
IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization


 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transgenic Mice
2.2. Protein Extraction
2.3. Western Blot Analysis
2.4. Histology and Immunostaining
2.5. Confocal Imaging and Image Analysis
2.6. Antibodies for Immunostaining and Immunoblotting
2.7. RNA Extraction, cDNA Synthesis, and qRT-PCR
2.8. Primary Cell Isolation and Cell Culture
2.9. Statistical Analysis
3. Results
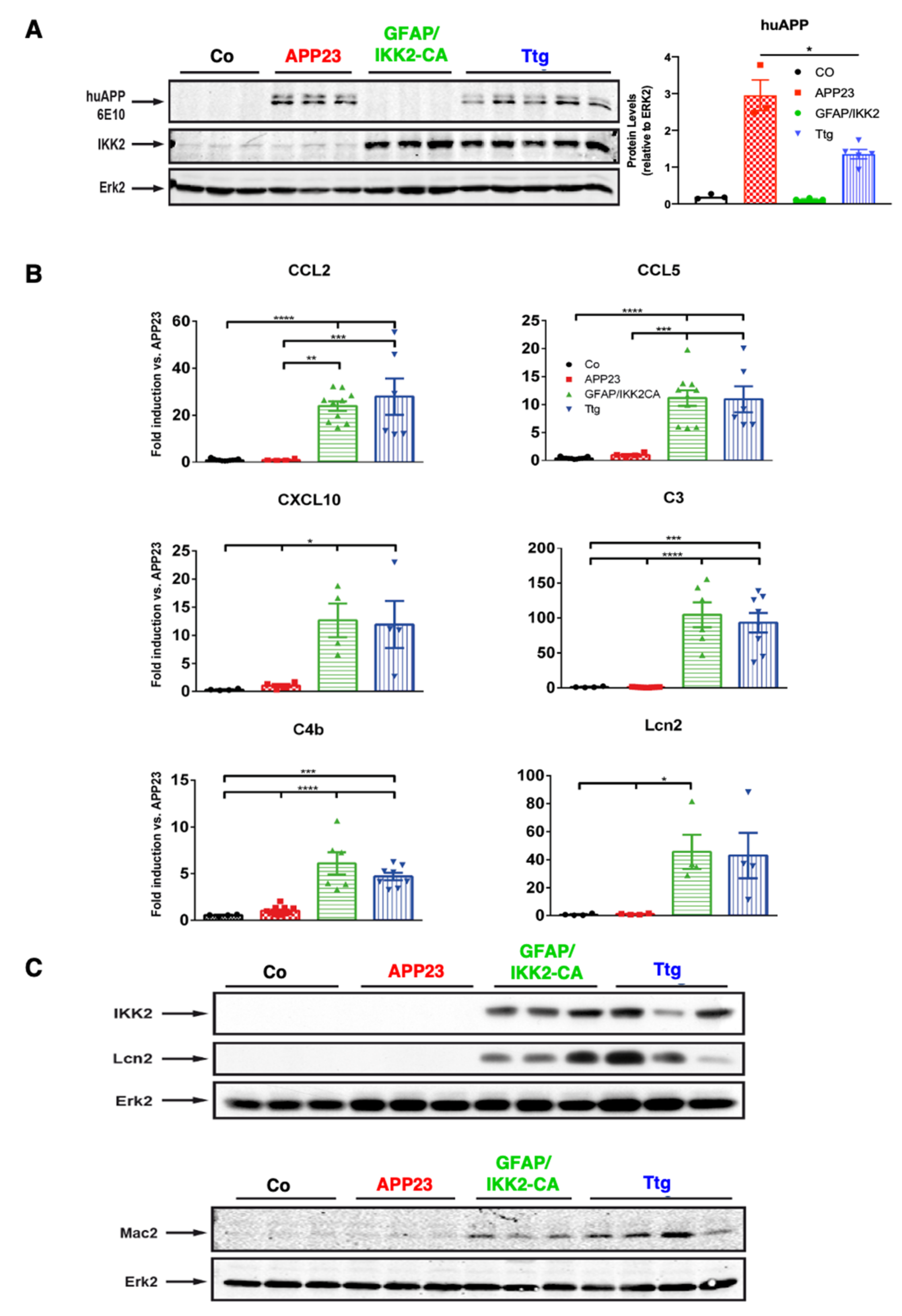
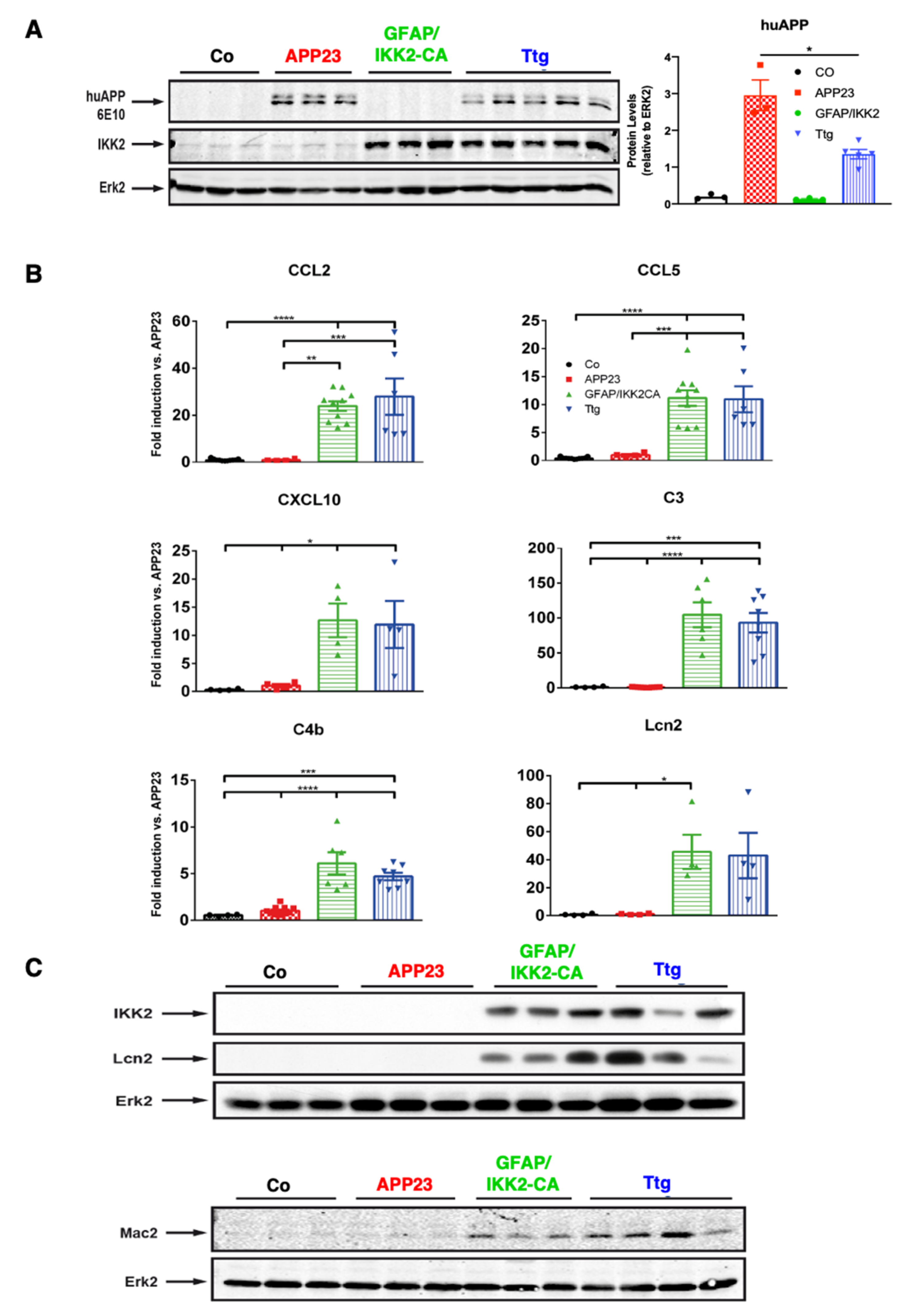
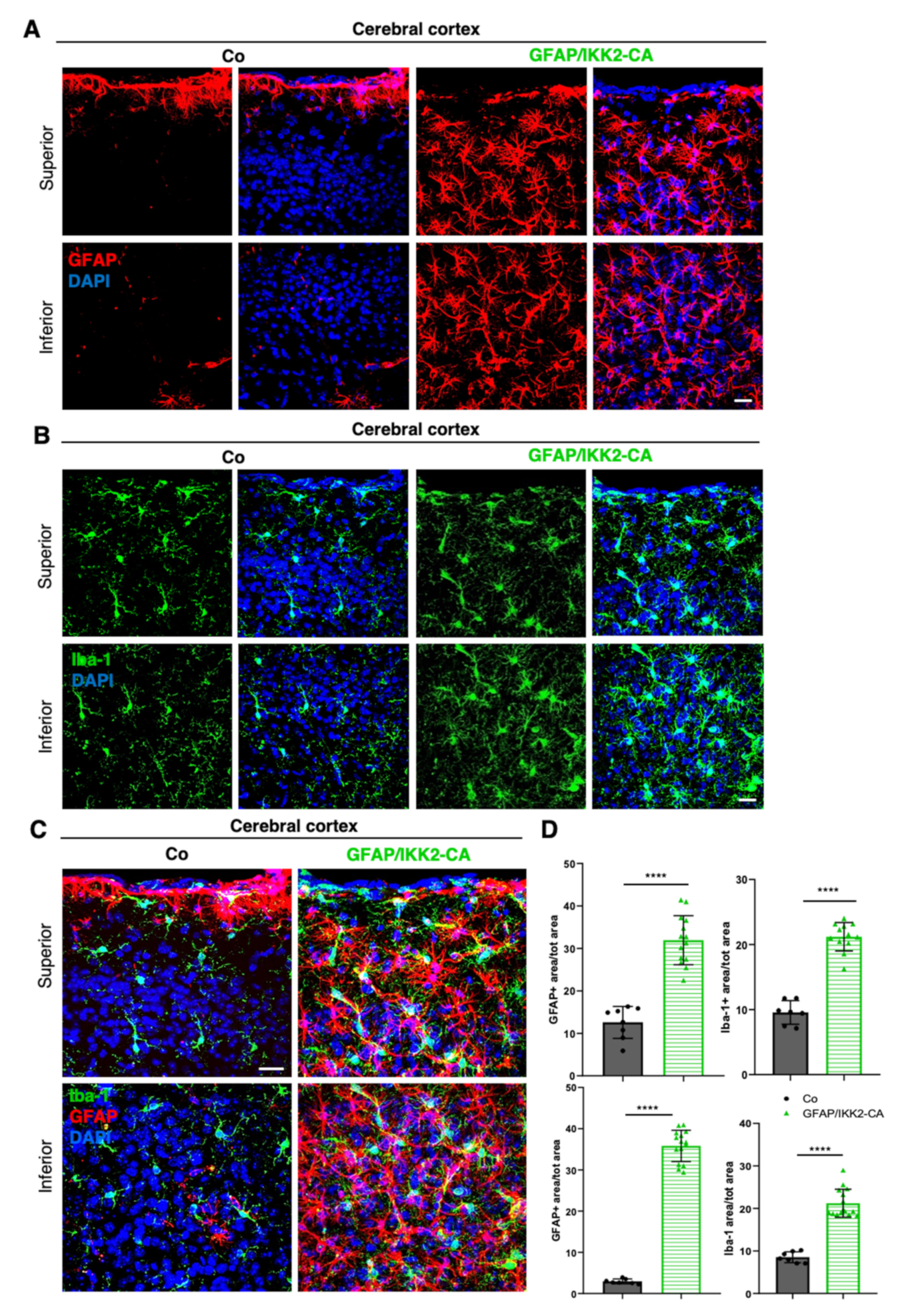
3.1. Establishment of a Novel APP23 Mouse Model of AD (Ttg) Combined with Conditional Activation of Neuroinflammation
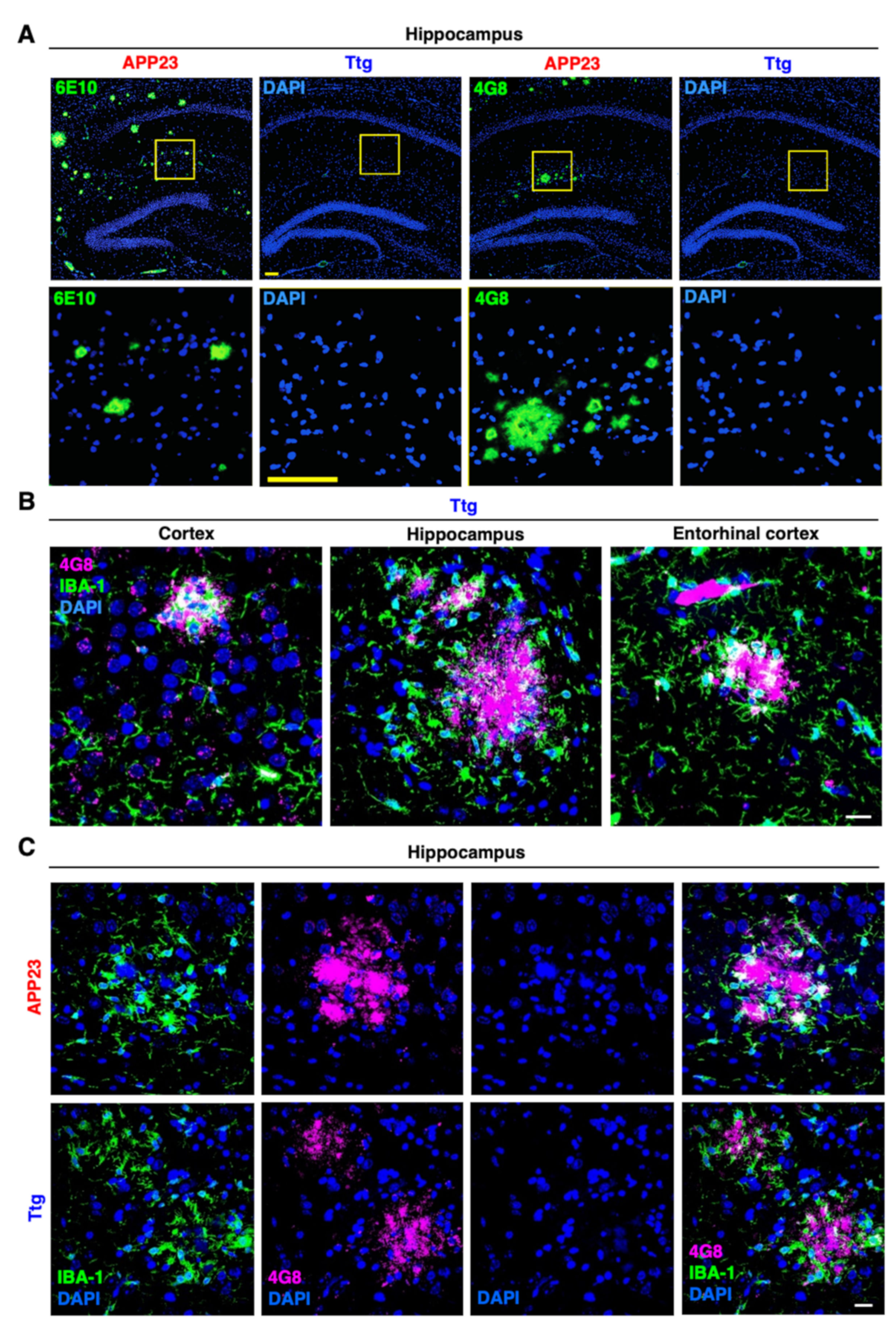
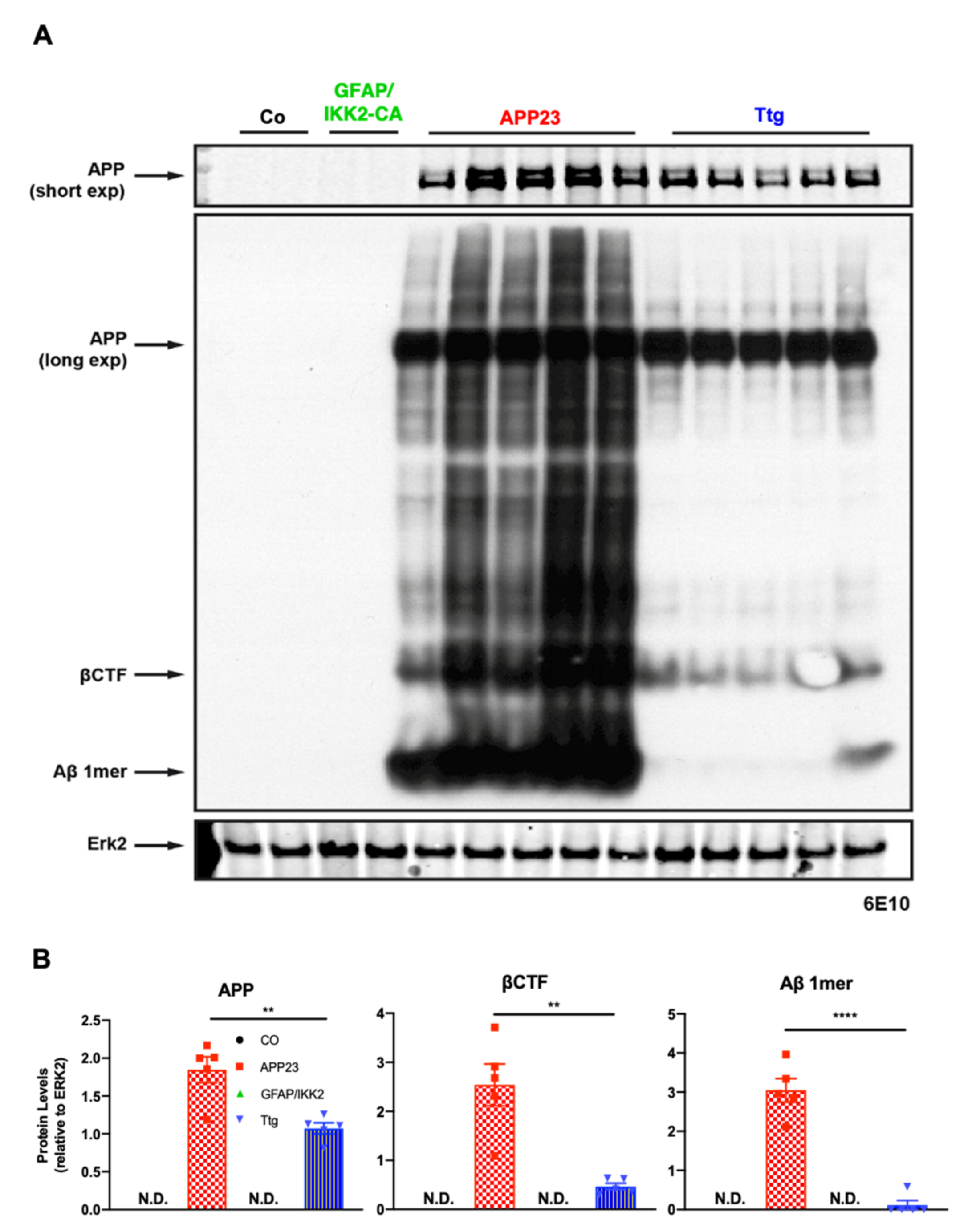
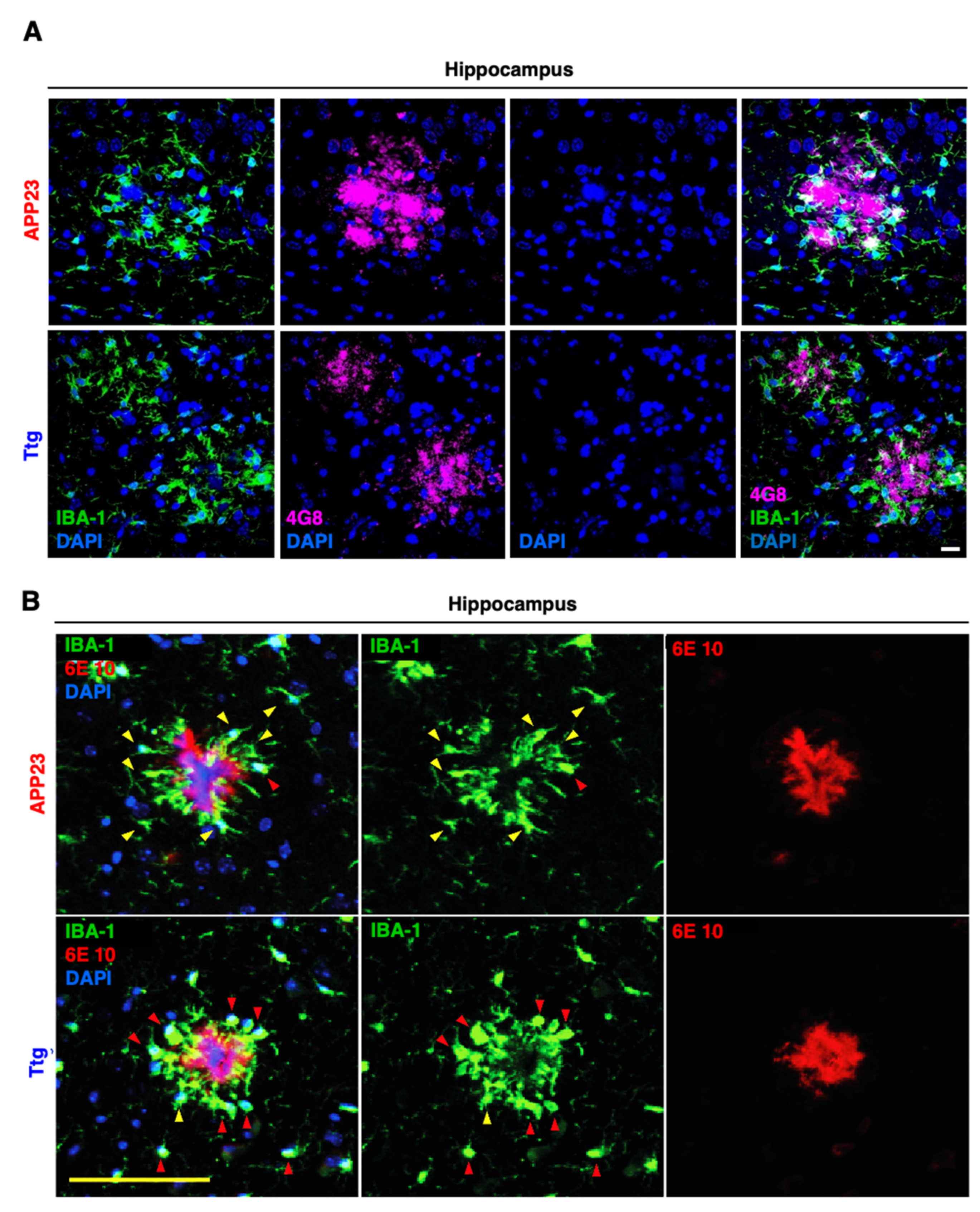
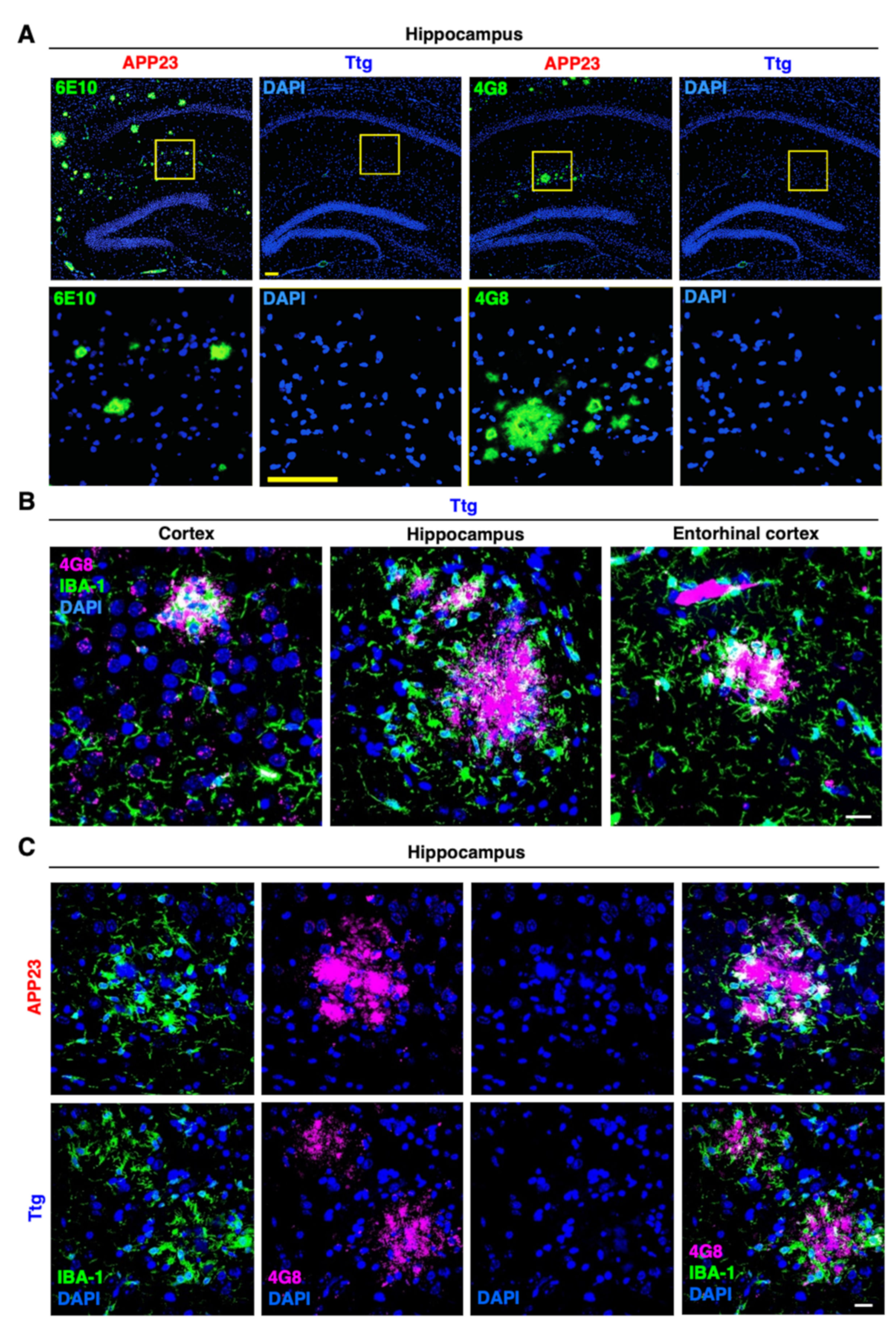
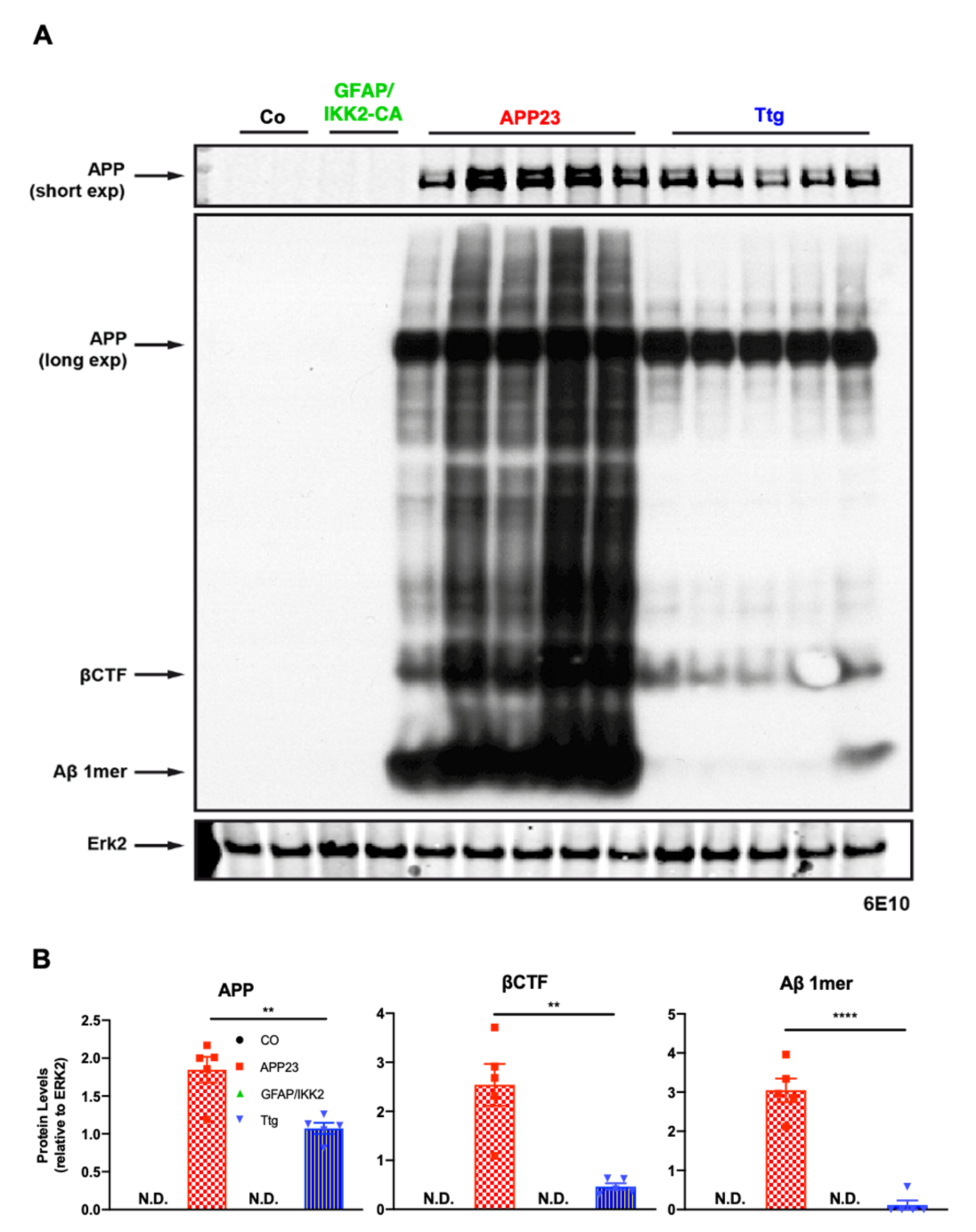
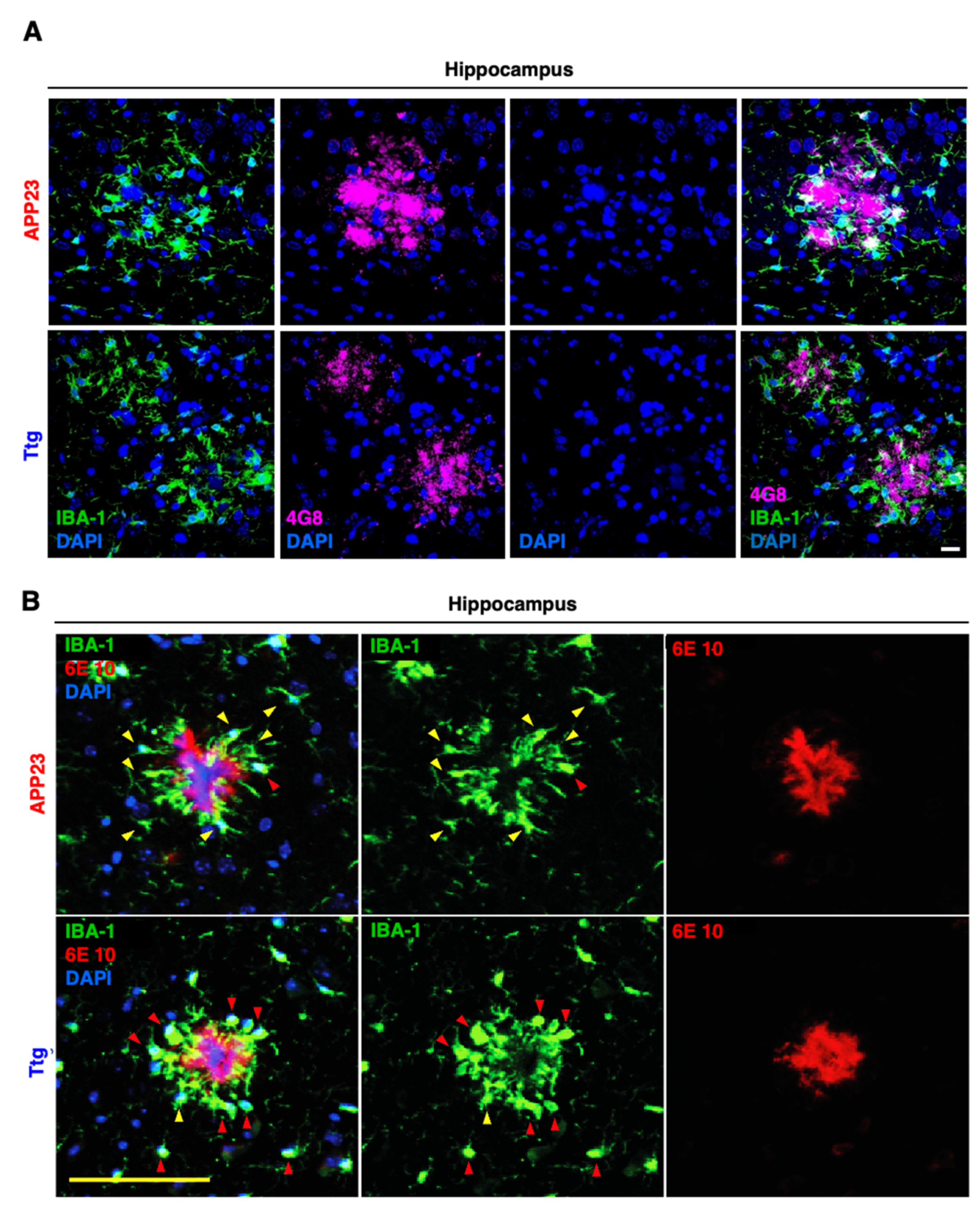
3.2. Astrocyte-Driven Neuroinflammation in APP23 Mice Reduces Plaque Number and Ameliorates Amyloid Plaque Burden
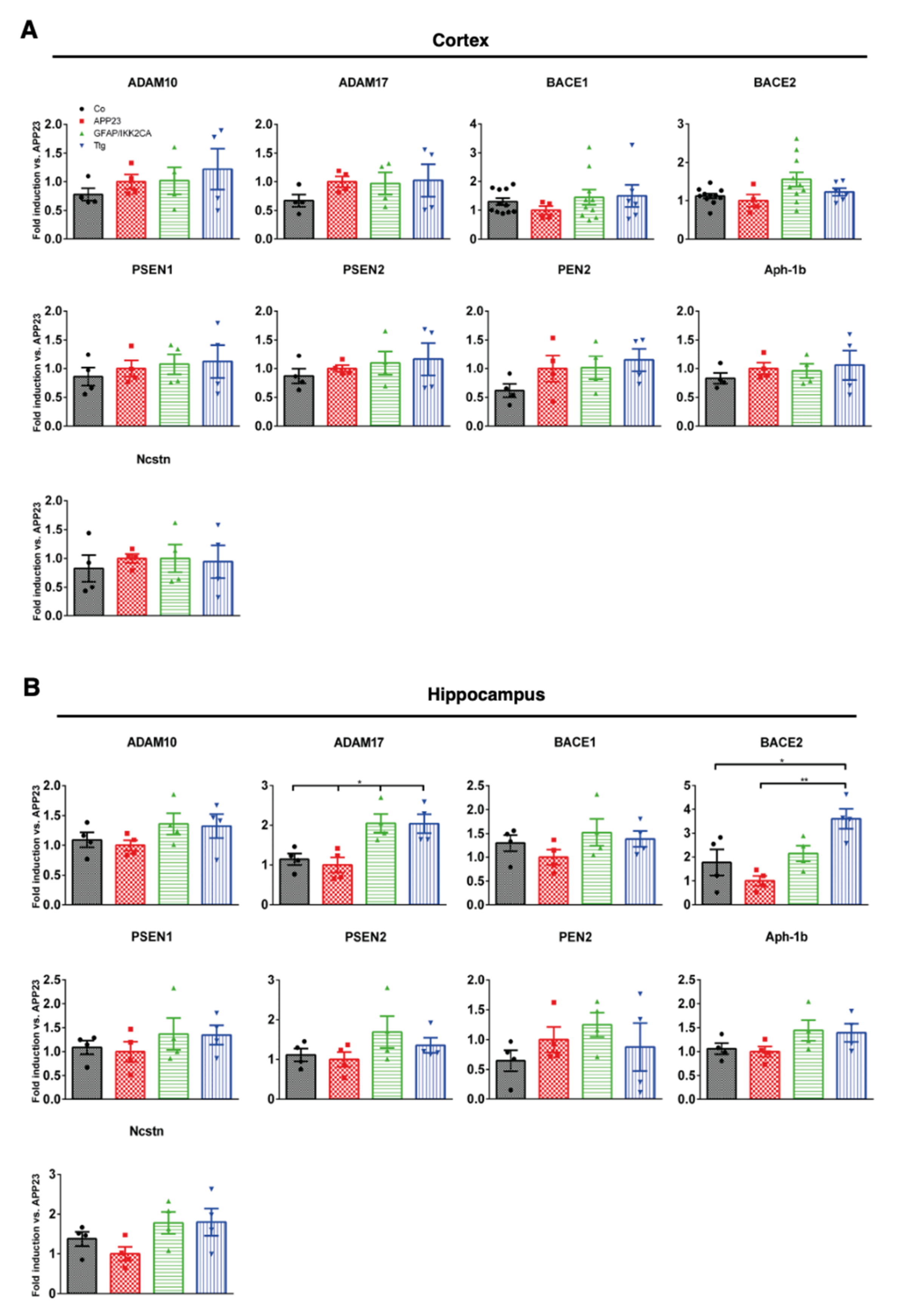
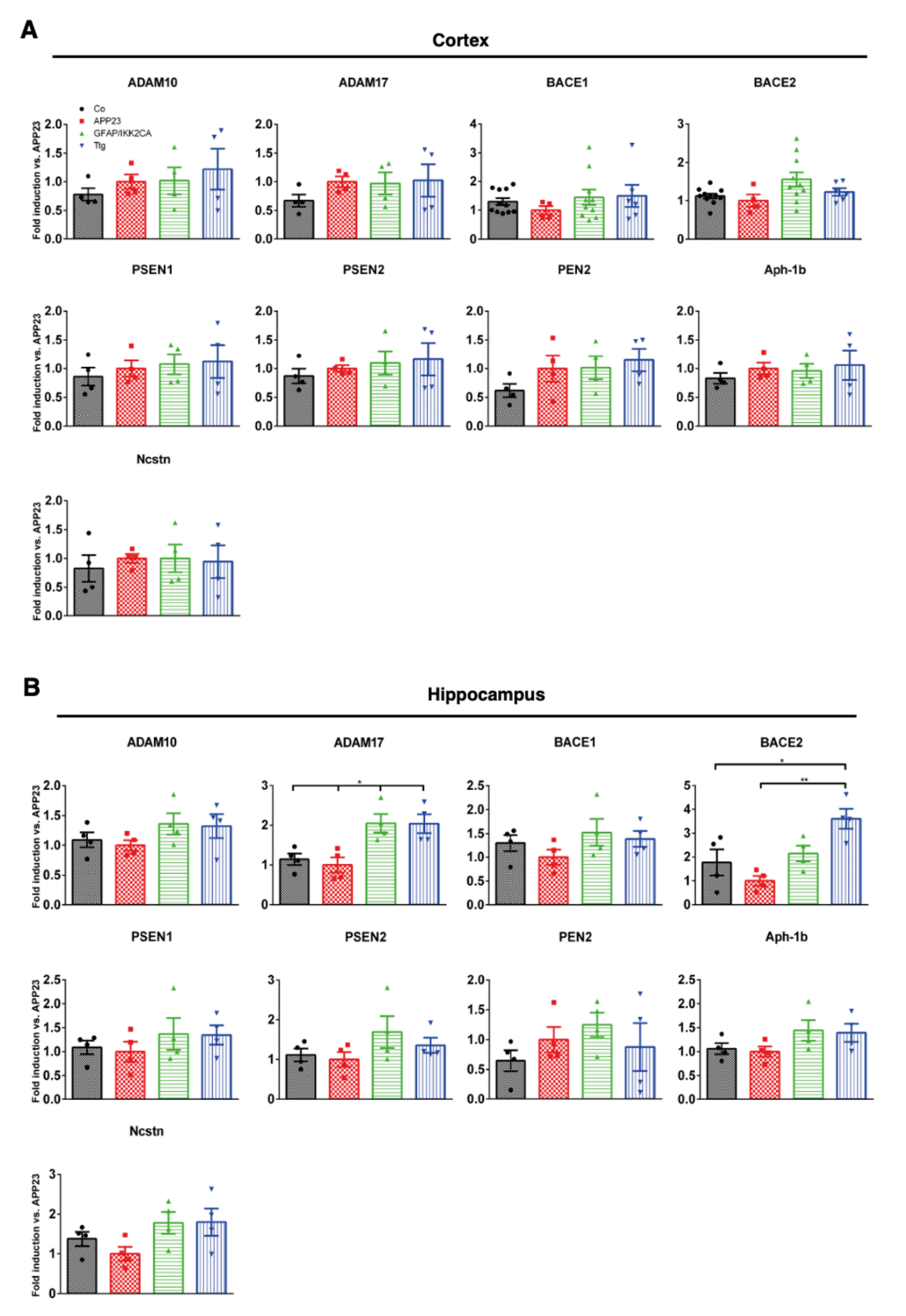
3.3. Expression of Enzymes Involved in APP Processing and Proteolytic Aβ Degradation Is Not Prominently Altered by Astrocytic IKK2/NF-κB Activation
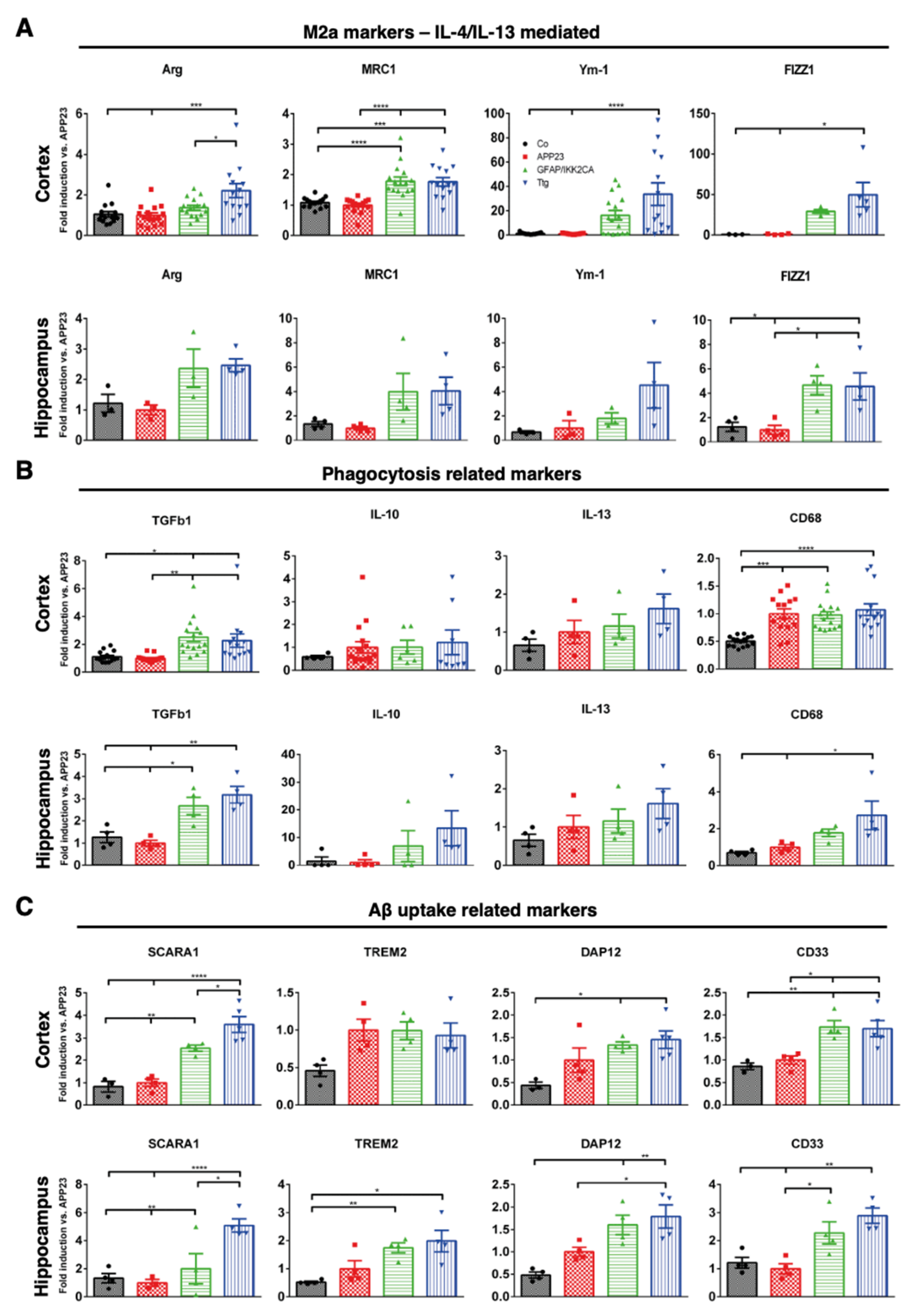
