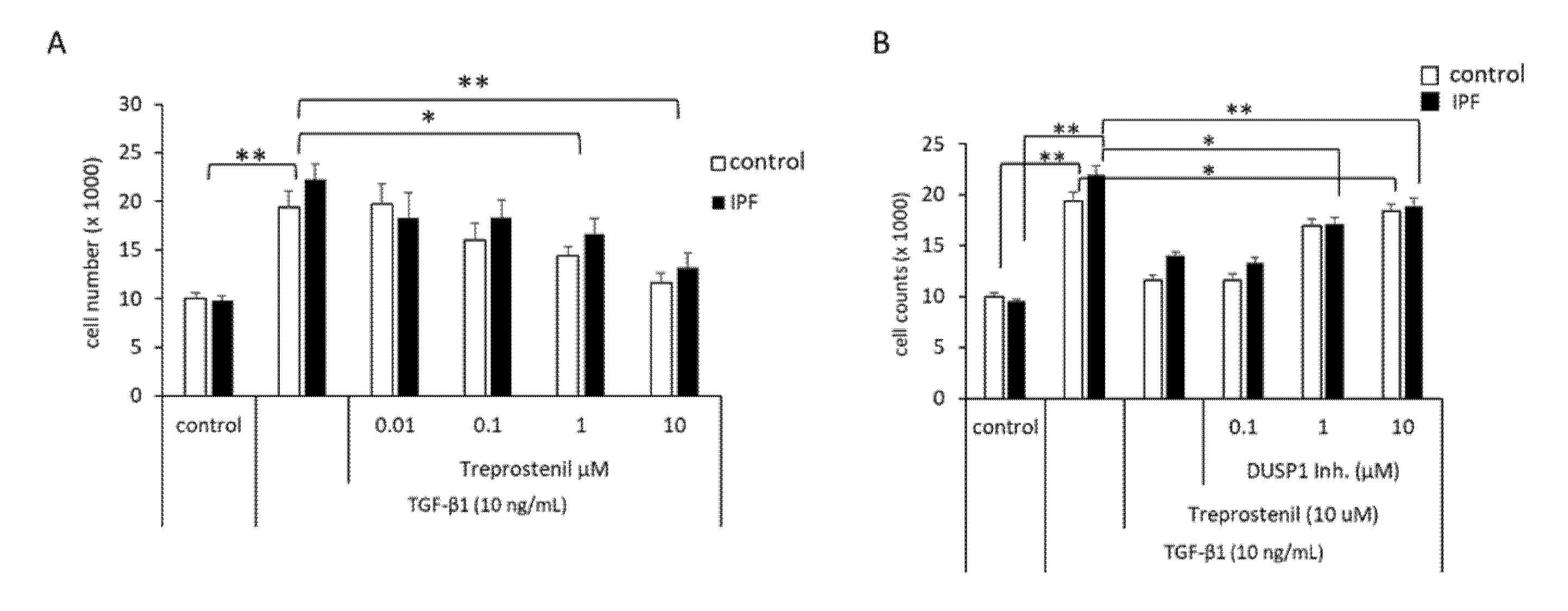
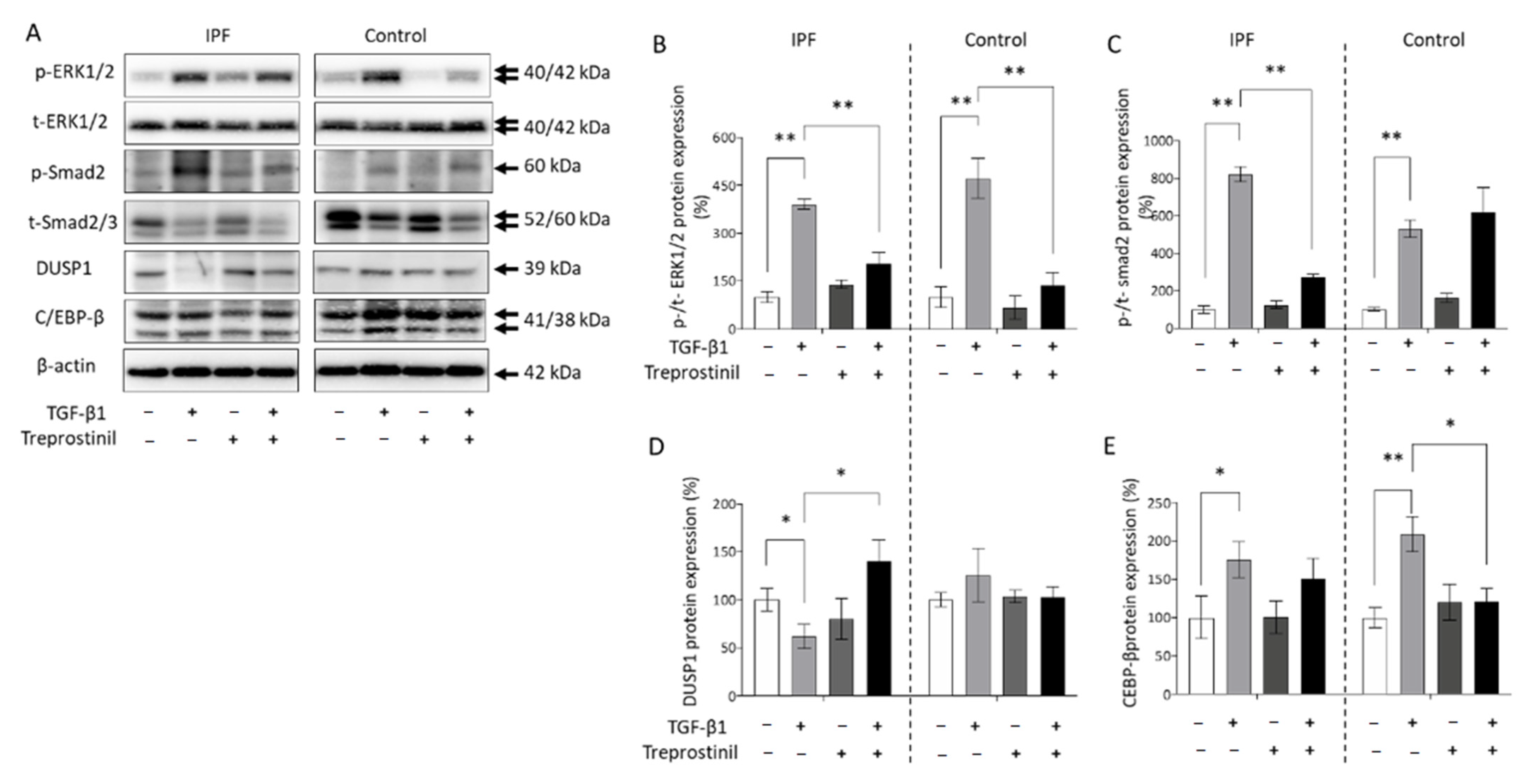
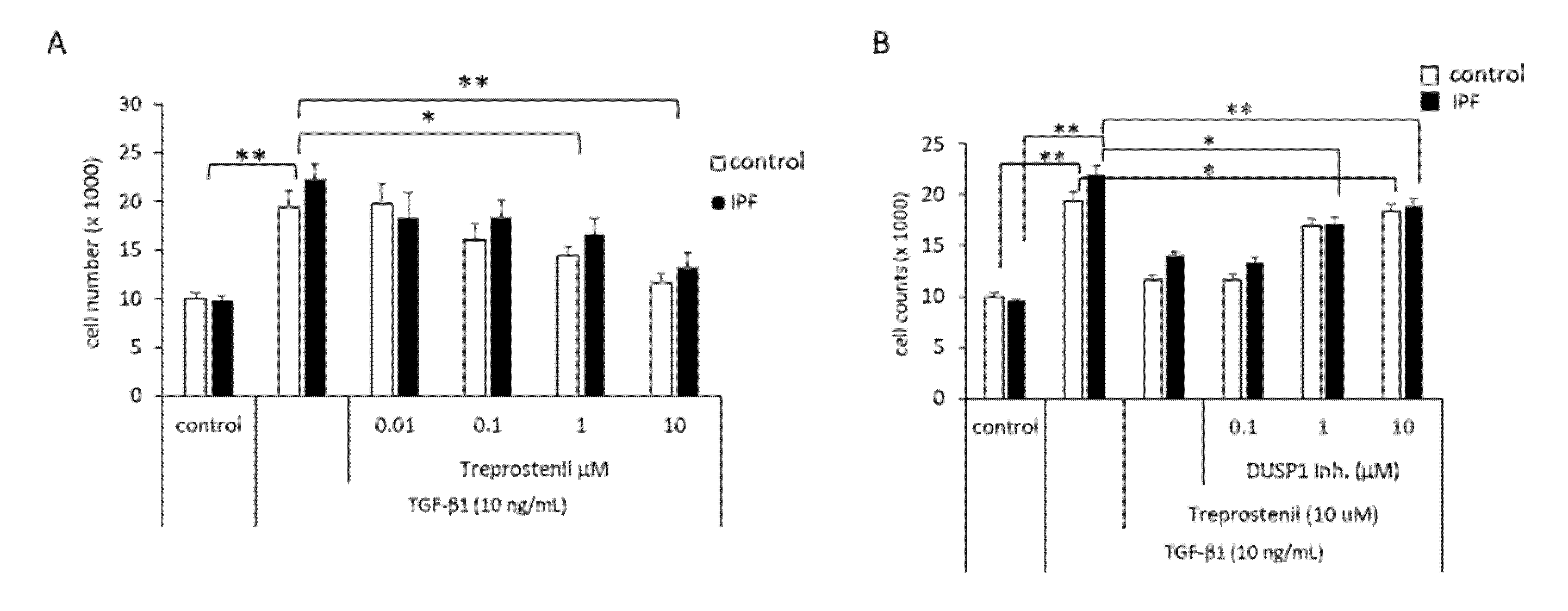
3.4. Proliferation Control of IPF-Fibroblasts Is Reduced by Treprostinil-Dependent DUSP1 Inhibition of Erk1/2
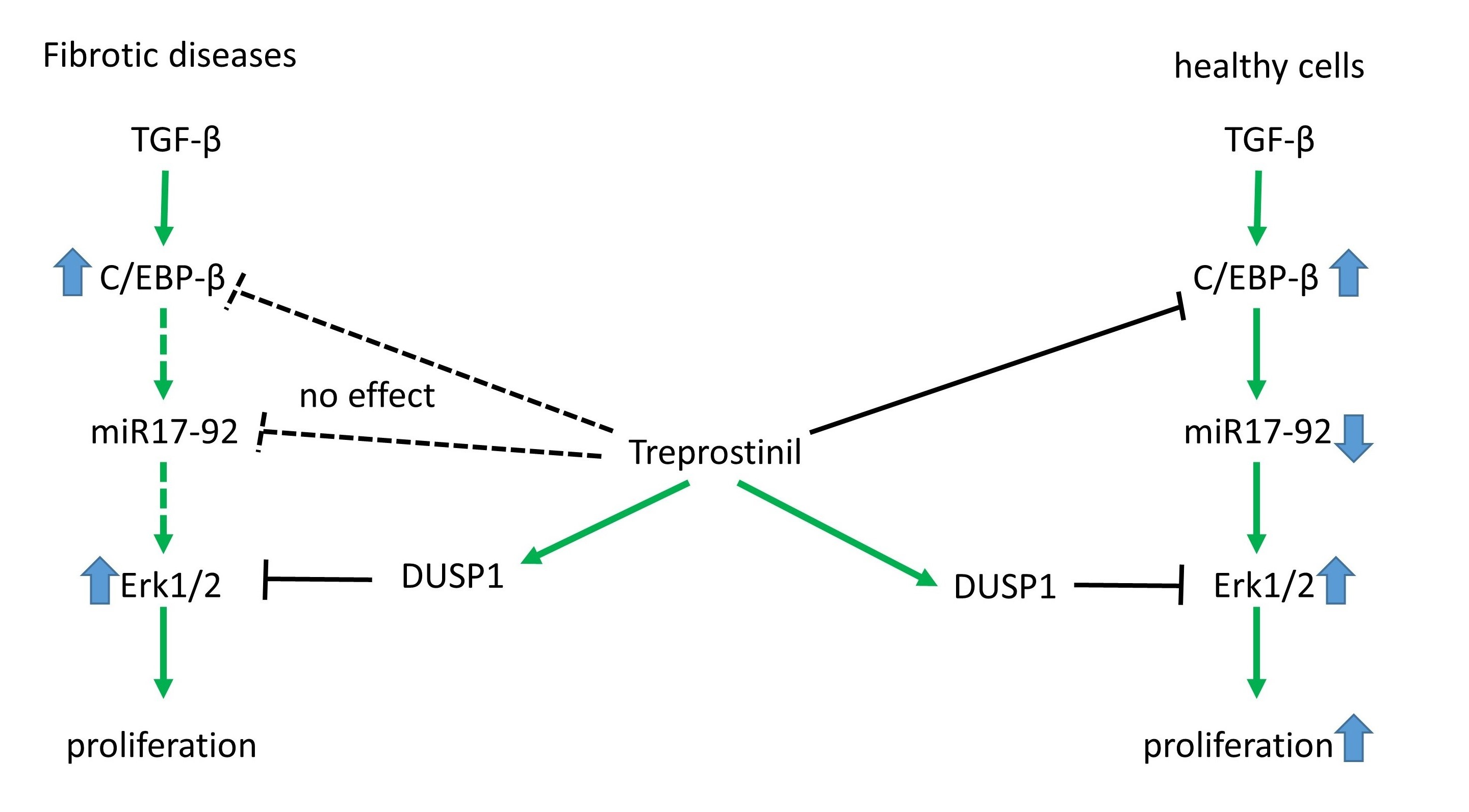
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knudsen, L.; Ruppert, C.; Ochs, M. Tissue remodelling in pulmonary fibrosis. Cell Tissue Res. 2017, 367, 607–626. [Google Scholar] [CrossRef]
- Sauleda, J.; Núñez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Patterson, C.M.; Reed, A.K.; Thillai, M. Lung transplantation for idiopathic pulmonary fibrosis. Lancet Respir. Med. 2019, 7, 271–282. [Google Scholar] [CrossRef]
- Richeldi, L.; Varone, F.; Bergna, M.; de Andrade, J.; Falk, J.; Hallowell, R.; Jouneau, S.; Kondoh, Y.; Morrow, L.; Randerath, W.; et al. Pharmacological management of progressive-fibrosing interstitial lung diseases: A review of the current evidence. Eur. Respir. Rev. 2018, 27, 180074. [Google Scholar] [CrossRef] [Green Version]
- Winters, N.I.; Burman, A.; Kropski, J.A.; Blackwell, T.S. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Kato, M.; Takahashi, F.; Sato, T.; Mitsuishi, Y.; Tajima, K.; Ihara, H.; Nurwidya, F.; Baskoro, H.; Murakami, A.; Kobayashi, I.; et al. Tranilast Inhibits Pulmonary Fibrosis by Suppressing TGFβ/SMAD2 Pathway. Drug Des. Dev. Ther. 2020, 14, 4593–4603. [Google Scholar] [CrossRef]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Chen, B.Y.; Nie, J.; Zhao, G.H.; Zhuo, J.Y.; Yuan, J.; Li, Y.C.; Wang, L.L.; Chen, Z.W. Polydatin prevents bleomycin-induced pulmonary fibrosis by inhibiting the TGF-β/Smad/ERK signaling pathway. Exp. Ther. Med. 2020, 20, 62. [Google Scholar] [CrossRef]
