
Maternal B Cell-Intrinsic MyD88 Signaling Mediates LPS-Driven Intrauterine Fetal Death


 and
and
Abstract
:
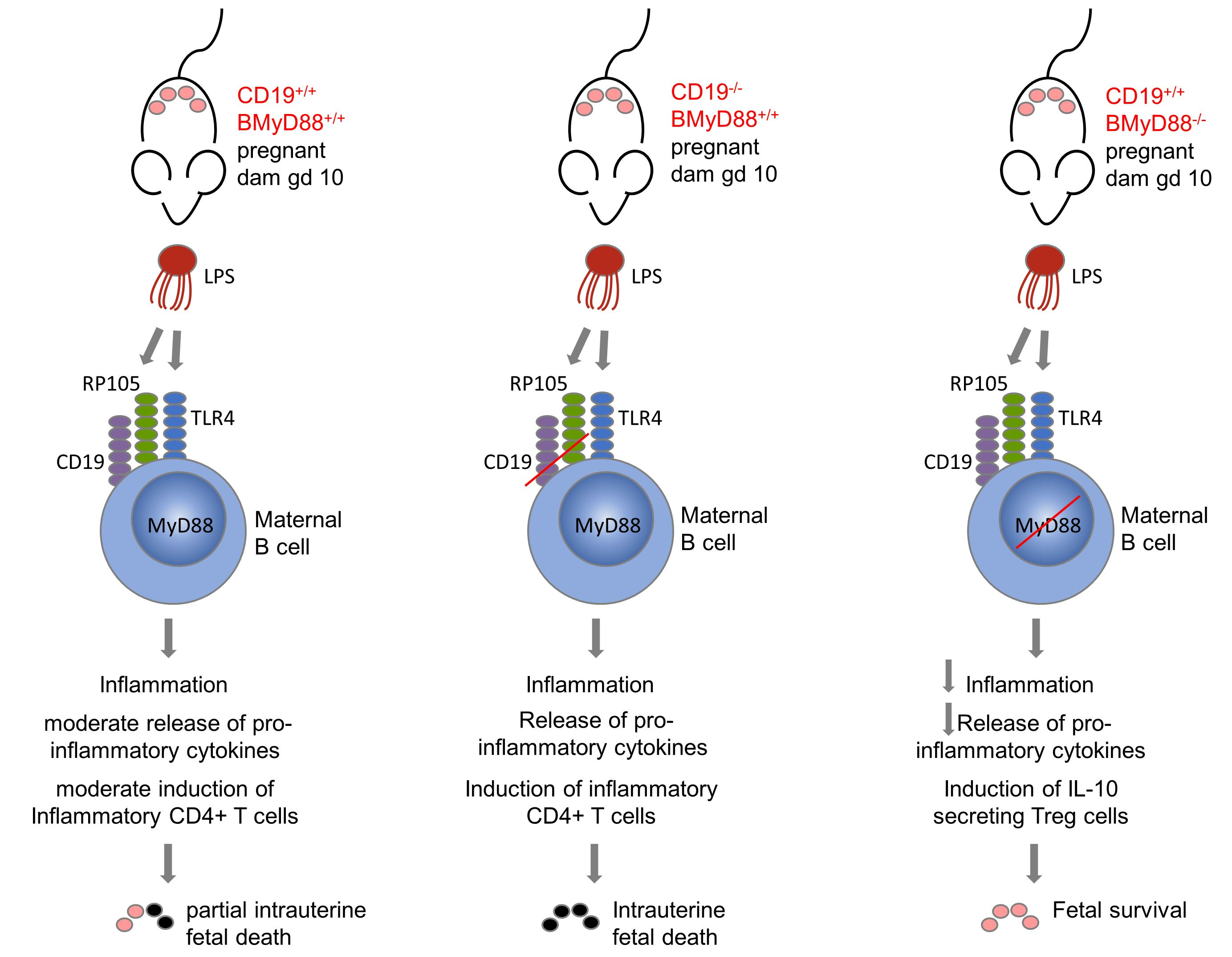{kind=link}
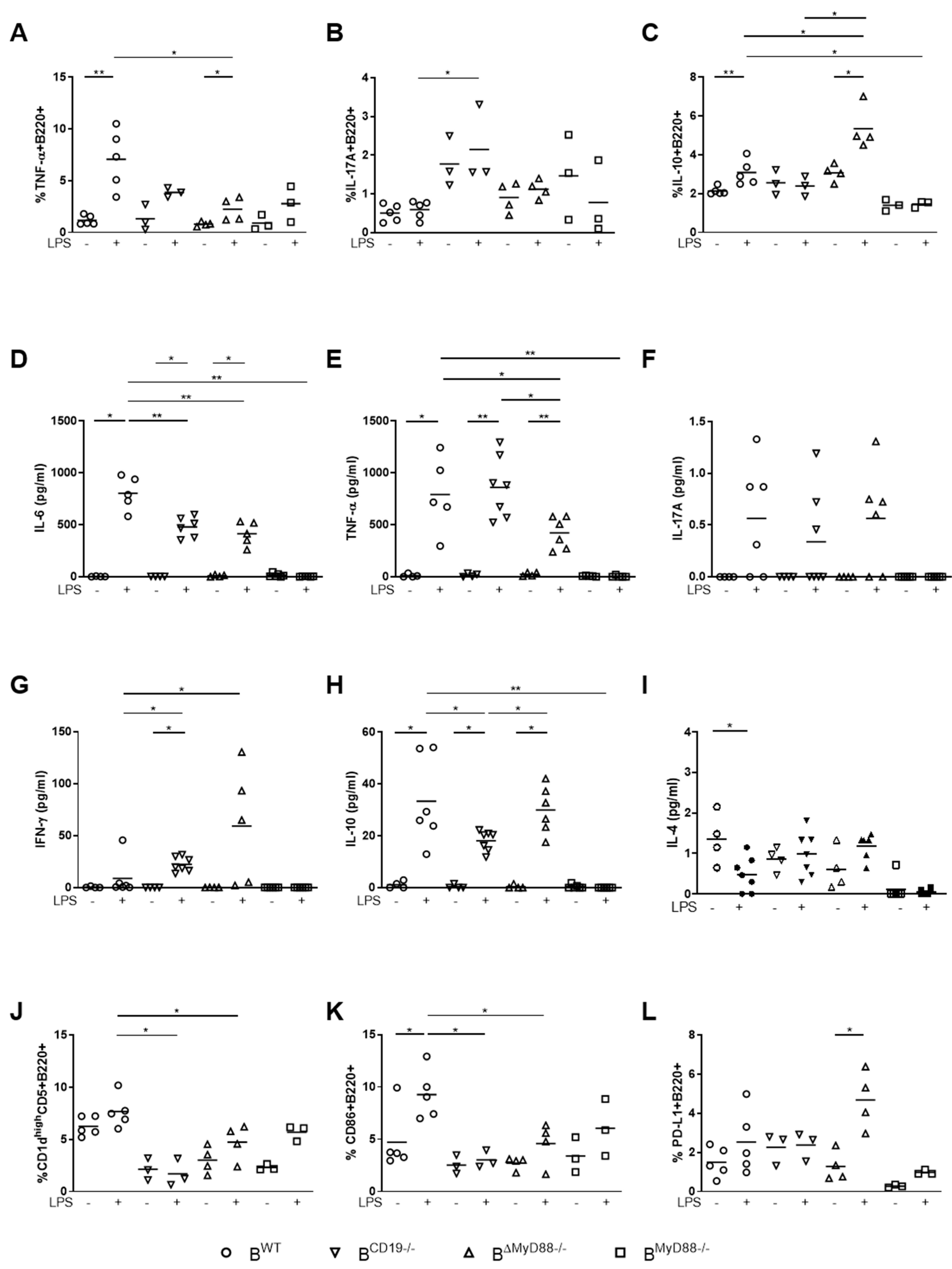{kind=link}
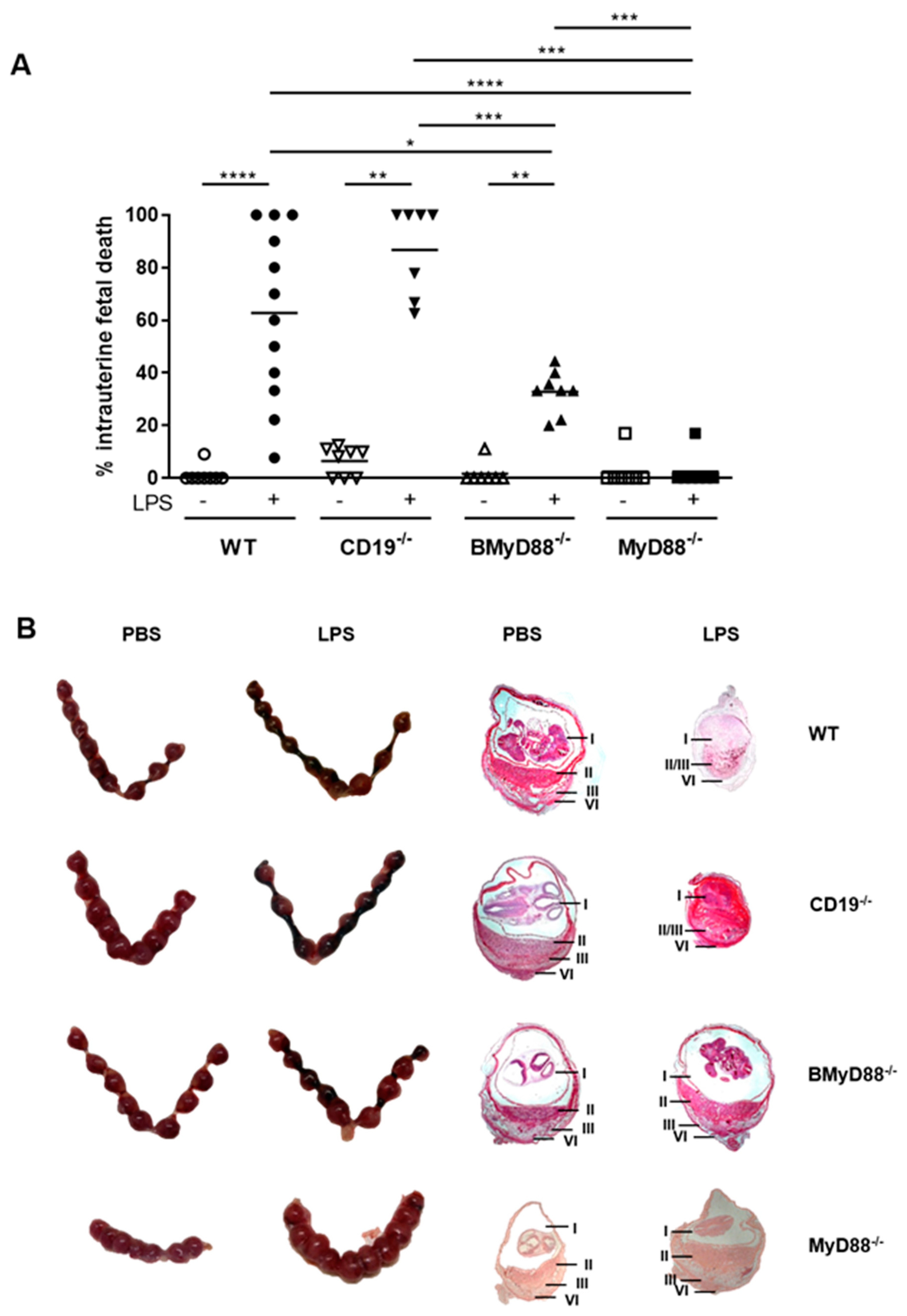{kind=link}
{kind=link}
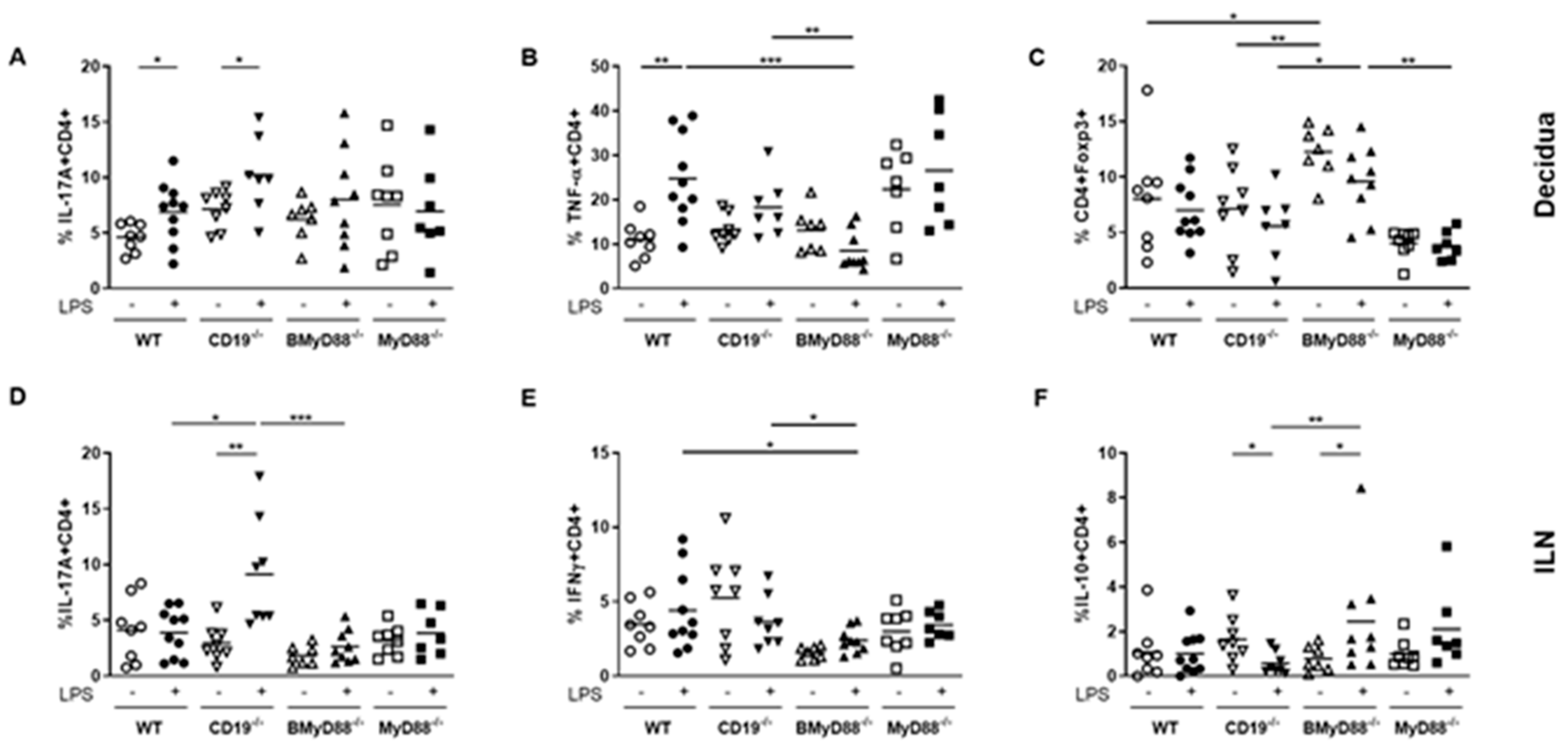{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Husbandry and Model Generation
2.2. Sample Collection and Histology
2.3. Quantitative Histological Measurements
2.4. B Cell Isolation and Stimulation
2.5. Cell Staining and Flow Cytometry
2.6. Cytokine Detection in Sera and Supernatants
2.7. Statistics
3. Results
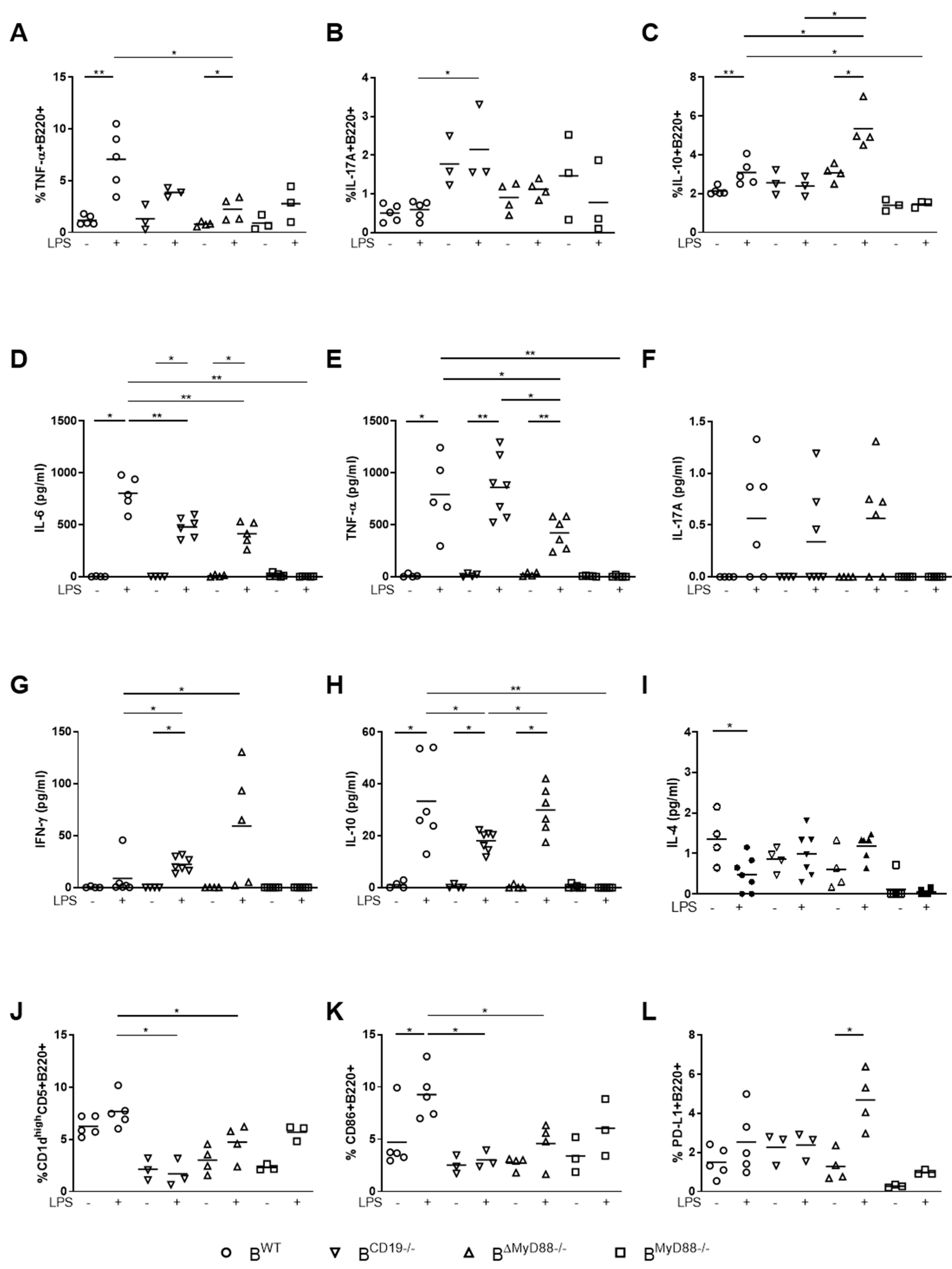
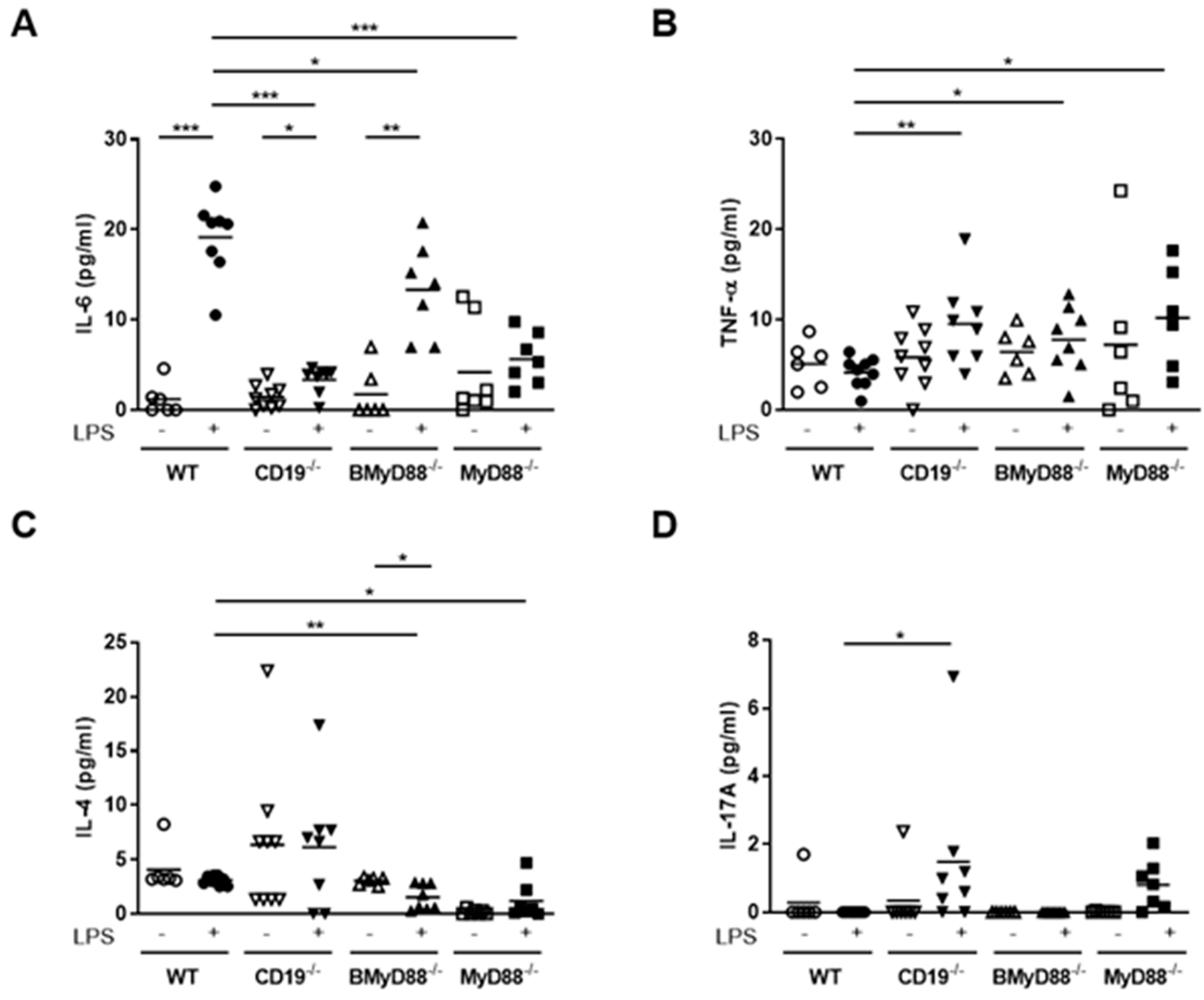
3.1. MyD88-Deficient B Cells and CD19-Deficient B Cells from Pregnant Mice Have a Defective LPS Response
3.2. Deficient B Cell-Specific MyD88 Signaling Hinders IUFD after LPS Treatment
3.3. Systemic Maternal Imbalance between Pro-Inflammatory and Anti-Inflammatory Processes Alters the Function of Decidual Tissue, Leading to IUFD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bukowski, R.; Carpenter, M.; Conway, D.; Coustan, D.; Dudley, D.J.; Goldenberg, R.L.; Hogue, C.J.R.; Koch, M.A.; Parker, C.B.; Pinar, H.; et al. Causes of Death Among Stillbirths. JAMA 2011, 306, 2459–2468. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, C.R.; Montalbano, A.P.; Gao, L. Fetal-to-maternal signaling in the timing of birth. J. Steroid Biochem. Mol. Biol. 2017, 170, 19–27. [Google Scholar] [CrossRef]
- Howell, K.R.; Powell, T.L. Effects of maternal obesity on placental function and fetal development. Reproduction 2017, 153, R97–R108. [Google Scholar] [CrossRef] [PubMed]
