Fer and FerT Govern Mitochondrial Susceptibility to Metformin and Hypoxic Stress in Colon and Lung Carcinoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Culture and Metformin Treatment
2.2. Generating SW/HΔFer/FerT and SW/HΔFer Cells
2.3. Subjecting Cells to Hypoxia
2.4. Immunoblot Analysis
2.5. Cell Death Analysis
2.6. Quantification of Cellular ATP and NAD+ Levels
2.7. Determination of Mitochondrial Activity
2.8. Statistical Analysis
3. Results
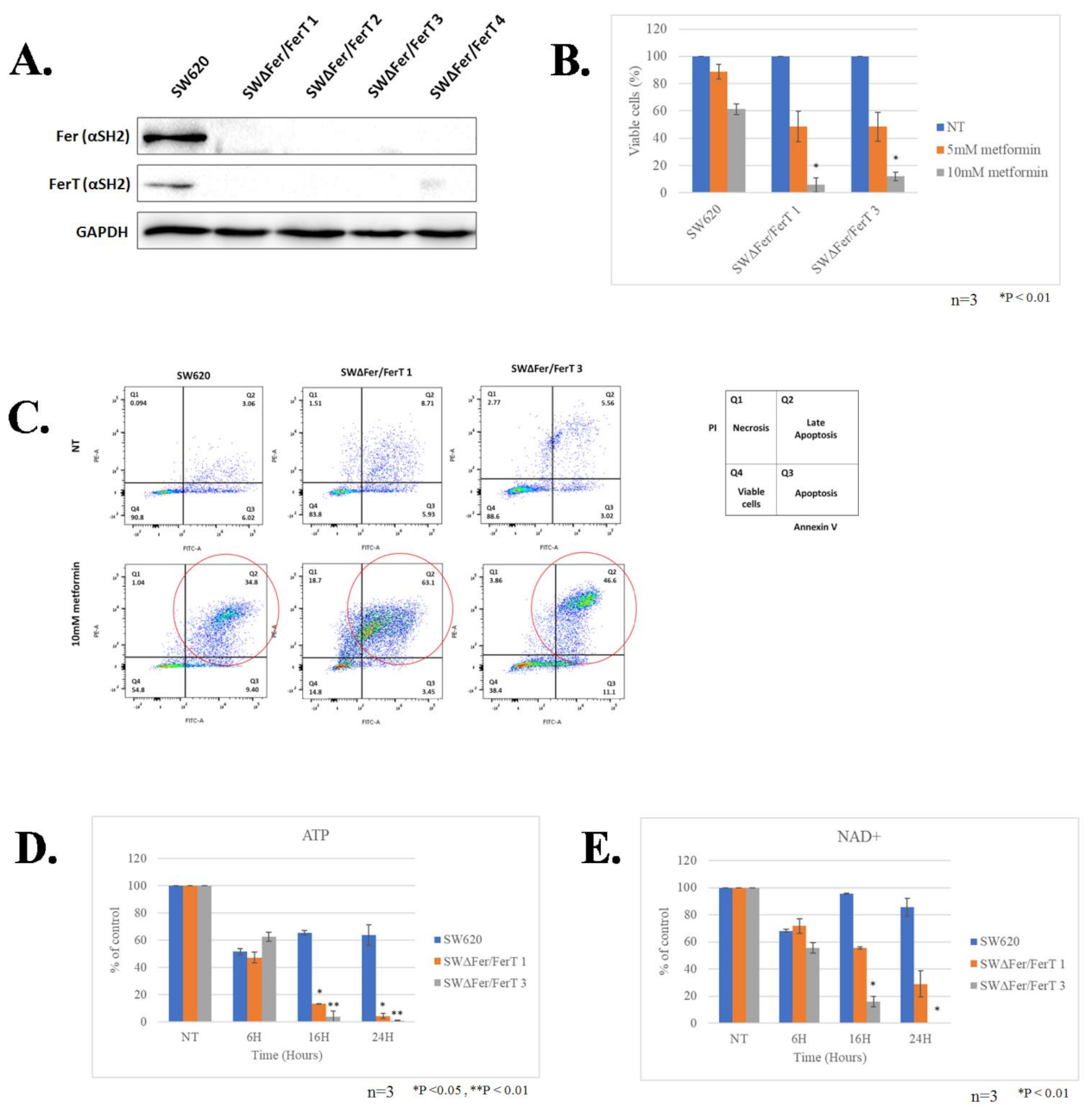
3.1. Fer/FerT Deficiency Exacerbates Susceptibility of SW620 and H1299 Cells to Metformin
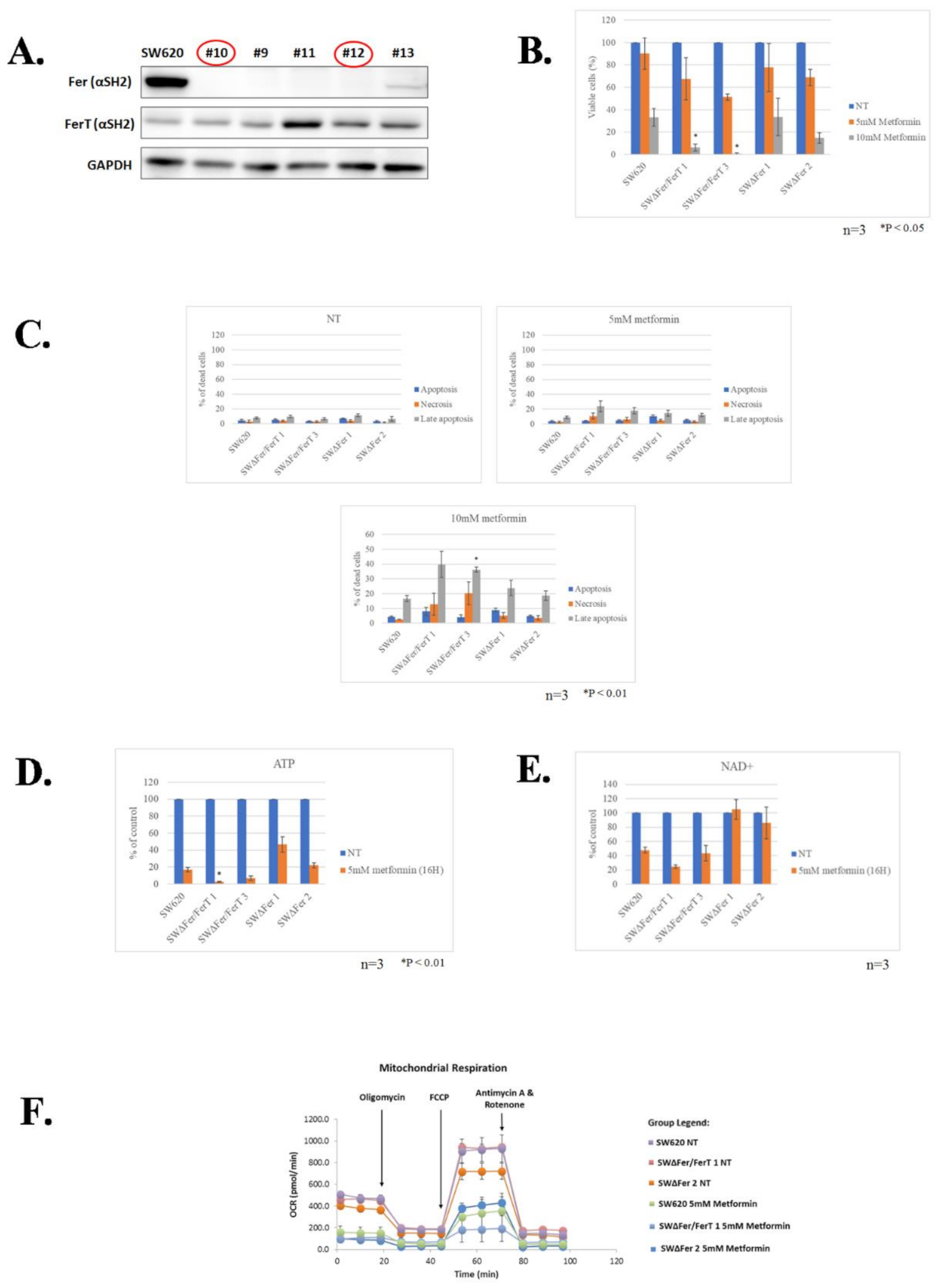
3.2. FerT Governs the Vulnerability of SW620 Cells to Metformin
3.3. FerT Governs The Sensitivity of SW620 Cells to Hypoxic Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Marin de Mas, I.; Zodda, E.; Marin, S.; Morrish, F.; Selivanov, V.; Meca-Cortes, O.; Delowar, H.; Pons, M.; Izquierdo, I.; et al. Metabolic reprogramming and dependencies associated with epithelial cancer stem cells independent of the Epithelial-Mesenchymal Transition Program. Stem Cells 2016, 34, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Budczies, J.; Brockmoller, S.F.; Muller, B.M.; Barupal, D.K.; Richter-Ehrenstein, C.; Kleine-Tebbe, A.; Griffin, J.L.; Oresic, M.; Dietel, M.; Denkert, C.; et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: Alterations in glutamine and beta-alanine metabolism. J. Proteom. 2013, 94, 279–288. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Yaffe, E.; Hikri, E.; Elkis, Y.; Cohen, O.; Segal, A.; Makovski, A.; Varvak, A.; Shpungin, S.; Nir, U. Oncogenic properties of a spermatogenic meiotic variant of fer kinase expressed in somatic cells. Cancer Res. 2014, 74, 6474–6485. [Google Scholar] [CrossRef]
