The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Eosinophil Isolation
2.3. Confocal Fluorescence Microscopy
2.4. Signal Processing and Statistical Analysis
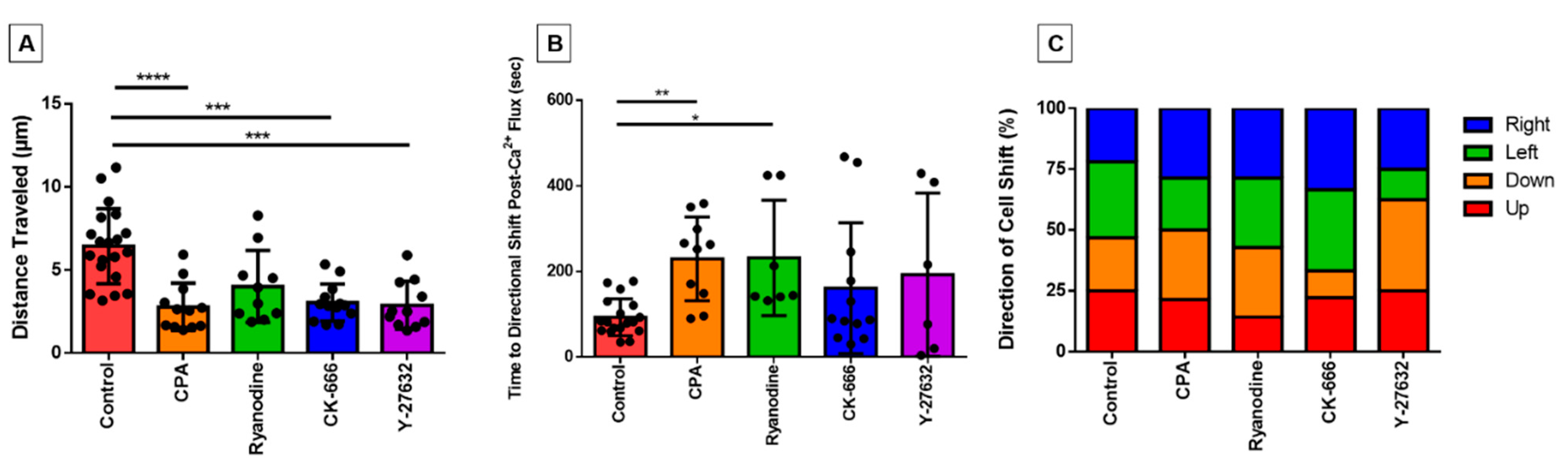
3. Results
3.1. Pharmacological Agents
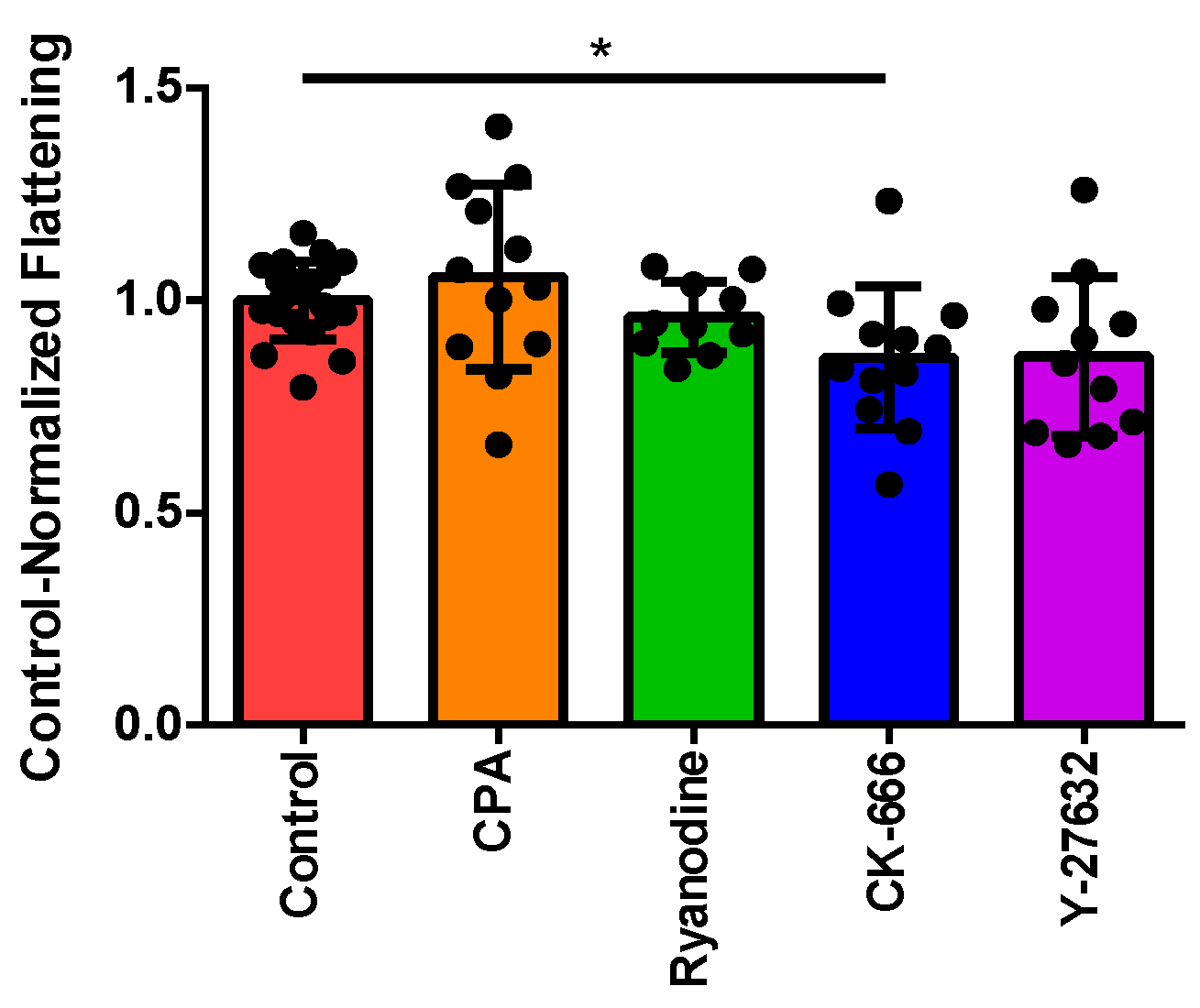
3.2. Normalizing Treatment Groups to the Control for Area Ratio
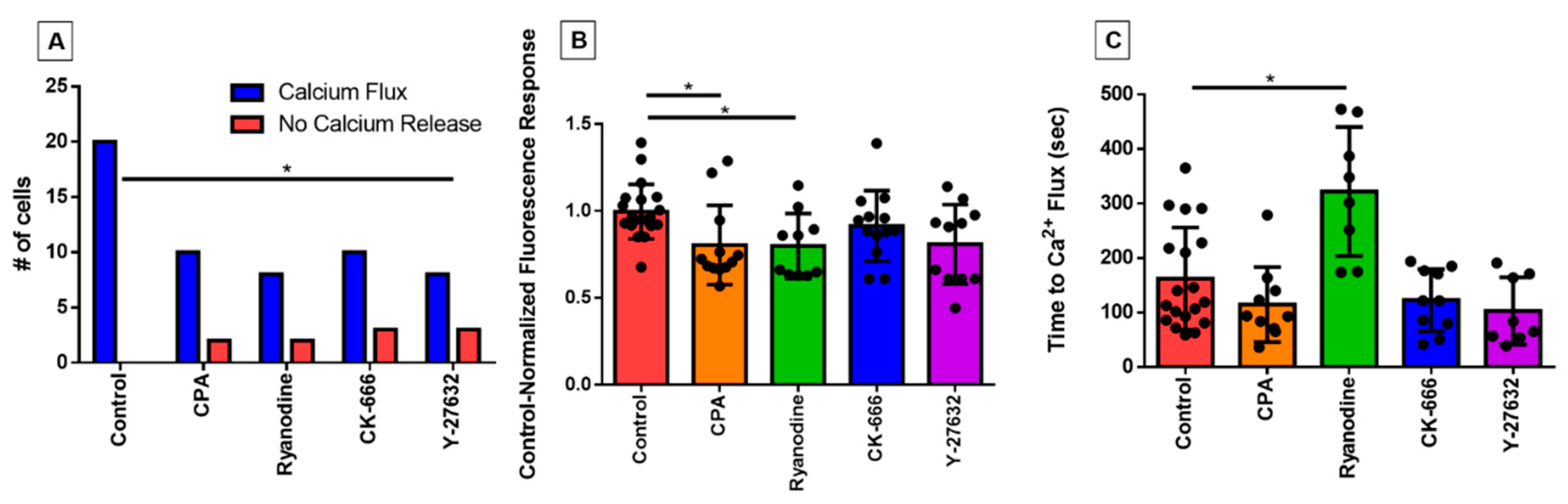
3.3. Characterizing the Effect of Pharmacological Agents on Shear Stress-Induced Calcium Release
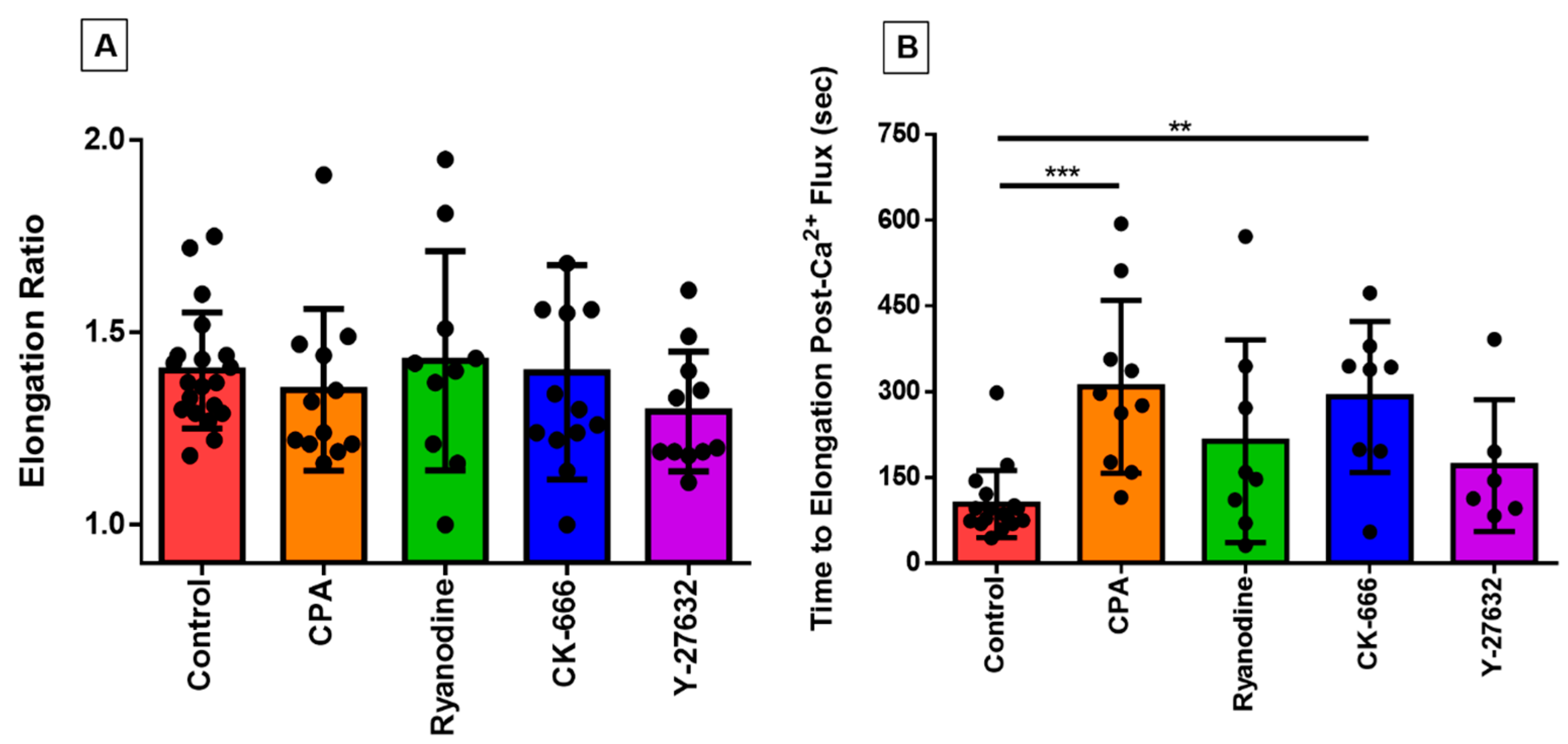
3.4. Investigating the Effect of Fluid Shear Stress on Loss of Cell Circularity
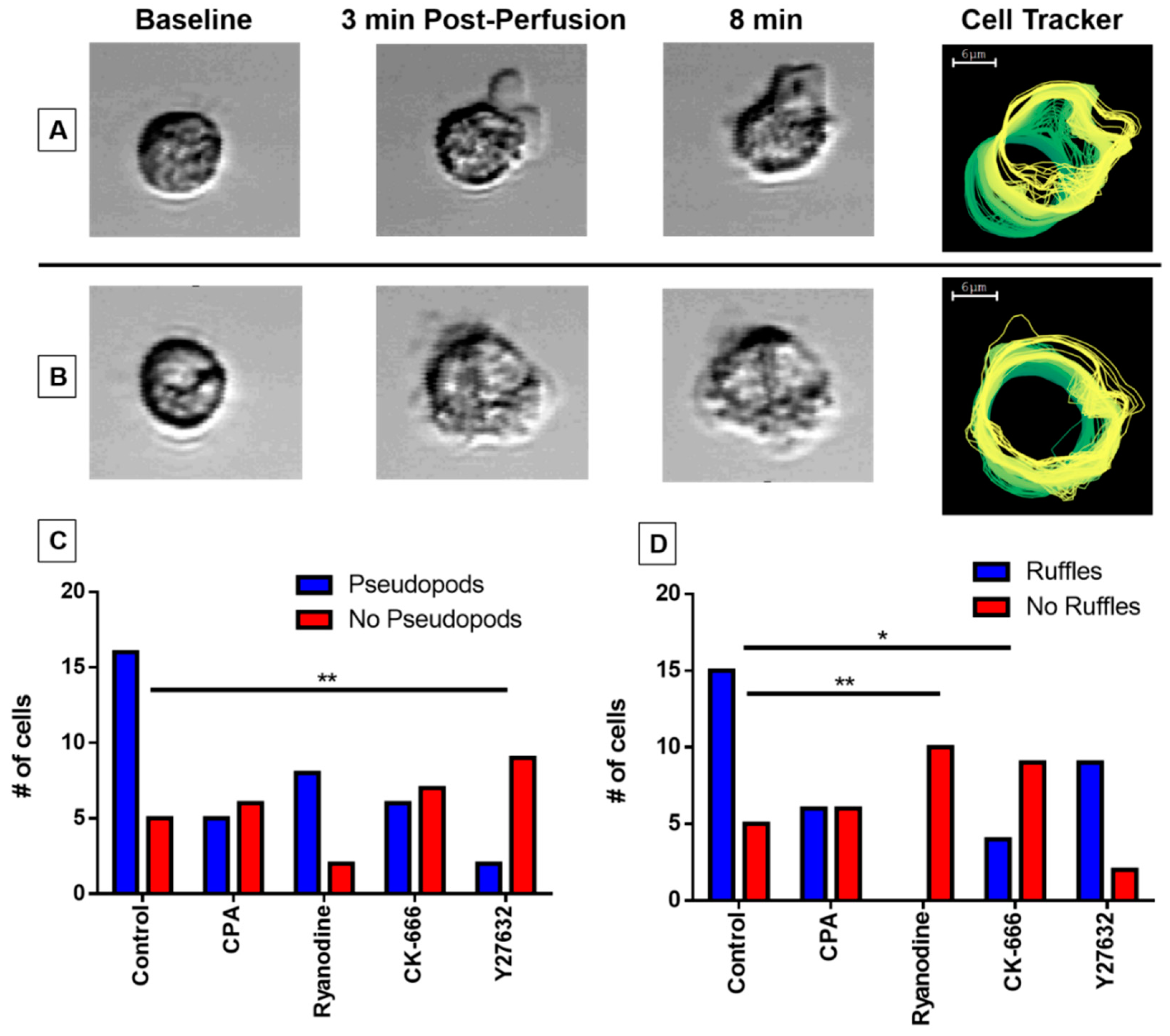
3.5. Pseudopodia Formation and Membrane Ruffling
3.6. Directionality of Eosinophil Motility
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, J.A.; Bocher, B.S. Eosinophils and eosinophil-associated diseases: An update. J. Allergy Clin. Immunol. 2018, 141, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Akuthota, P.; Jayne, D.; Khoury, P.; Klion, A.; Langford, C.A.; Merkel, P.A.; Moosig, F.; Specks, U.; Cid, M.C.; et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 376, 1921–1932. [Google Scholar] [CrossRef]
- Busse, W.W.; Ring, J.; Huss-Marp, J.; Kahn, J.-E. A review of treatment with mepolizumab, an anti-IL-5 mAb, in hypereosinophilic syndromes and asthma. J. Allergy Clin. Immunol. 