- Gammill, H.S.; Stephenson, M.D.; Aydelotte, T.M.; Nelson, J.L. Microchimerism in recurrent miscarriage. Cell. Mol. Immunol. 2014, 11, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartha, J.L.; Comino-Delgado, R. Lymphocyte subpopulations in intrauterine growth retardation in women with or without previous pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 82, 23–27. [Google Scholar] [CrossRef]
- Busse, M.; Campe, K.J.; Nowak, D.; Schumacher, A.; Plenagl, S.; Langwisch, S.; Tiegs, G.; Reinhold, A.; Zenclussen, A.C. IL-10 producing B cells rescue mouse fetuses from inflammation-driven fetal death and are able to modulate T cell immune responses. Sci. Rep. 2019, 9, 9335. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F.; Muzzio, D.; Soldati, R.; Fest, S.; Zenclussen, A.C. Regulatory B10 cells restore pregnancy tolerance in a mouse model. Biol. Reprod. 2013, 89, 90. [Google Scholar] [CrossRef] [Green Version]
- Rolle, L.; Memarzadeh Tehran, M.; Morell-Garcia, A.; Raeva, Y.; Schumacher, A.; Hartig, R.; Costa, S.D.; Jensen, F.; Zenclussen, A.C. Cutting edge: IL-10-producing regulatory B cells in early human pregnancy. Am. J. Reprod. Immunol. 2013, 70, 448–453. [Google Scholar] [CrossRef]
- Busse, M.; Campe, K.J.; Redlich, A.; Oettel, A.; Hartig, R.; Costa, S.D.; Zenclussen, A.C. Regulatory B Cells Are Decreased and Impaired in Their Function in Peripheral Maternal Blood in Pre-term Birth. Front. Immunol. 2020, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, A.; Ehrentraut, S.; Scharm, M.; Wang, H.S.; Hartig, R.; Morse, H.C.; Zenclussen, A.C. Plasma Cell Alloantigen 1 and IL-10 Secretion Define Two Distinct Peritoneal B1a B Cell Subsets With Opposite Functions, PC1(high) Cells Being Protective and PC1(low) Cells Harmful for the Growing Fetus. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tedder, T.F. B10 Cells: A Functionally Defined Regulatory B Cell Subset. J. Immunol. 