- Lambers, C.; Kornauth, C.; Oberndorfer, F.; Boehm, P.M.; Tamm, M.; Klepetko, W.; Roth, M. Mechanism of anti-remodelling action of treprostinil in human pulmonary arterial smooth muscle cells. PLoS ONE 2018, 13, e0205195. [Google Scholar] [CrossRef] [Green Version]
- Lambers, C.; Boehm, P.M.; Karabacak, Y.; Samaha, E.; Benazzo, A.; Jaksch, P.; Roth, M. Combined Activation of Guanylate Cyclase and Cyclic AMP in Lung Fibroblasts as a Novel Therapeutic Concept for Lung Fibrosis. Biomed. Res. Int. 2019, 2019, 1345402. [Google Scholar] [CrossRef]
- Sun, Q.; Fang, L.; Roth, M.; Tang, X.; Papakonstantinou, E.; Zhai, W.; Louis, R.; Heinen, V.; Schleich, F.N.; Lu, S.; et al. Bronchial thermoplasty decreases airway remodelling by blocking epithelium-derived heat shock protein-60 secretion and protein arginine methyltransferase-1 in fibroblasts. Eur. Respir. J. 2019, 54, 1900300. [Google Scholar] [CrossRef]
- Bagnato, G.; Roberts, W.N.; Roman, J.; Gangemi, S. A systematic review of overlapping microRNA patterns in systemic sclerosis and idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 160125. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Huang, W.; Zhang, L.; Chen, Q.; Zhao, H. Molecular pathogenesis involved in human idiopathic pulmonary fibrosis based on an integrated microRNA-mRNA interaction network. Mol. Med. Rep. 2018, 18, 4365–4373. [Google Scholar] [CrossRef] [Green Version]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewicz, D.; Zakrzewicz, A.; Didiasova, M.; Korencak, M.; Kosanovic, D.; Schermuly, T.; Markart, P.; Wygrecka, M. Elevated protein arginine methyltransferase 1 expression regulates fibroblast motility in pulmonary fibrosis. Biochim. Biophys. Acta 2015, 1852, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Chen, H.; Sun, J.; Lu, D.; Chen, C.; Liu, D. PRMT1 promotes glucose toxicity-induced β cell dysfunction by regulating the nucleo-cytoplasmic trafficking of PDX-1 in a FOXO1-dependent manner in INS-1 cells. Endocrine 2015, 49, 669–682. [Google Scholar] [CrossRef]