2010, 125, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Lima-Matos, A.; Ponte, E.V.; de Jesus, J.P.V.; Almeida, P.C.A.; Lima, V.B.; Kwon, N.; Riley, J.; de Mello, L.M.; Cruz, A.A. Eosinophilic asthma, according to a blood eosinophil criterion, is associated with disease severity and lack of control among underprivileged urban Brazilians. Respir. Med. 2018, 145, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Sehmi, R.; Nair, P. Anti-IL5 therapy for asthma and beyond. World Allergy Organ. J. 2014, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.; Chupp, G.; Bardin, P.; Bourdin, A.; Garcia, G.; Harley, B.; Yancey, S.; Humbert, M. The role of mepolizumab in atopic and nonatopic severe asthma with persistent eosinophilia. Eur. Respir. J. 2014, 44, 239–241. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; O’Byrne, P.M.; Boulet, L.-P.; Wang, Y.; Cockcroft, D.; Bigler, J.; FitzGerald, J.M.; Boedigheimer, M.; Davis, B.E.; Dias, C.; et al. Effects of an Anti-TSLP Antibody on Allergen-Induced Asthmatic Responses. N. Engl. J. Med. 2014, 370, 2102–2110. [Google Scholar] [CrossRef]
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef]
- Johansson, M.W. Activation states of blood eosinophils in asthma. Clin. Exp. Allergy 2014, 44, 482–498. [Google Scholar]
- Barthel, S.R.; Johansson, M.W.; McNamee, D.M.; Mosher, D.F. Roles of integrin activation in eosinophil function and the eosinophilic inflammation of asthma. J. Leukoc. Biol. 2008, 83, 1–12. [Google Scholar] [CrossRef]