2015, 194, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClure, E.M.; Goldenberg, R.L. Infection and stillbirth. Semin. Fetal. Neonat Med. 2009, 14, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surgers, L.; Bleibtreu, A.; Burdet, C.; Clermont, O.; Laouenan, C.; Lefort, A.; Mentre, F.; Carbonne, B.; Bingen, E.; Meynard, J.L.; et al. Escherichia coli bacteraemia in pregnant women is life-threatening for foetuses. Clin. Microbiol. Infect. 2014, 20, O1035–O1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolockiene, E.; Morsing, E.; Holst, E.; Herbst, A.; Ljungh, A.; Svenningsen, N.; Hagerstrand, I.; Nystrom, L. Intrauterine infection may be a major cause of stillbirth in Sweden. Acta Obstet. Gyn Scan 2001, 80, 511–518. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Yazawa, N.; Fujimoto, M.; Sato, S.; Miyake, K.; Asano, N.; Nagai, Y.; Takeuchi, O.; Takeda, K.; Okochi, H.; Akira, S.; et al. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood 2003, 102, 1374–1380. [Google Scholar] [CrossRef]
- Busse, M.; Langwisch, S.; Tedford, K.; Fischer, K.D.; Zenclussen, A.C. Maternal B cell signaling orchestrates fetal development in mice. Development 2021, 16, 199783. [Google Scholar] [CrossRef]
- Granato, A.; Hayashi, E.A.; Baptista, B.J.; Bellio, M.; Nobrega, A. IL-4 regulates Bim expression and promotes B cell maturation in synergy with BAFF conferring resistance to cell death at negative selection checkpoints. J. Immunol. 2014, 192, 5761–5775. [Google Scholar] [CrossRef] [Green Version]
- Wurster, A.L.; Rodgers, V.L.; White, M.F.; Rothstein, T.L.; Grusby, M.J. Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J. Biol. Chem. 2002, 277, 27169–27175. [Google Scholar] [CrossRef] [Green Version]
- Lebman, D.A.; Coffman, R.L. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J. Exp. Med. 1988, 168, 853–862. [Google Scholar] [CrossRef]
