
Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
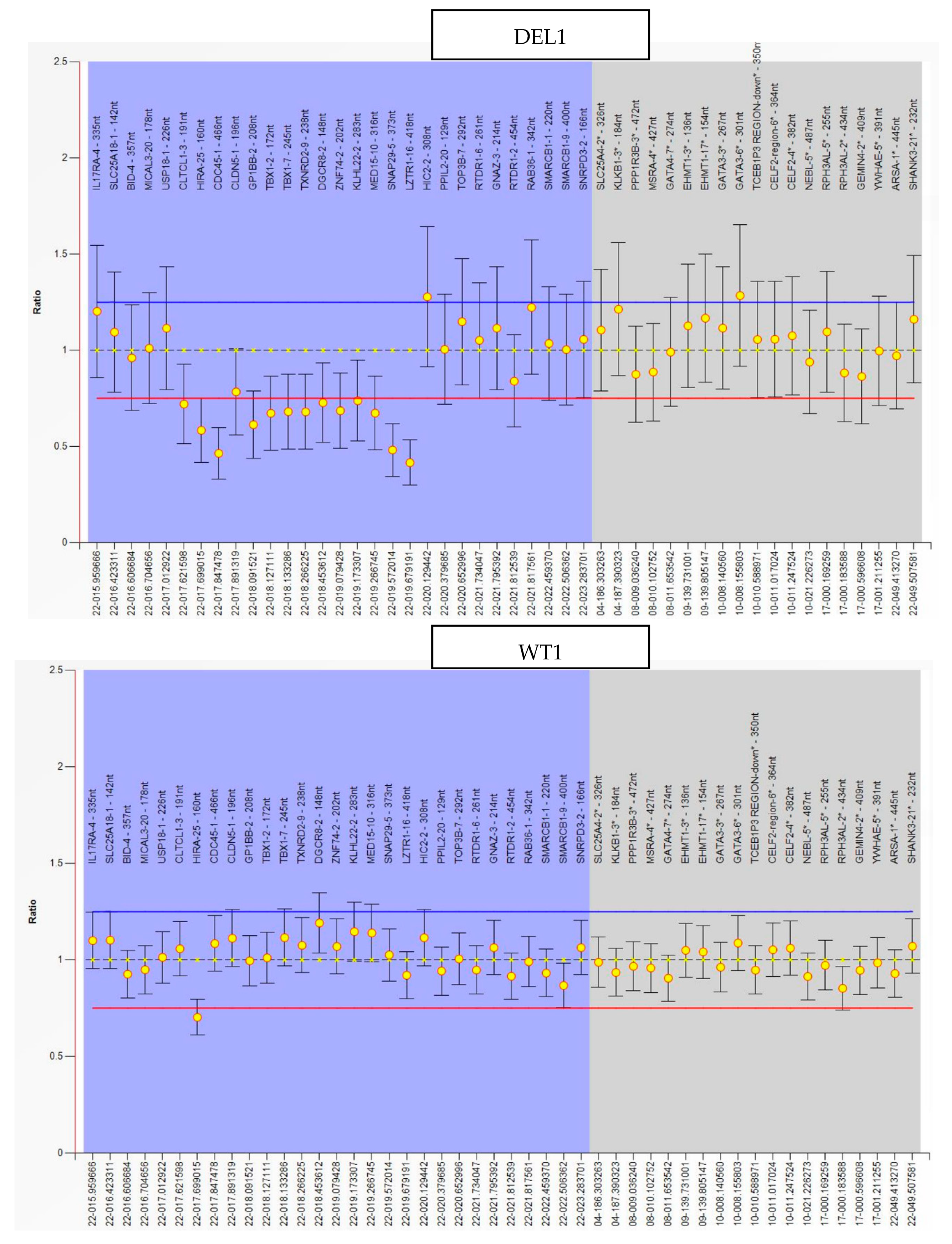
2.1. hiPSC Generation and Characterization
2.2. HBMEC Differentiation
2.3. Generation of Induced BBB (iBBB) and Trans-Endothelial Electrical Resistance (TEER) Measurement
2.4. Measurement of Solute Permeability (P)
2.5. Immunostaining of Tight Junction Proteins and the Endothelial Surface Glycocalyx (ESG)
2.6. Confocal Microscopy and Quantification of Tight Junction Proteins and the ESG
2.7. Total RNA Isolation and Bulk RNA Sequencing
2.8. Differential Expression Analysis
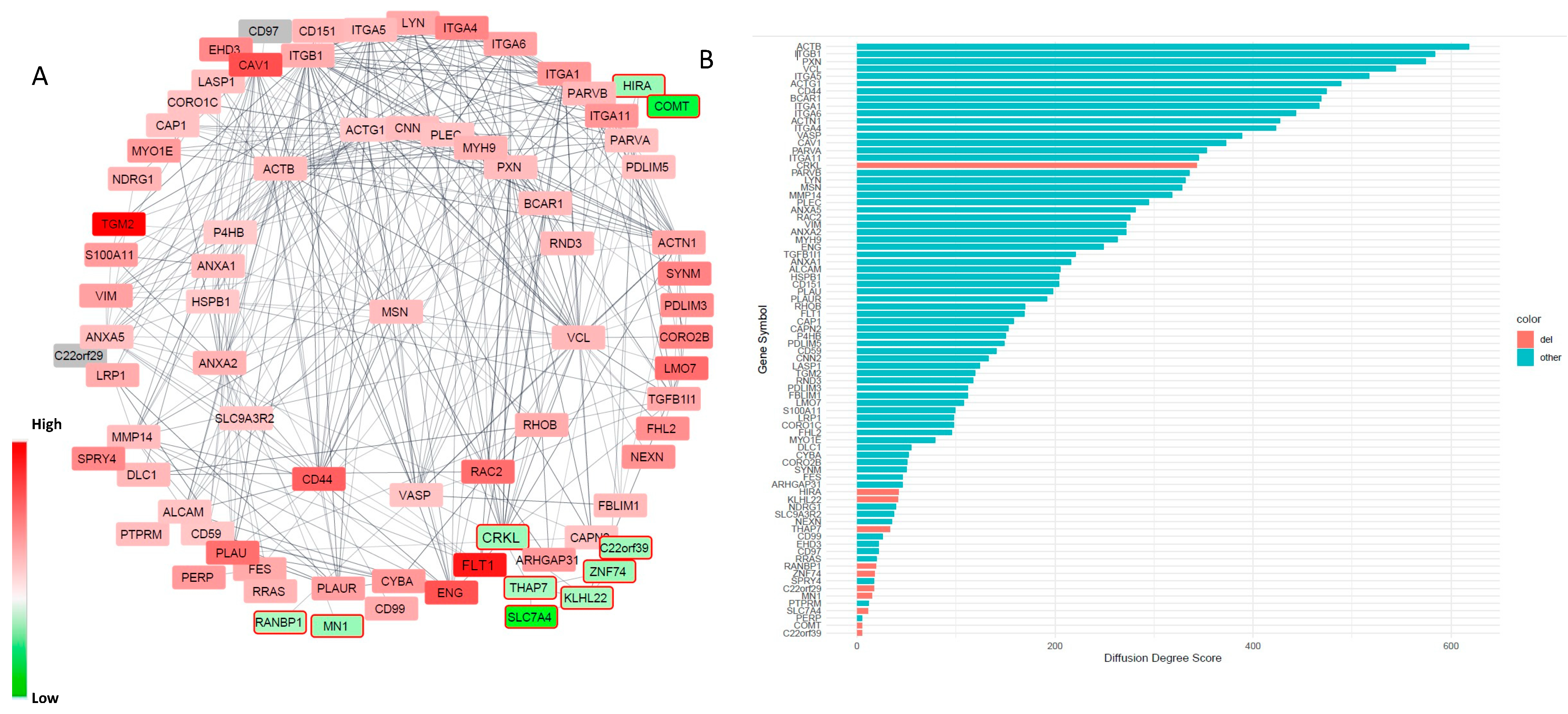
2.9. Gene Ontology and Protein–Protein Interaction Network Analysis
2.10. Topology Analysis of the Node Contribution to the PPI Network
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Blood–Brain Barrier Function is Impaired in the 22q11.2DS-SCZ iBBB
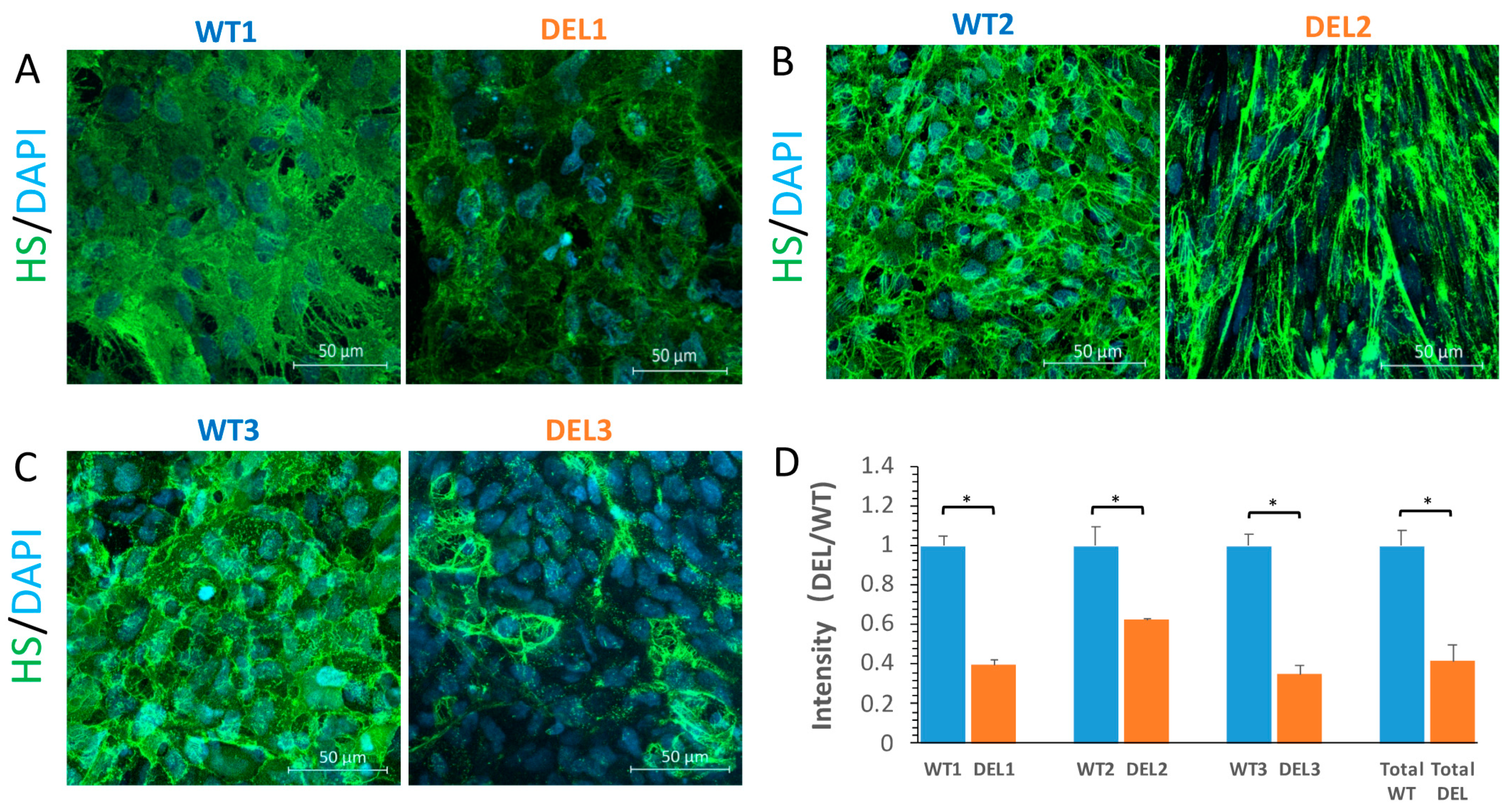
3.2. Tight Junction Proteins and the Endothelial Surface Glycocalyx (ESG) are Disrupted in 22q11.2DS-SCZ iBBB
3.3. Alterations in Focal Adhesion and Vascular Endothelial Growth Factor (VEGF)-Associated Junction Signaling Pathways in the 22q11.2DS-SCZ iBBB
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








References
- Scambler, P.J. The 22q11 deletion syndromes. Hum. Mol. Genet. 2000, 9, 2421–2426. [Google Scholar] [CrossRef]
- Pulver, A.E.; Nestadt, G.; Goldberg, R.; Shprintzen, R.J.; Lamacz, M.; Wolyniec, P.S.; Morrow, B.; Karayiorgou, M.; Antonarakis, S.E.; Housman, D. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 1994, 182, 476–478. [Google Scholar] [CrossRef]
- Tang, S.; Yi, J.; Calkins, M.; Whinna, D.; Kohler, C.; Souders, M.; McDonald-McGinn, D.; Zackai, E.; Emanuel, B.; Gur, R. Psychiatric disorders in 22q11. 2 deletion syndrome are prevalent but undertreated. Psychol. Med. 2014, 44, 1267. [Google Scholar] [CrossRef] [PubMed]
- Shalev, H.; Serlin, Y.; Friedman, A. Breaching the blood-brain barrier as a gate to psychiatric disorder. Cardiovasc. Psychiatry Neurol. 2009, 2009, 278531. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.A.; Zimina, I.S.; Vikhreva, O.V.; Krukov, N.O.; Rachmanova, V.I.; Orlovskaya, D.D. Ultrastructural damage of capillaries in the neocortex in schizophrenia. World J. Biol. Psychiatry 2010, 11, 567–578. [Google Scholar] [CrossRef]