- Ali, M.; Heyob, K.; Jacob, N.K.; Rogers, L.K. Alterative Expression and Localization of Profilin 1/VASPpS157 and Cofilin 1/VASPpS239 Regulates Metastatic Growth and Is Modified by DHA Supplementation. Mol. Cancer Ther. 2016, 15, 2220–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, S.D.; Waxman, A.; Rajagopal, S.; Case, A.; Johri, S.; DuBrock, H.; De La Zerda, D.J.; Sahay, S.; King, C.; Melendres-Groves, L.; et al. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: A post-hoc analysis of the INCREASE study. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Gottlieb, J.; Smits, J.; Schramm, R.; Langer, F.; Buhl, R.; Witt, C. Lung Transplantation in Germany Since the Introduction of the Lung Allocation Score. Dtsch. Arztebl. Int. 2017, 114, 179–185. [Google Scholar]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Fang, L.; Wang, X.; Sun, Q.; Papakonstantinou, E.; S’ng, C.; Tamm, M.; Stolz, D.; Roth, M. IgE Downregulates PTEN through MicroRNA-21-5p and Stimulates Airway Smooth Muscle Cell Remodeling. Int. J. Mol. Sci. 2019, 20, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Li, B.; Ma, X.; Huang, C.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Zhu, J.; et al. Folic Acid Protected Neural Cells Against Aluminum-Maltolate-Induced Apoptosis by Preventing miR-19 Downregulation. Neurochem. Res. 2016, 41, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Yu, X.; Huang, Z.; Zheng, S.; Zhou, Y.; Lv, H.; Zeng, Y.; Xu, J.F.; Zhu, X.; Yi, X. Analysis of Microarray-Identified Genes and MicroRNAs Associated with Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2017, 2017, 1804240. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, Y.; Ma, J.; Liu, Y.; Bi, J.; Zhang, L.; Chen, L.; Yao, C.; Lv, W.; Chang, G.; et al. MiR-17-5p suppresses cell proliferation and invasion by targeting ETV1 in triple-negative breast cancer. BMC Cancer 2017, 17, 745. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the miR-17-92 Cluster of miRNAs in Cancer. Front. Med. 2015, 2, 64. [Google Scholar] [CrossRef] [Green Version]
- Mirzamohammadi, F.; Kozlova, A.; Papaioannou, G.; Paltrinieri, E.; Ayturk, U.M.; Kobayashi, T. Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models. Nat. Commun. 2018, 9, 1352. [Google Scholar] [CrossRef]
- Yan, Y.; Hanse, E.A.; Stedman, K.; Benson, J.M.; Lowman, X.H.; Subramanian, S.; Kelekar, A. Transcription factor C/EBP-β induces tumor-suppressor phosphatase PHLPP2 through repression of the miR-17-92 cluster in differentiating AML cells. Cell Death Differ. 2016, 23, 1232–1242. [Google Scholar] [CrossRef]
- Sun, Q.; Fang, L.; Tang, X.; Lu, S.; Tamm, M.; Stolz, D.; Roth, M. TGF-β Upregulated Mitochondria Mass through the SMAD2/3→C/EBPβ→PRMT1 Signal Pathway in Primary Human Lung Fibroblasts. J. Immunol. 2019, 202, 37–47. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Qi, P.; Ma, Z. Biology of MiR-17-92 Cluster and Its Progress in Lung Cancer. Int. J. Med. Sci. 2018, 15, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Liu, L.; Mandal, J.; Molino, A.; Stolz, D.; Tamm, M.; Lu, S.; Roth, M. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal 2016, 28, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, L.; Wang, H.; Mandal, J.; Khan, P.; Hostettler, K.E.; Stolz, D.; Tamm, M.; Molino, A.; Lardinois, D.; et al. Constitutive high expression of protein arginine methyltransferase 1 in asthmatic airway smooth muscle cells is caused by reduced microRNA-19a expression and leads to enhanced remodeling. J. Allergy Clin. Immunol. 