- Yanagihara, Y.; Ikizawa, K.; Kajiwara, K.; Koshio, T.; Basaki, Y.; Akiyama, K. Functional significance of IL-4 receptor on B cells in IL-4-induced human IgE production. J. Allergy Clin. Immunol. 1995, 96, 1145–1151. [Google Scholar] [CrossRef]
- Goldstein, H.; Volkman, D.J.; Ambrus, J.L., Jr.; Fauci, A.S. Characterization of a T4+/Leu-8+ T cell clone that directly helps B cell Ig production by secreting B cell differentiation factor. J. Immunol. 1985, 135, 339–343. [Google Scholar]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. The IL-4 receptor: Signaling mechanisms and biologic functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauri, C.; Ehrenstein, M.R. The ‘short’ history of regulatory B cells. Trends Immunol. 2008, 29, 34–40. [Google Scholar] [CrossRef]
- Zacca, E.R.; Onofrio, L.I.; Acosta, C.D.V.; Ferrero, P.V.; Alonso, S.M.; Ramello, M.C.; Mussano, E.; Onetti, L.; Cadile, I.I.; Stancich, M.I.; et al. PD-L1(+) Regulatory B Cells Are Significantly Decreased in Rheumatoid Arthritis Patients and Increase After Successful Treatment. Front. Immunol. 2018, 9, 2241. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Hams, E.; Floudas, A.; Sparwasser, T.; Weaver, C.T.; Fallon, P.G. PD-L1hi B cells are critical regulators of humoral immunity. Nat. Commun. 2015, 6, 5997. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Dey, S.K.; Fisher, S.J. Preterm labor: One syndrome, many causes. Science 2014, 345, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodger, J.C. Lack of a requirement for a maternal humoral immune response to establish or maintain successful allogeneic pregnancy. Transplantation 1985, 40, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pinto, D.; Moreno, J. B cells can prime naive CD4+ T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur. J. Immunol. 2005, 35, 1097–1105. [Google Scholar] [CrossRef]