- Barthel, S.R.; Annis, D.S.; Mosher, D.F.; Johansson, M.W. Differential Engagement of Modules 1 and 4 of Vascular Cell Adhesion Molecule-1 (CD106) by Integrins α4β1 (CD49d/29) and αMβ2 (CD11b/18) of Eosinophils. J. Biol. Chem. 2006, 281, 32175–32187. [Google Scholar] [CrossRef] [PubMed]
- Weller, P.F.; Rand, T.H.; Goelz, S.E.; Chi-Rosso, G.; Lobb, R.R. Human eosinophil adherence to vascular endothelium mediated by binding to vascular cell adhesion molecule 1 and endothelial leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 7430–7433. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzai, M.; Small, M.; Sehmi, R.; Gauvreau, G.; Janssen, L. Integrins are Mechanosensors That Modulate Human Eosinophil Activation. Front. Immunol. 2015, 6, 525. [Google Scholar] [CrossRef]
- Son, K.; Small, M.; Sehmi, R.; Janssen, L. The eosinophil actin cytoskeleton undergoes rapid rearrangement in response to fluid shear stress. J. Leukoc. Biol. 2020, 108, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Son, K.; Mukherjee, M.; McIntyre, B.A.; Eguez, J.C.; Radford, K.; LaVigne, N.; Ethier, C.; Davoine, F.; Janssen, L.; Lacy, P.; et al. Improved recovery of functionally active eosinophils and neutrophils using novel immunomagnetic technology. J. Immunol. Methods 2017, 449, 44–55. [Google Scholar] [CrossRef]
- Ballermann, B.J.; Dardik, A.; Eng, E.; Liu, A. Shear stress and the endothelium. Kidney Int. 1998, 54, S100–S108. [Google Scholar] [CrossRef]