- Uranova, N.; Zimina, I.; Vikhreva, O.; Rachmanova, V.; Klintsova, A.; Orlovskaya, D. Reduced capillary density in the prefrontal cortex in schizophrenia. Am. J. Med. Sci. Med. 2013, 1, 45–51. [Google Scholar] [CrossRef][Green Version]
- Vostrikov, V.; Orlovskaya, D.; Uranova, N. Deficit of pericapillary oligodendrocytes in the prefrontal cortex in schizophrenia. World J. Biol. Psychiatry 2008, 9, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Azmitia, E.; Saccomano, Z.; Alzoobaee, M.; Boldrini, M.; Whitaker-Azmitia, P. Persistent angiogenesis in the autism brain: An immunocytochemical study of postmortem cortex, brainstem and cerebellum. J. Autism Dev. Disord. 2016, 46, 1307–1318. [Google Scholar] [CrossRef]
- Enwright, J.F., III; Huo, Z.; Arion, D.; Corradi, J.P.; Tseng, G.; Lewis, D.A. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol. Psychiatry 2018, 23, 1606–1613. [Google Scholar] [CrossRef]
- Cioffi, S.; Martucciello, S.; Fulcoli, F.G.; Bilio, M.; Ferrentino, R.; Nusco, E.; Illingworth, E. Tbx1 regulates brain vascularization. Hum. Mol. Genet. 2014, 23, 78–89. [Google Scholar] [CrossRef]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef]
- Nishiura, K.; Ichikawa-Tomikawa, N.; Sugimoto, K.; Kunii, Y.; Kashiwagi, K.; Tanaka, M.; Yokoyama, Y.; Hino, M.; Sugino, T.; Yabe, H. PKA activation and endothelial claudin-5 breakdown in the schizophrenic prefrontal cortex. Oncotarget 2017, 8, 93382. [Google Scholar] [CrossRef]
- Fu, B.M. Transport Across the Blood-Brain Barrier. Adv. Exp. Med. Biol. 2018, 1097, 235–259. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Saint-Laurent, O.; Godschalk, A.; Terouz, S.; Briels, C.; Larouche, S.; Bourbonnière, L.; Larochelle, C.; Prat, A. Focal disturbances in the blood–brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol. Dis. 2015, 74, 14–24. [Google Scholar] [CrossRef]
- Muldoon, L.L.; Alvarez, J.I.; Begley, D.J.; Boado, R.J.; Del Zoppo, G.J.; Doolittle, N.D.; Engelhardt, B.; Hallenbeck, J.M.; Lonser, R.R.; Ohlfest, J.R. Immunologic privilege in the central nervous system and the blood–brain barrier. J. Cereb. Blood Flow Metab. 2013, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.A.; Fan, J.; Azar, T.T.; Yen, W.; Zeng, M.; Chen, J.; Ratliff, B.B.; Song, J.; Tarbell, J.M.; Goligorsky, M.S. Exocytosis of endothelial lysosome-related organelles hair-triggers a patchy loss of glycocalyx at the onset of sepsis. Am. J. Pathol. 2016, 186, 248–258. [Google Scholar] [CrossRef]