- Ismail, A.S.; Severson, K.M.; Vaishnava, S.; Behrendt, C.L.; Yu, X.F.; Benjamin, J.L.; Ruhn, K.A.; Hou, B.D.; DeFranco, A.L.; Yarovinsky, F.; et al. gamma delta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc. Natl. Acad. Sci. USA 2011, 108, 8743–8748. [Google Scholar] [CrossRef] [Green Version]
- Brandl, K.; Sun, L.; Neppl, C.; Siggs, O.M.; Le Gall, S.M.; Tomisato, W.; Li, X.H.; Du, X.; Maennel, D.N.; Blobel, C.P.; et al. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 19967–19972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelman, A.E.; Zhang, J.D.; Choi, Y.; Turka, L.A. Toll-like receptor ligands directly promote activated CD4(+) T cell survival. J. Immunol. 2004, 172, 6065–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, B.D.; Saudan, P.; Ott, G.; Wheeler, M.L.; Ji, M.; Kuzmich, L.; Lee, L.M.; Coffman, R.L.; Bachmann, M.F.; DeFranco, A.L. Selective Utilization of Toll-like Receptor and MyD88 Signaling in B Cells for Enhancement of the Antiviral Germinal Center Response. Immunity 2011, 34, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Bahlburg, A.; Khim, S.; Rawlings, D.J. B cell-intrinsic TLR signals amplify but are not required for humoral immunity. J. Exp. Med. 2007, 204, 3095–3101. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.; Kearney, J.F. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity 2000, 12, 39–49. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Zhao, H.; Wang, Y.; Carter, R.H. Cutting edge: Primary and secondary effects of CD19 deficiency on cells of the marginal zone. J. Immunol. 2009, 182, 7343–7347. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Ono, N.; Steeber, D.A.; Pisetsky, D.S.; Tedder, T.F. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J. Immunol. 1996, 157, 4371–4378. [Google Scholar]