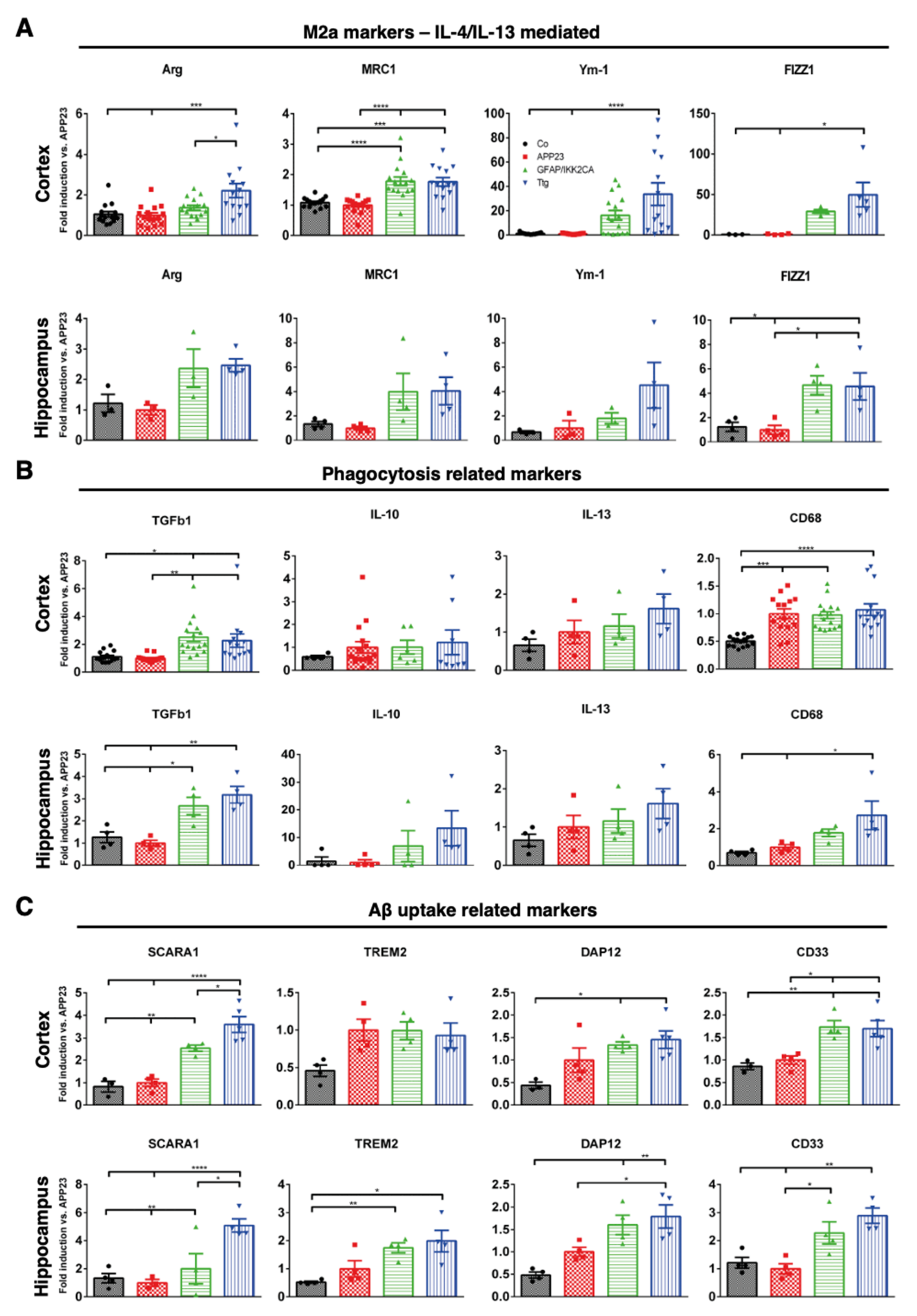
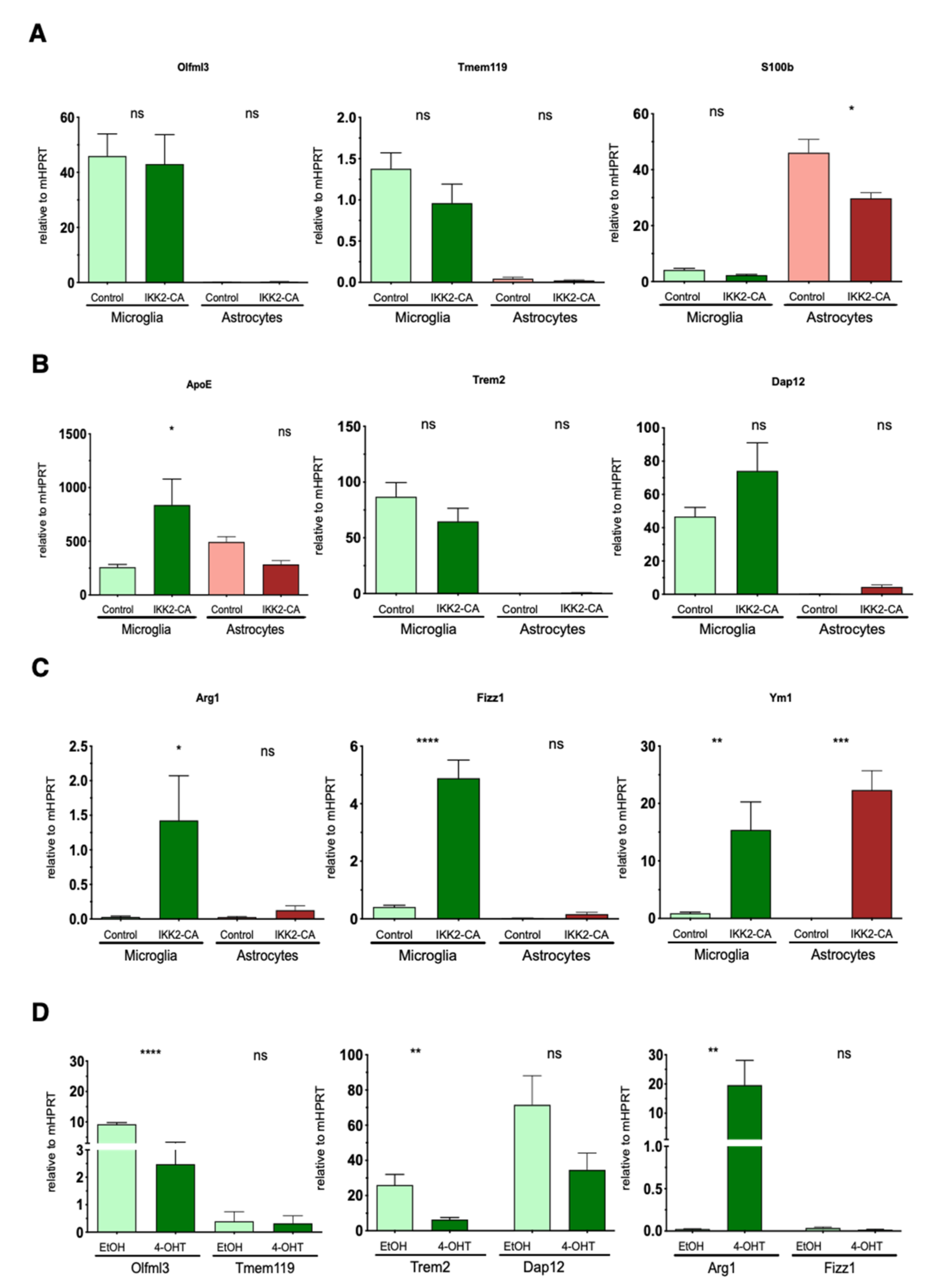
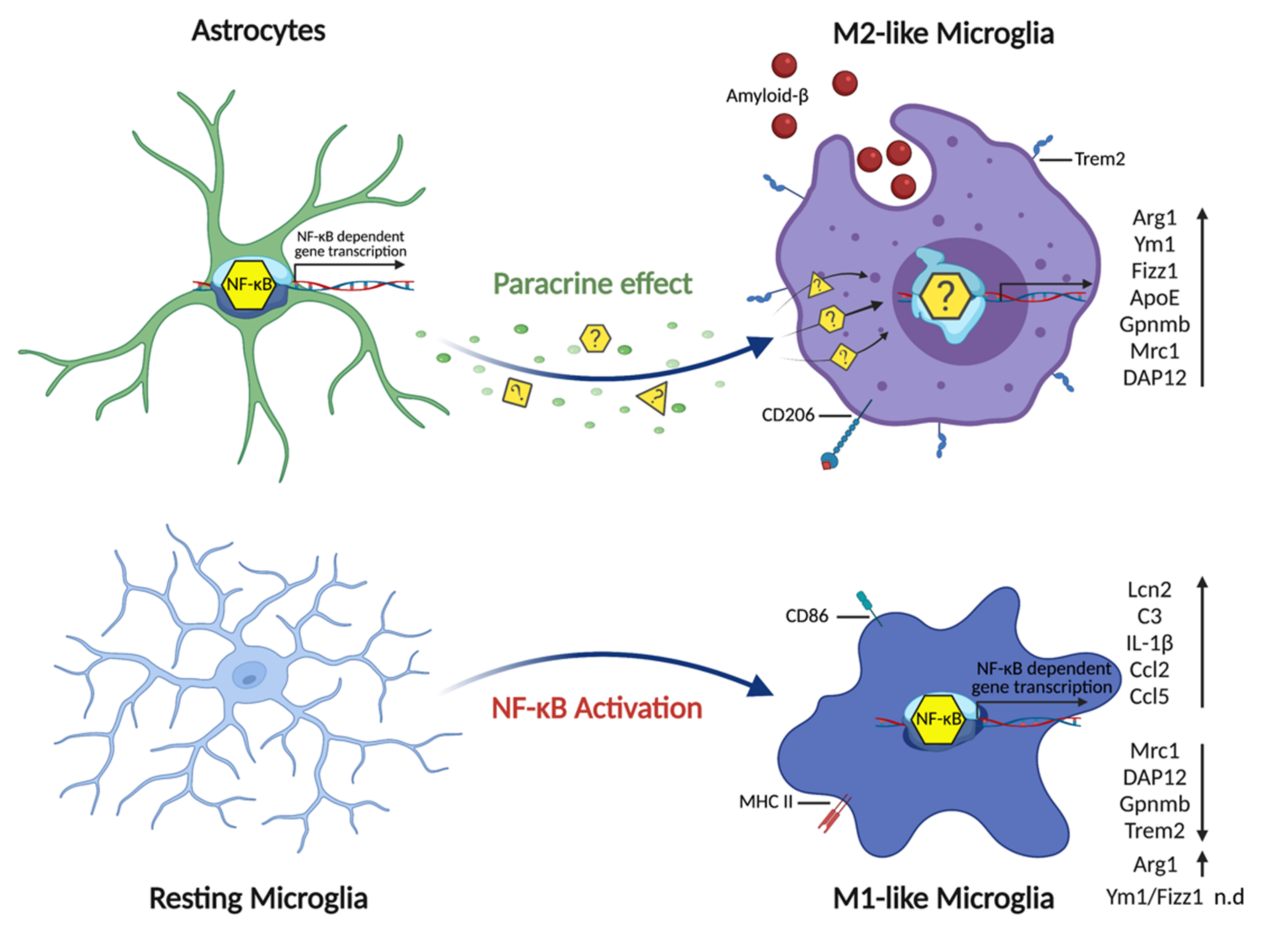
3.4. Astrocytic NF-κB Activation Promotes Microglia Polarization toward a Phagocytic-Proficient Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.C.; Kinghorn, K.J.; Woodling, N.S. Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural. Regen. Res. 2020, 15, 1208–1219. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltschmidt, C.; Greiner, J.F.W.; Kaltschmidt, B. The Transcription Factor NF-kappaB in Stem Cells and Development. Cells 2021, 10, 2042. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Pra, I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, M.R.; Cole, G.M.; Ard, M.D. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol. Aging 2004, 25, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Franco-Bocanegra, D.K.; George, B.; Lau, L.C.; Holmes, C.; Nicoll, J.A.R.; Boche, D. Microglial motility in Alzheimer’s disease and after Abeta42 immunotherapy: A human post-mortem study. Acta Neuropathol. Commun. 2019, 7, 174. [Google Scholar] [CrossRef]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Bocanegra, D.K.; Gourari, Y.; McAuley, C.; Chatelet, D.S.; Johnston, D.A.; Nicoll, J.A.R.; Boche, D. Microglial morphology in Alzheimer’s disease and after Abeta immunotherapy. Sci. Rep. 2021, 11, 15955. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