- Ueno, M.; Sakamoto, H.; Liao, Y.-J.; Onodera, M.; Huang, C.-L.; Miyanaka, H.; Nakagawa, T. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem. Cell Biol. 2004, 122, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.M.; Tarbell, J.M. Mechano-sensing and transduction by endothelial surface glycocalyx: Composition, structure, and function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Haeren, R.; van de Ven, S.; van Zandvoort, M.; Vink, H.; van Overbeeke, J.J.; Hoogland, G.; Rijkers, K. Assessment and Imaging of the Cerebrovascular Glycocalyx. Curr. Neurovasc. Res. 2016, 13, 249–260. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Lee, E.-S.; Jeong, Y. In vivo imaging of the cerebral endothelial glycocalyx in mice. J. Vasc. Res. 2017, 54, 59–67. [Google Scholar] [CrossRef]
- Abbott, K.L.; Pierce, J.M. Lectin-based glycoproteomic techniques for the enrichment and identification of potential biomarkers. Methods Enzymol. 2010, 480, 461–476. [Google Scholar] [PubMed]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Coisne, C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 2011, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, W.; Fu, B.M. A model for the blood–brain barrier permeability to water and small solutes. J. Biomech. 2010, 43, 2133–2140. [Google Scholar] [CrossRef]
- Yuan, W.; Li, G.; Fu, B.M. Effect of surface charge of immortalized mouse cerebral endothelial cell monolayer on transport of charged solutes. Ann. Biomed. Eng. 2010, 38, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.W.; Fan, J.; Luu, E.; Khalid, W.; Xia, Y.; Khadka, N.; Bikson, M.; Fu, B.M. In Vivo Modulation of the Blood–Brain Barrier Permeability by Transcranial Direct Current Stimulation (tDCS). Ann. Biomed. Eng. 2020, 48, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef]