2017, 140, 510–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakner, A.M.; Steuerwald, N.M.; Walling, T.L.; Ghosh, S.; Li, T.; McKillop, I.H.; Russo, M.W.; Bonkovsky, H.L.; Schrum, L.W. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 2012, 56, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Chen, J.; Qin, J.; Chen, R.; Yi, Z. TGF-β-induced α-SMA expression is mediated by C/EBPβ acetylation in human alveolar epithelial cells. Mol. Med. 2021, 27, 22. [Google Scholar] [CrossRef] [PubMed]
- Berschneider, B.; Ellwanger, D.C.; Baarsma, H.A.; Thiel, C.; Shimbori, C.; White, E.S.; Kolb, M.; Neth, P.; Königshoff, M. miR-92a regulates TGF-β1-induced WISP1 expression in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2014, 53, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.Y.; Xie, C.Q.; Jiang, C.L.; Sun, J.T.; Feng, H.C.; Li, C.; Huang, X.W. MiR-92a inhibits fibroblast-like synoviocyte proliferation and migration in rheumatoid arthritis by targeting AKT2. J. Biosci. 2018, 43, 911–919. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. MiR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Li, P.; Li, J.; Chen, T.; Wang, H.; Chu, H.; Chang, J.; Zang, W.; Wang, Y.; Ma, Y.; Du, Y.; et al. Expression analysis of serum microRNAs in idiopathic pulmonary fibrosis. Int. J. Mol. Med. 2014, 33, 1554–1562. [Google Scholar] [CrossRef]
- Makiguchi, T.; Yamada, M.; Yoshioka, Y.; Sugiura, H.; Koarai, A.; Chiba, S.; Fujino, N.; Tojo, Y.; Ota, C.; Kubo, H.; et al. Serum extracellular vesicular miR-21-5p is a predictor of the prognosis in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 110. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Xu, F.; Xie, S.; Zuo, W.; Wen, G.; Zhao, T.; Wan, X. MicroRNA-448 overexpression inhibits fibroblast proliferation and collagen synthesis and promotes cell apoptosis via targeting ABCC3 through the JNK signaling pathway. J. Cell Physiol. 2020, 235, 1374–1385. [Google Scholar] [CrossRef]