- Yadav, R.; Larbi, K.Y.; Young, R.E.; Nourshargh, S. Migration of leukocytes through the vessel wall and beyond. Thromb. Haemost. 2003, 90, 598–606. [Google Scholar] [CrossRef]
- Muller, W.A.; Luscinskas, F.W. Assays of transendothelial migration in vitro. Methods Enzym. 2008, 443, 155–176. [Google Scholar]
- Ohkawara, Y.; Yamauchi, K.; Maruyama, N.; Hoshi, H.; Ohno, I.; Honma, M.; Tanno, Y.; Tamura, G.; Shirato, K.; Ohtani, H. In situ expression of the cell adhesion molecules in bronchial tissues from asthmatics with air flow limitation: In vivo evidence of VCAM-1/VLA-4 interaction in selective eosinophil infiltration. Am. J. Respir. Cell Mol. Biol. 1995, 12, 4–12. [Google Scholar] [CrossRef]
- Wu, C.; Fields, J.; Kapteijn, B.A.; McDonald, J.A. The role of alpha 4 beta 1 integrin in cell motility and fibronectin matrix assembly. J. Cell Sci. 1995, 108, 821–829. [Google Scholar]
- Price, L.S.; Leng, J.; Schwartz, M.A.; Bokoch, G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell 1998, 9, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerdá, J.; Chorev, D.S.; Geiger, B.; Cossart, P. The Diverse Family of Arp2/3 Complexes. Trends Cell Biol. 2017, 27, 93–100. [Google Scholar] [CrossRef]
- Henson, J.H.; Yeterian, M.; Weeks, R.M.; Medrano, A.E.; Brown, B.L.; Geist, H.L.; Pais, M.D.; Oldenbourg, R.; Shuster, C.B. Arp2/3 complex inhibition radically alters lamellipodial actin architecture, suspended cell shape, and the cell spreading process. Mol. Biol. Cell 2015, 26, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Wehrle-Haller, B.; Imhof, B.A. The inner lives of focal adhesions. Trends Cell Biol. 2002, 12, 382–389. [Google Scholar] [CrossRef]