- Yanaba, K.; Yoshizaki, A.; Asano, Y.; Kadono, T.; Tedder, T.F.; Sato, S. IL-10-producing regulatory B10 cells inhibit intestinal injury in a mouse model. Am. J. Pathol. 2011, 178, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Ishiura, N.; Nakashima, H.; Kuwano, Y.; Okochi, H.; Tamaki, K.; Sato, S.; Tedder, T.F.; Fujimoto, M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol 2010, 184, 4801–4809. [Google Scholar] [CrossRef]
- Talukdar, A.; Rai, R.; Aparna Sharma, K.; Rao, D.N.; Sharma, A. Peripheral Gamma Delta T cells secrete inflammatory cytokines in women with idiopathic recurrent pregnancy loss. Cytokine 2018, 102, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.M.; Xiao, Z.N.; Wang, X.B.; Huang, Y. IL-17 Induces Fetal Loss in a CBA/JxBALB/c Mouse Model, and an Anti-IL-17 Antibody Prevents Fetal Loss in a CBA/JxDBA/2 Mouse Model. Am. J. Reprod. Immunol. 2016, 75, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.L.; Yin, T.L.; Yan, W.J.; Li, J.; He, F.; Yang, J. Molecular detection of uterine innate lymphoid cells in the immunological mouse model of pregnancy loss. Int. Immunopharmacol. 2019, 68, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, X.; Liu, X.; Zhang, R.; Fu, Q.; Xu, X. The Treg/Th17 imbalance in Toxoplasma gondii-infected pregnant mice. Am J Reprod Immunol 2012, 67, 112–121. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, J.Y.; Hur, S.E.; Kim, C.J.; Na, B.J.; Lee, M.; Gilman-Sachs, A.; Kwak-Kim, J. An imbalance in interleukin-17-producing T and Foxp3(+) regulatory T cells in women with idiopathic recurrent pregnancy loss. Hum Reprod 2011, 26, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Lee, S.K.; Kim, J.Y.; Na, B.J.; Hur, S.E.; Lee, M.; Kwak-Kim, J. Intravenous immunoglobulin G modulates peripheral blood Th17 and Foxp3(+) regulatory T cells in pregnant women with recurrent pregnancy loss. Am. J. Reprod. Immunol. 