- Sheu, C.C.; Chang, W.A.; Tsai, M.J.; Liao, S.H.; Chong, I.W.; Kuo, P.L. Bioinformatic analysis of next-generation sequencing data to identify dysregulated genes in fibroblasts of idiopathic pulmonary fibrosis. Int. J. Mol. Med. 2019, 43, 1643–1656. [Google Scholar] [CrossRef]
- Wang, C.; Cao, H.; Gu, S.; Shi, C.; Chen, X.; Han, X. Expression analysis of microRNAs and mRNAs in myofibroblast differentiation of lung resident mesenchymal stem cells. Differentiation 2020, 112, 10–16. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, T.; Hu, X.; Liu, Z.; Zhao, L.; Liu, H.; Liu, Z.; Ma, L. Downregulation of microRNA-30a in bronchoalveolar lavage fluid from idiopathic pulmonary fibrosis patients. Mol. Med. Rep. 2018, 18, 5799–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Wang, Y.; Song, X.; Sun, W.; Zhang, J.; Liu, Y.; Li, H.; Meng, C.; Zhang, J.; Zheng, Q.; et al. Potential regulatory role of circular RNA in idiopathic pulmonary fibrosis. Int. J. Mol. Med. 2018, 42, 3256–3268. [Google Scholar] [CrossRef] [Green Version]
- Bibaki, E.; Tsitoura, E.; Vasarmidi, E.; Margaritopoulos, G.; Trachalaki, A.; Koutoulaki, C.; Georgopoulou, T.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. MiR-185 and miR-29a are similarly expressed in the bronchoalveolar lavage cells in IPF and lung cancer but common targets DNMT1 and COL1A1 show disease specific patterns. Mol. Med. Rep. 2018, 17, 7105–7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goda, C.; Balli, D.; Black, M.; Milewski, D.; Le, T.; Ustiyan, V.; Ren, X.; Kalinichenko, V.V.; Kalin, T.V. Loss of FOXM1 in macrophages promotes pulmonary fibrosis by activating p38 MAPK signaling pathway. PLoS Genet. 