- Stebbins, M.J.; Wilson, H.K.; Canfield, S.G.; Qian, T.; Palecek, S.P.; Shusta, E.V. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods 2016, 101, 93–102. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Human blood-brain barrier endothelial cells derived from pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11. 2 deletion. BMC Syst. Biol. 2016, 10, 105. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Liang, J.; Qu, B.; Suganthan, P.; Hernández-Díaz, A.G. Problem definitions and evaluation criteria for the CEC 2013 special session on real-parameter optimization. Comput. Intell. Lab. Zhengzhou Univ. Zhengzhou China Nanyang Technol. Univ. Singap. Tech. Rep. 2013, 201212, 281–295. [Google Scholar]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I. Endothelial LRP1 transports amyloid-β 1–42 across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, B.M. Quantification of malignant breast cancer cell MDA-MB-231 transmigration across brain and lung microvascular endothelium. Ann. Biomed. Eng. 2016, 44, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Sun, Y.; Xia, Y.; Tarbell, J.M.; Fu, B.M. Endothelial surface glycocalyx (ESG) components and ultra-structure revealed by stochastic optical reconstruction microscopy (STORM). Biorheology 2019, 56, 77–88. [Google Scholar] [CrossRef]
- Fu, B.M.; Yang, J.; Cai, B.; Fan, J.; Zhang, L.; Zeng, M. Reinforcing endothelial junctions prevents microvessel permeability increase and tumor cell adhesion in microvessels in vivo. Sci. Rep. 2015, 5, 15697. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Chislock, E.M.; Pendergast, A.M. Abl Family Kinases Regulate Endothelial Barrier Function In Vitro and in Mice. PLoS ONE 2013, 8, e85231. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Stalmans, I.; Lambrechts, D.; De smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 2003, 9, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Chen, T.-C.; Chau, G.-Y.; Jan, Y.-H.; Chen, C.-H.; Hsu, C.-N.; Lin, K.-T.; Juang, Y.-L.; Lu, P.-J.; Cheng, H.-C. Analysis of protein-protein interactions in cross-talk pathways reveals CRKL protein as a novel prognostic marker in hepatocellular carcinoma. Mol. Cell. Proteom. 2013, 12, 1335–1349. [Google Scholar] [CrossRef]
- Takeda, N.; Maemura, K.; Imai, Y.; Harada, T.; Kawanami, D.; Nojiri, T.; Manabe, I.; Nagai, R. Endothelial PAS domain protein 1 gene promotes angiogenesis through the transactivation of both vascular endothelial growth factor and its receptor, Flt-1. Circ. Res. 2004, 95, 146–153. [Google Scholar] [CrossRef]