- Zotova, E.; Holmes, C.; Johnston, D.; Neal, J.W.; Nicoll, J.A.; Boche, D. Microglial alterations in human Alzheimer’s disease following Abeta42 immunization. Neuropathol. Appl. Neurobiol. 2011, 37, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattke, M.; Magnutzki, A.; Walther, P.; Wirth, T.; Baumann, B. Nuclear factor kappaB activation impairs ependymal ciliogenesis and links neuroinflammation to hydrocephalus formation. J. Neurosci. 2012, 32, 11511–11523. [Google Scholar] [CrossRef] [Green Version]
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-kappaB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018, 37, e98697. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Burki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Oeckl, P.; Lattke, M.; Wirth, T.; Baumann, B.; Ferger, B. Astrocyte-specific IKK2 activation in mice is sufficient to induce neuroinflammation but does not increase susceptibility to MPTP. Neurobiol. Dis. 2012, 48, 481–487. [Google Scholar] [CrossRef]
- Baumann, B.; Wagner, M.; Aleksic, T.; von Wichert, G.; Weber, C.K.; Adler, G.; Wirth, T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J. Clin. Invest. 2007, 117, 1502–1513. [Google Scholar] [CrossRef]
- Hipp, N.I.; Christner, L.; Wirth, T.; Mueller-Klieser, W.; Walenta, S.; Schrock, E.; Debatin, K.M.; Beltinger, C. MYCN and survivin cooperatively contribute to malignant transformation of fibroblasts. Carcinogenesis 2014, 35, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, A.; Lattke, M.; Wirth, T.; Baumann, B. Sustained, neuron-specific IKK/NF-kappaB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener. 2013, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]
- Lattke, M.; Reichel, S.N.; Magnutzki, A.; Abaei, A.; Rasche, V.; Walther, P.; Calado, D.P.; Ferger, B.; Wirth, T.; Baumann, B. Transient IKK2 activation in astrocytes initiates selective non-cell-autonomous neurodegeneration. Mol. Neurodegener. 2017, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Lee, S.; Park, D.H.; Kook, H.; Park, K.G.; Lee, I.K.; Suk, K. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav. Rev. 2015, 49, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci. 2009, 39, 99–103. [Google Scholar] [CrossRef]
- Reichert, F.; Rotshenker, S. Galectin-3 (MAC-2) Controls Microglia Phenotype Whether Amoeboid and Phagocytic or Branched and Non-phagocytic by Regulating the Cytoskeleton. Front. Cell Neurosci. 