2020, 16, e1008692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, J.E.; Oh, E.K.; Kim, D.H.; Choi, S.H. Differential expression profiles and roles of inducible DUSPs and ERK1/2-specific constitutive DUSP6 and DUSP7 in microglia. Biochem. Biophys. Res. Commun. 2015, 467, 254–260. [Google Scholar] [CrossRef]
- Shen, J.; Xing, W.; Liu, R.; Zhang, Y.; Xie, C.; Gong, F. MiR-32-5p influences high glucose-induced cardiac fibroblast proliferation and phenotypic alteration by inhibiting DUSP1. BMC Mol. Biol. 2019, 20, 21. [Google Scholar] [CrossRef]
- Ramkissoon, A.; Chaney, K.E.; Milewski, D.; Williams, K.B.; Williams, R.L.; Choi, K.; Miller, A.; Kalin, T.V.; Pressey, J.G.; Szabo, S.; et al. Targeted Inhibition of the Dual Specificity Phosphatases DUSP1 and DUSP6 Suppress MPNST Growth via JNK. Clin. Cancer Res. 2019, 25, 4117–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, A.J.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Delafontaine, P.; Chandrasekar, B. Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell Signal 2012, 24, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penke, L.R.K.; Speth, J.; Wettlaufer, S.; Draijer, C.; Peters-Golden, M. Bortezomib Inhibits Lung Fibrosis and Fibroblast Activation Without Proteasome Inhibition. Am. J. Respir. Cell Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Juhl, P.; Bondesen, S.; Hawkins, C.L.; Karsdal, M.A.; Bay-Jensen, A.C.; Davies, M.J.; Siebuhr, A.S. Dermal fibroblasts have different extracellular matrix profiles induced by TGF-β, PDGF and IL-6 in a model for skin fibrosis. Sci. Rep. 2020, 10, 17300. [Google Scholar] [CrossRef] [PubMed]
- Landi, C.; Carleo, A.; Vantaggioato, L.; Bergantini, L.; d’Alessandro, M.; Cameli, P.; Sebastiani, G.; Dotta, F.; Bargagli, E. Common molecular pathways targeted by nintedanib in cancer and IPF: A bioinformatic study. Pulm. Pharmacol. Ther. 2020, 64, 101941. [Google Scholar] [CrossRef]