- Chen, Y.; Takikawa, M.; Tsutsumi, S.; Yamaguchi, Y.; Okabe, A.; Shimada, M.; Kawase, T.; Sada, A.; Ezawa, I.; Takano, Y.; et al. PHLDA1, another PHLDA family protein that inhibits Akt. Cancer Sci. 2018, 109, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Bellou, S.; Hink, M.A.; Bagli, E.; Panopoulou, E.; Bastiaens, P.I.H.; Murphy, C.; Fotsis, T. VEGF autoregulates its proliferative and migratory ERK1/2 and p38 cascades by enhancing the expression of DUSP1 and DUSP5 phosphatases in endothelial cells. Am. J. Physiol-Cell Physiol. 2009, 297, C1477–C1489. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Tian, F.; Zhang, L.; Xue, C.; Linhardt, R.J. Glycosaminoglycanomics of cultured cells using a rapid and sensitive LC-MS/MS approach. ACS Chem. Biol. 2015, 10, 1303–1310. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zeng, M.; Sun, Y.; Fu, B.M. Quantification of blood-brain barrier solute permeability and brain transport by multiphoton microscopy. J. Biomech. Eng. 2014, 136, 031005. [Google Scholar] [CrossRef] [PubMed]
- Linville, R.M.; DeStefano, J.G.; Sklar, M.B.; Xu, Z.; Farrell, A.M.; Bogorad, M.I.; Chu, C.; Walczak, P.; Cheng, L.; Mahairaki, V. Human iPSC-derived blood-brain barrier microvessels: Validation of barrier function and endothelial cell behavior. Biomaterials 2019, 190, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood–brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Sato, S.; Yamaguchi, H.; Kamoi, M.; Asashima, T.; Terasaki, T. Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J. Cell. Physiol. 2007, 210, 81–86. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Yamaguchi, H.; Katsukura, Y.; Asashima, T.; Terasaki, T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J. Neurochem. 2008, 104, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Arinami, T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J. Hum. Genet. 2006, 51, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Kealy, J.; Humphries, M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.; Martiniano, R.; Shashi, V.; Hooper, S. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Crockett, A.M.; Ryan, S.K.; Vasquez, A.H.; Canning, C.; Kanyuch, N.; Kebir, H.; Ceja, G.; Gesualdi, J.; Viaene, A.; Kapoor, R. Disruption of the Blood-Brain Barrier in 22q11. 2 Deletion Syndrome. Brain 2021, 144, 1351–1360. [Google Scholar]