2019, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.S.; Grishaev, A.; Bax, A. Monomeric alpha-synuclein binds Congo Red micelles in a disordered manner. Biochemistry 2012, 51, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Tripoli, D.L.; Czerniewski, L.; Ballabio, A.; et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J. Neurosci. 2015, 35, 12137–12151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, N.K.; Jha, S.K.; Kumar, D.; Kejriwal, N.; Sharma, R.; Ambasta, R.K.; Kumar, P. Impact of Insulin Degrading Enzyme and Neprilysin in Alzheimer’s Disease Biology: Characterization of Putative Cognates for Therapeutic Applications. J. Alzheimers Dis. 2015, 48, 891–917. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar] [CrossRef]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, D.; Wilkinson, K.; Zhao, L.; Hickman, S.E.; Means, T.K.; Puckett, L.; Farfara, D.; Kingery, N.D.; Weiner, H.L.; El Khoury, J. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 2013, 4, 2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenrauch, M.; Ogorek, I.; Klafki, H.; Otto, M.; Stadelmann, C.; Weggen, S.; Wiltfang, J.; Wirths, O. Glycoprotein NMB: A novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Checler, F.; Afram, E.; Pardossi-Piquard, R.; Lauritzen, I. Is gamma-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99? J. Biol. Chem. 2021, 296, 100489. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Alcolea, D.; Sanchez-Valle, R.; Guardia-Laguarta, C.; Colom-Cadena, M.; Badiola, N.; Suarez-Calvet, M.; Llado, A.; Barrera-Ocampo, A.A.; Sepulveda-Falla, D.; et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013, 125, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Friesen, M.; Meyer-Luehmann, M. Abeta Seeding as a Tool to Study Cerebral Amyloidosis and Associated Pathology. Front. Mol. Neurosci. 2019, 12, 233. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, M.; Shen, X.; Wang, H. The Distinct Role of ADAM17 in APP Proteolysis and Microglial Activation Related to Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 471–482. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a conditional beta-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight 2019, 4, e123431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis: A Focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef]
- Wildsmith, K.R.; Holley, M.; Savage, J.C.; Skerrett, R.; Landreth, G.E. Evidence for impaired amyloid beta clearance in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, G.M.; Tarasov, V.V.; Makhmutova, A.; Chubarev, V.N.; Avila-Rodriguez, M.; Bachurin, S.O.; Aliev, G. The Possibility of an Infectious Etiology of Alzheimer Disease. Mol. Neurobiol. 2019, 56, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Sait, A.; Angeli, C.; Doig, A.J.; Day, P.J.R. Viral Involvement in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1049–1060. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microglia | In Vivo | Ex Vivo | ||||
|---|---|---|---|---|---|---|
| Gene Name | Mean Gene Expression | Significance | Mean Gene Expression | Significance | ||
| (a) Polarization-associated | Control (Co) | IKK2-CA | EtOH | 4-OHT | ||
| Cd14 | 4.859 | 7.054 | ns | 37.68 | 80.44 | * |
| Cd86 | 11.22 | 13.46 | ns | 4.213 | 1.589 | ** |
| Cd163 | 0.9658 | 0.3654 | ** | 0.02 | 0.0866 | * |
| Inos | 0.06810 | 0.05243 | ns | na | na | na |
| Mrc1 | 1.075 | 2.323 | ns | 8.459 | 0.647 | * |
| Ym1 | 0.9075 | 15.39 | ** | nd | nd | nd |
| (b) Inflammation-associated | Control (Co) | IKK2-CA | EtOH | 4-OHT | ||
| C3 | 2.893 | 29.29 | *** | 2.15 | 155.26 | ** |
| Ccl2 | 1.287 | 3.525 | ** | 0.6 | 3.32 | * |
| Ccl5 | 0.1399 | 0.867 | * | 0.05 | 5.19 | ** |
| Ccl6 | 17.96 | 36.8633 | ns | 44.95 | 5.992 | **** |
| Gpnmb | 0.1434 | 0.8237 | ** | 3.436 | 1.156 | ns |
| Gbp2 | 0.6915 | 3.618 | *** | 0.09 | 3.28 | *** |
| Il1b | 7.363 | 26.35 | * | 1.22 | 92.112 | * |
| Lcn2 | 4.681 | 1.710 | * | 0.06 | 262.59 | * |
| Spp1 | 6.622 | 11.75 | ns | 111.4 | 27.02 | ** |