- Silva, C.I.; Müller, N.L.; Hansell, D.M.; Lee, K.S.; Nicholson, A.G.; Wells, A.U. Nonspecific interstitial pneumonia and idiopathic pulmonary fibrosis: Changes in pattern and distribution of disease over time. Radiology 2008, 247, 251–259. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Gender | Age | Therapy |
|---|---|---|---|
| IPF patients | Male | 50 | Pirfenidone |
| Male | 63 | Nintedanib | |
| Male | 50 | Nintedanib | |
| Male | 65 | Nintedanib | |
| Male | 59 | steroids | |
| Male | 48 | Nintedanib | |
| Male | 61 | none | |
| Male | 65 | none | |
| Mean ± SEM | 57.6 ± 2.5 | ||
| Interstitial Fibrosis patients | Female | 52 | steroids |
| Male | 48 | none | |
| Male | 60 | steroids | |
| Male | 69 | none | |
| Female | 68 | steroids | |
| Mean ± SEM | 59.4 ± 4.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blumer, S.; Fang, L.; Chen, W.-C.; Khan, P.; Hostettler, K.; Tamm, M.; Roth, M.; Lambers, C. IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1. Cells 2021, 10, 2836. https://doi.org/10.3390/cells10112836
Blumer S, Fang L, Chen W-C, Khan P, Hostettler K, Tamm M, Roth M, Lambers C. IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1. Cells. 2021; 10(11):2836. https://doi.org/10.3390/cells10112836
Chicago/Turabian StyleBlumer, Sabrina, Lei Fang, Wei-Chih Chen, Petra Khan, Katrin Hostettler, Michael Tamm, Michael Roth, and Christopher Lambers. 2021. "IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1" Cells 10, no. 11: 2836. https://doi.org/10.3390/cells10112836
APA StyleBlumer, S., Fang, L., Chen, W.-C., Khan, P., Hostettler, K., Tamm, M., Roth, M., & Lambers, C. (2021). IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1. Cells, 10(11), 2836. https://doi.org/10.3390/cells10112836