- Lu, T.M.; Houghton, S.; Magdeldin, T.; Durán, J.G.B.; Minotti, A.P.; Snead, A.; Sproul, A.; Nguyen, D.-H.T.; Xiang, J.; Fine, H.A.; et al. Pluripotent stem cell-derived epithelium misidentified as brain microvascular endothelium requires ETS factors to acquire vascular fate. Proc. Natl. Acad. Sci. USA 2021, 118, e2016950118. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin β4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng. 2011, 39, 2223–2241. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.R.; Armstrong, L.; Baker, S.; Wong, D.W.; Wylie, E.C.; Ramnath, R.; Jenkins, R.; Singh, A.; Steadman, R.; Welsh, G.I. Glycosaminoglycan regulation by VEGFA and VEGFC of the glomerular microvascular endothelial cell glycocalyx in vitro. Am. J. Pathol. 2013, 183, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Motahari, Z.; Moody, S.A.; Maynard, T.M.; LaMantia, A.S. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? J. Neurodev. Disord. 2019, 11, 7. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Pollak, T.A.; Drndarski, S.; Stone, J.M.; David, A.S.; McGuire, P.; Abbott, N.J. The blood-brain barrier in psychosis. Lancet Psychiatry 2018, 5, 79–92. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Xia, Y.; Zhu, H.; Luu, E.; Huang, G.; Sun, Y.; Sun, K.; Markx, S.; Leong, K.W.; Xu, B.; et al. Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model. Cells 2021, 10, 2576. https://doi.org/10.3390/cells10102576
Li Y, Xia Y, Zhu H, Luu E, Huang G, Sun Y, Sun K, Markx S, Leong KW, Xu B, et al. Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model. Cells. 2021; 10(10):2576. https://doi.org/10.3390/cells10102576
Chicago/Turabian StyleLi, Yunfei, Yifan Xia, Huixiang Zhu, Eric Luu, Guangyao Huang, Yan Sun, Kevin Sun, Sander Markx, Kam W. Leong, Bin Xu, and et al. 2021. "Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model" Cells 10, no. 10: 2576. https://doi.org/10.3390/cells10102576
APA StyleLi, Y., Xia, Y., Zhu, H., Luu, E., Huang, G., Sun, Y., Sun, K., Markx, S., Leong, K. W., Xu, B., & Fu, B. M. (2021). Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model. Cells, 10(10), 2576. https://doi.org/10.3390/cells10102576