- Elkis, Y.; Cohen, M.; Yaffe, E.; Satmary-Tusk, S.; Feldman, T.; Hikri, E.; Nyska, A.; Feiglin, A.; Ofran, Y.; Shpungin, S.; et al. A novel Fer/FerT targeting compound selectively evokes metabolic stress and necrotic death in malignant cells. Nat. Commun. 2017, 8, 940. [Google Scholar] [CrossRef]
- Craig, A.W.; Greer, P.A. Fer kinase is required for sustained p38 kinase activation and maximal chemotaxis of activated mast cells. Mol. Cell. Biol. 2002, 22, 6363–6374. [Google Scholar]
- Craig, A.W.; Zirngibl, R.; Greer, P. Disruption of coiled-coil domains in Fer protein-tyrosine kinase abolishes trimerization but not kinase activation. J. Biol. Chem. 1999, 274, 19934–19942. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A. Coiled coils: New structures and new functions. Trends Biochem. Sci. 1996, 21, 375–382. [Google Scholar] [CrossRef]
- Hazan, B.; Bern, O.; Carmel, M.; Lejbkowicz, F.; Goldstein, R.S.; Nir, U. ferT encodes a meiosis-specific nuclear tyrosine kinase. Cell Growth Differ. 1993, 4, 443–449. [Google Scholar] [PubMed]
- Fischman, K.; Edman, J.C.; Shackleford, G.M.; Turner, J.A.; Rutter, W.J.; Nir, U. A murine fer testis-specific transcript (ferT) encodes a truncated Fer protein. Mol. Cell. Biol. 1990, 10, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Halachmy, S.; Bern, O.; Schreiber, L.; Carmel, M.; Sharabi, Y.; Shoham, J.; Nir, U. p94fer facilitates cellular recovery of gamma irradiated pre-T cells. Oncogene 1997, 14, 2871–2880. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Makovski, A.; Yaffe, E.; Shpungin, S.; Nir, U. Intronic promoter drives the BORIS-regulated expression of FerT in colon carcinoma cells. J. Biol. Chem. 2012, 287, 6100–6112. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Truesdell, P.; Meens, J.; Kadish, C.; Yang, X.; Boag, A.H.; Craig, A.W. Fer protein-tyrosine kinase promotes lung adenocarcinoma cell invasion and tumor metastasis. Mol. Cancer Res. 2013, 11, 952–963. [Google Scholar] [CrossRef]