- DeMali, K.A.; Barlow, C.A.; Burridge, K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002, 159, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef]
- Santulli, S.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Van Petegem, F. Ryanodine receptors: Structure and function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef]
- Vukcevic, M.; Zorzato, F.; Keck, S.; Tsakiris, D.A.; Keiser, A.; Maizels, R.M.; Treves, S. Gain of function in the immune system caused by a ryanodine receptor 1 mutation. J. Cell Sci. 2013, 126, 3485–3492. [Google Scholar] [CrossRef]
- Cohen, G.; Raupachova, J.; Wimmer, T.; Deicher, R.; Hörl, W.H. The uraemic retention solute para-hydroxy-hippuric acid attenuates apoptosis of polymorphonuclear leukocytes from healthy subjects but not from haemodialysis patients. Nephrol. Dial. Transpl. 2008, 23, 2512–2519. [Google Scholar] [CrossRef]
- Schwartz, A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar]
- Horwitz, A.F.; Lauffenburger, D.A. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar]
- Borm, B.; Requardt, R.P.; Herzog, V.; Kirfel, G. Membrane ruffles in cell migration: Indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp. Cell Res. 2005, 302, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, M. New insights into the formation and the function of lamellipodia and ruffles in mesenchymal cell migration. Cell Adhes. Migr. 2018, 12, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Xanthis, I.; Souilhol, C.; Serbanovic-Canic, J.; Roddie, H.; Kalli, A.C.; Fragiadaki, M.; Wong, R.; Shah, D.R.; Askari, J.A.; Canham, L.; et al. β1 integrin is a sensor of blood flow direction. J. Cell Sci. 2019, 132, jcs229542. [Google Scholar] [CrossRef]
- Matthews, B.D.; Overby, D.R.; Mannix, R.; Ingber, D.E. Cellular adaptation to mechanical stress: Role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J. Cell Sci. 2006, 119, 508–518. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, K.; Hussain, A.; Sehmi, R.; Janssen, L. The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils. Cells 2021, 10, 157. https://doi.org/10.3390/cells10010157
Son K, Hussain A, Sehmi R, Janssen L. The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils. Cells. 2021; 10(1):157. https://doi.org/10.3390/cells10010157
Chicago/Turabian StyleSon, Kiho, Amer Hussain, Roma Sehmi, and Luke Janssen. 2021. "The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils" Cells 10, no. 1: 157. https://doi.org/10.3390/cells10010157
APA StyleSon, K., Hussain, A., Sehmi, R., & Janssen, L. (2021). The Cycling of Intracellular Calcium Released in Response to Fluid Shear Stress Is Critical for Migration-Associated Actin Reorganization in Eosinophils. Cells, 10(1), 157. https://doi.org/10.3390/cells10010157