2014, 71, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, J.W.; Kasahara, S.; Clark, E.A.; Ledbetter, J.A. Anti-CD180 (RP105) activates B cells to rapidly produce polyclonal Ig via a T cell and MyD88-independent pathway. J. Immunol. 2011, 187, 4199–4209. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Liew, L.N.; Kuo, I.C.; Huang, C.H.; Goh, D.L.; Chua, K.Y. The modulatory effects of lipopolysaccharide-stimulated B cells on differential T-cell polarization. Immunology 2008, 125, 218–228. [Google Scholar] [CrossRef]
- Nagai, Y.; Shimazu, R.; Ogata, H.; Akashi, S.; Sudo, K.; Yamasaki, H.; Hayashi, S.; Iwakura, Y.; Kimoto, M.; Miyake, K. Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood 2002, 99, 1699–1705. [Google Scholar] [CrossRef]
- Morbach, H.; Schickel, J.N.; Cunningham-Rundles, C.; Conley, M.E.; Reisli, I.; Franco, J.L.; Meffre, E. CD19 controls Toll-like receptor 9 responses in human B cells. J. Allergy Clin. Immunol. 2016, 137, 889–898.e6. [Google Scholar] [CrossRef] [Green Version]
- Barr, T.A.; Brown, S.; Mastroeni, P.; Gray, D. B cell intrinsic MyD88 signals drive IFN-gamma production from T cells and control switching to IgG2c. J. Immunol. 2009, 183, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Into, T.; Takigawa, T.; Niida, S.; Shibata, K. MyD88 deficiency alters expression of antimicrobial factors in mouse salivary glands. PLoS ONE 2014, 9, e113333. [Google Scholar] [CrossRef] [Green Version]