- Itoh, T.; Hasegawa, J.; Tsujita, K.; Kanaho, Y.; Takenawa, T. The tyrosine kinase Fer is a downstream target of the PLD-PA pathway that regulates cell migration. Sci. Signal 2009, 2, ra52. [Google Scholar] [CrossRef]
- Yoneyama, T.; Angata, K.; Bao, X.; Courtneidge, S.; Chanda, S.K.; Fukuda, M. Fer kinase regulates cell migration through alpha-dystroglycan glycosylation. Mol. Biol. Cell 2012, 23, 771–780. [Google Scholar] [CrossRef]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, Y.; Wang, J.; Liu, K.; Wang, X. The effect of metformin on mortality among diabetic cancer patients: A systematic review and meta-analysis. JNCI Cancer Spectr. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Xue, W.H.; Ding, X.F.; Li, L.F.; Dou, M.M.; Zhang, W.J.; Lv, Z.; Fan, Z.R.; Zhao, J.; Wang, L.X. Association between metformin and the risk of gastric cancer in patients with type 2 diabetes mellitus: A meta-analysis of cohort studies. Oncotarget 2017, 8, 55622–55631. [Google Scholar] [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e44. [Google Scholar] [CrossRef]
- Schulten, H.J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Joshua, A.M.; Zannella, V.E.; Downes, M.R.; Bowes, B.; Hersey, K.; Koritzinsky, M.; Schwab, M.; Hofmann, U.; Evans, A.; van der Kwast, T.; et al. A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. 2014, 17, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Schuler, K.M.; Rambally, B.S.; DiFurio, M.J.; Sampey, B.P.; Gehrig, P.A.; Makowski, L.; Bae-Jump, V.L. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015, 4, 161–173. [Google Scholar] [CrossRef]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef]
- Niraula, S.; Dowling, R.J.; Ennis, M.; Chang, M.C.; Done, S.J.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830. [Google Scholar] [CrossRef]