| Tgfb | 73.22 | 67.22 | ns | 53.25 | 52.49 | ns |
| Tgm1 | 0.1797 | 0.5212 | ** | 0.073 | 2.808 | ** |
| Tnfa | 1.22 | 3.037 | * | 2.134 | 9.581 | *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Magnutzki, A.; Alami, N.O.; Lattke, M.; Hein, T.M.; Scheller, J.S.; Kröger, C.; Oswald, F.; Yilmazer-Hanke, D.; Wirth, T.; et al. IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization. Cells 2021, 10, 2669. https://doi.org/10.3390/cells10102669
Yang S, Magnutzki A, Alami NO, Lattke M, Hein TM, Scheller JS, Kröger C, Oswald F, Yilmazer-Hanke D, Wirth T, et al. IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization. Cells. 2021; 10(10):2669. https://doi.org/10.3390/cells10102669
Chicago/Turabian StyleYang, Shu, Alexander Magnutzki, Najwa Ouali Alami, Michael Lattke, Tabea Melissa Hein, Judith Stefanie Scheller, Carsten Kröger, Franz Oswald, Deniz Yilmazer-Hanke, Thomas Wirth, and et al. 2021. "IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization" Cells 10, no. 10: 2669. https://doi.org/10.3390/cells10102669
APA StyleYang, S., Magnutzki, A., Alami, N. O., Lattke, M., Hein, T. M., Scheller, J. S., Kröger, C., Oswald, F., Yilmazer-Hanke, D., Wirth, T., & Baumann, B. (2021). IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization. Cells, 10(10), 2669. https://doi.org/10.3390/cells10102669