- Barbeiro, D.F.; Barbeiro, H.V.; Faintuch, J.; Ariga, S.K.; Mariano, M.; Popi, A.F.; de Souza, H.P.; Velasco, I.T.; Soriano, F.G. B-1 cells temper endotoxemic inflammatory responses. Immunobiology 2011, 216, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Flenady, V.; Koopmans, L.; Middleton, P.; Froen, J.F.; Smith, G.C.; Gibbons, K.; Coory, M.; Gordon, A.; Ellwood, D.; McIntyre, H.D.; et al. Major risk factors for stillbirth in high-income countries: A systematic review and meta-analysis. Lancet 2011, 377, 1331–1340. [Google Scholar] [CrossRef]
- Al-Azemi, M.; Raghupathy, R.; Azizieh, F. Pro-inflammatory and anti-inflammatory cytokine profiles in fetal growth restriction. Clin. Exp. Obstet. Gynecol. 2017, 44, 98–103. [Google Scholar] [PubMed]
- Raghupathy, R.; Al-Azemi, M.; Azizieh, F. Intrauterine growth restriction: Cytokine profiles of trophoblast antigen-stimulated maternal lymphocytes. Clin. Dev. Immunol. 2011, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busse, M.; Plenagl, S.; Campe, N.K.J.; Müller, A.J.; Tedford, K.; Schumacher, A.; Zenclussen, A.C. Maternal B Cell-Intrinsic MyD88 Signaling Mediates LPS-Driven Intrauterine Fetal Death. Cells 2021, 10, 2693. https://doi.org/10.3390/cells10102693
Busse M, Plenagl S, Campe NKJ, Müller AJ, Tedford K, Schumacher A, Zenclussen AC. Maternal B Cell-Intrinsic MyD88 Signaling Mediates LPS-Driven Intrauterine Fetal Death. Cells. 2021; 10(10):2693. https://doi.org/10.3390/cells10102693
Chicago/Turabian StyleBusse, Mandy, Susanne Plenagl, Norina Kim Jutta Campe, Andreas J. Müller, Kerry Tedford, Anne Schumacher, and Ana Claudia Zenclussen. 2021. "Maternal B Cell-Intrinsic MyD88 Signaling Mediates LPS-Driven Intrauterine Fetal Death" Cells 10, no. 10: 2693. https://doi.org/10.3390/cells10102693
APA StyleBusse, M., Plenagl, S., Campe, N. K. J., Müller, A. J., Tedford, K., Schumacher, A., & Zenclussen, A. C. (2021). Maternal B Cell-Intrinsic MyD88 Signaling Mediates LPS-Driven Intrauterine Fetal Death. Cells, 10(10), 2693. https://doi.org/10.3390/cells10102693