- Mitsuhashi, A.; Kiyokawa, T.; Sato, Y.; Shozu, M. Effects of metformin on endometrial cancer cell growth in vivo: A preoperative prospective trial. Cancer 2014, 120, 2986–2995. [Google Scholar] [CrossRef]
- Lord, S.R.; Cheng, W.C.; Liu, D.; Gaude, E.; Haider, S.; Metcalf, T.; Patel, N.; Teoh, E.J.; Gleeson, F.; Bradley, K.; et al. Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell Metab. 2018, 28, 679–688. [Google Scholar] [CrossRef]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef]
- Lee, M.; Hirpara, J.L.; Eu, J.Q.; Sethi, G.; Wang, L.; Goh, B.C.; Wong, A.L. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2019, 25, 101073. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.A.; Hagel, K.R.; Hawk, M.A.; Schafer, Z.T. Metabolism during ECM detachment: Achilles heel of cancer cells? Trends Cancer 2017, 3, 475–481. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marciano, O.; Mehazri, L.; Shpungin, S.; Varvak, A.; Zacksenhaus, E.; Nir, U. Fer and FerT Govern Mitochondrial Susceptibility to Metformin and Hypoxic Stress in Colon and Lung Carcinoma Cells. Cells 2021, 10, 97. https://doi.org/10.3390/cells10010097
Marciano O, Mehazri L, Shpungin S, Varvak A, Zacksenhaus E, Nir U. Fer and FerT Govern Mitochondrial Susceptibility to Metformin and Hypoxic Stress in Colon and Lung Carcinoma Cells. Cells. 2021; 10(1):97. https://doi.org/10.3390/cells10010097
Chicago/Turabian StyleMarciano, Odeya, Linoy Mehazri, Sally Shpungin, Alexander Varvak, Eldad Zacksenhaus, and Uri Nir. 2021. "Fer and FerT Govern Mitochondrial Susceptibility to Metformin and Hypoxic Stress in Colon and Lung Carcinoma Cells" Cells 10, no. 1: 97. https://doi.org/10.3390/cells10010097
APA StyleMarciano, O., Mehazri, L., Shpungin, S., Varvak, A., Zacksenhaus, E., & Nir, U. (2021). Fer and FerT Govern Mitochondrial Susceptibility to Metformin and Hypoxic Stress in Colon and Lung Carcinoma Cells. Cells, 10(1), 97. https://doi.org/10.3390/cells10010097