The Potential for Resident Lung Mesenchymal Stem Cells to Promote Functional Tissue Regeneration: Understanding Microenvironmental Cues

{kind=link}
Abstract
:1. Introduction
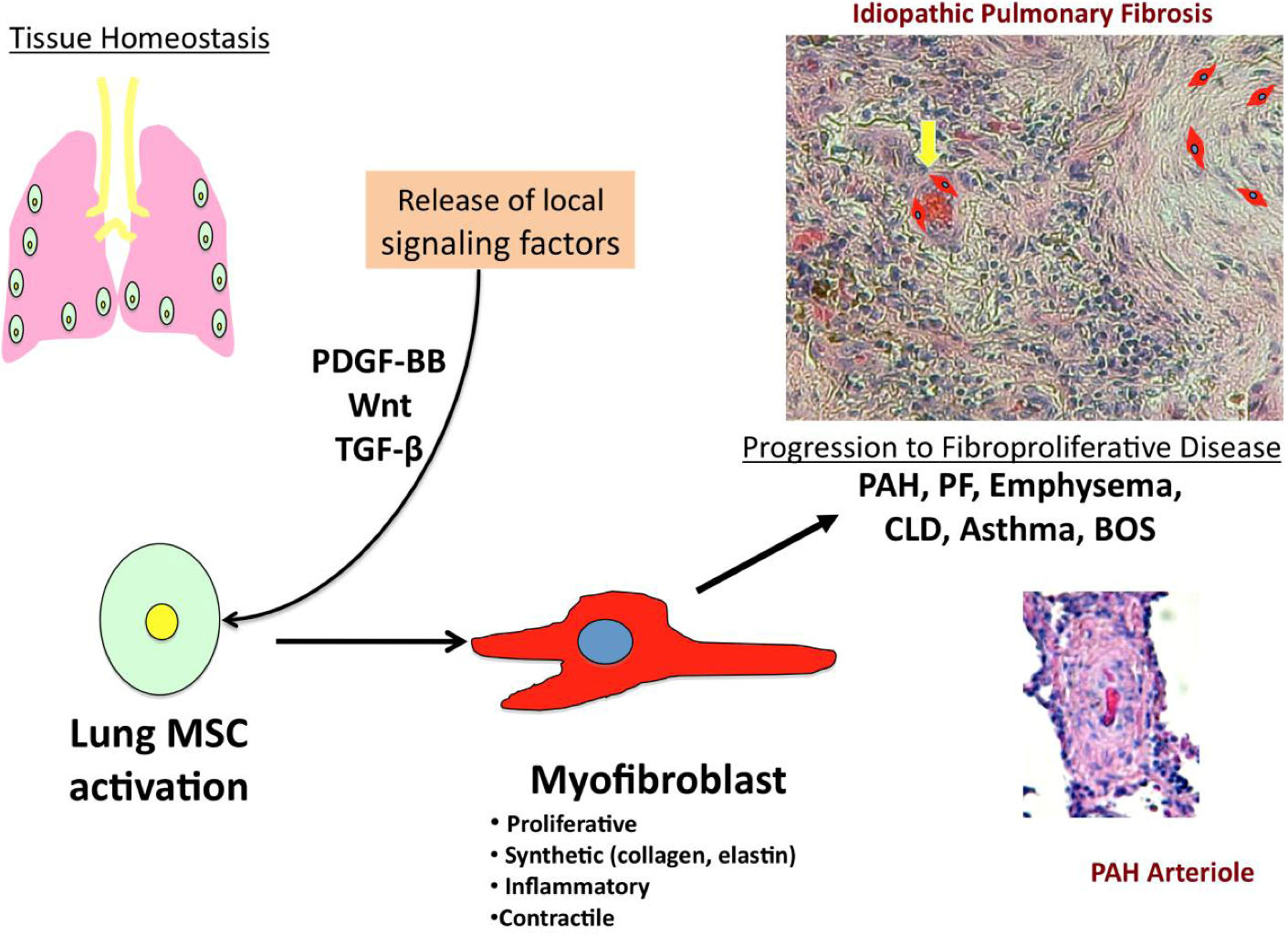
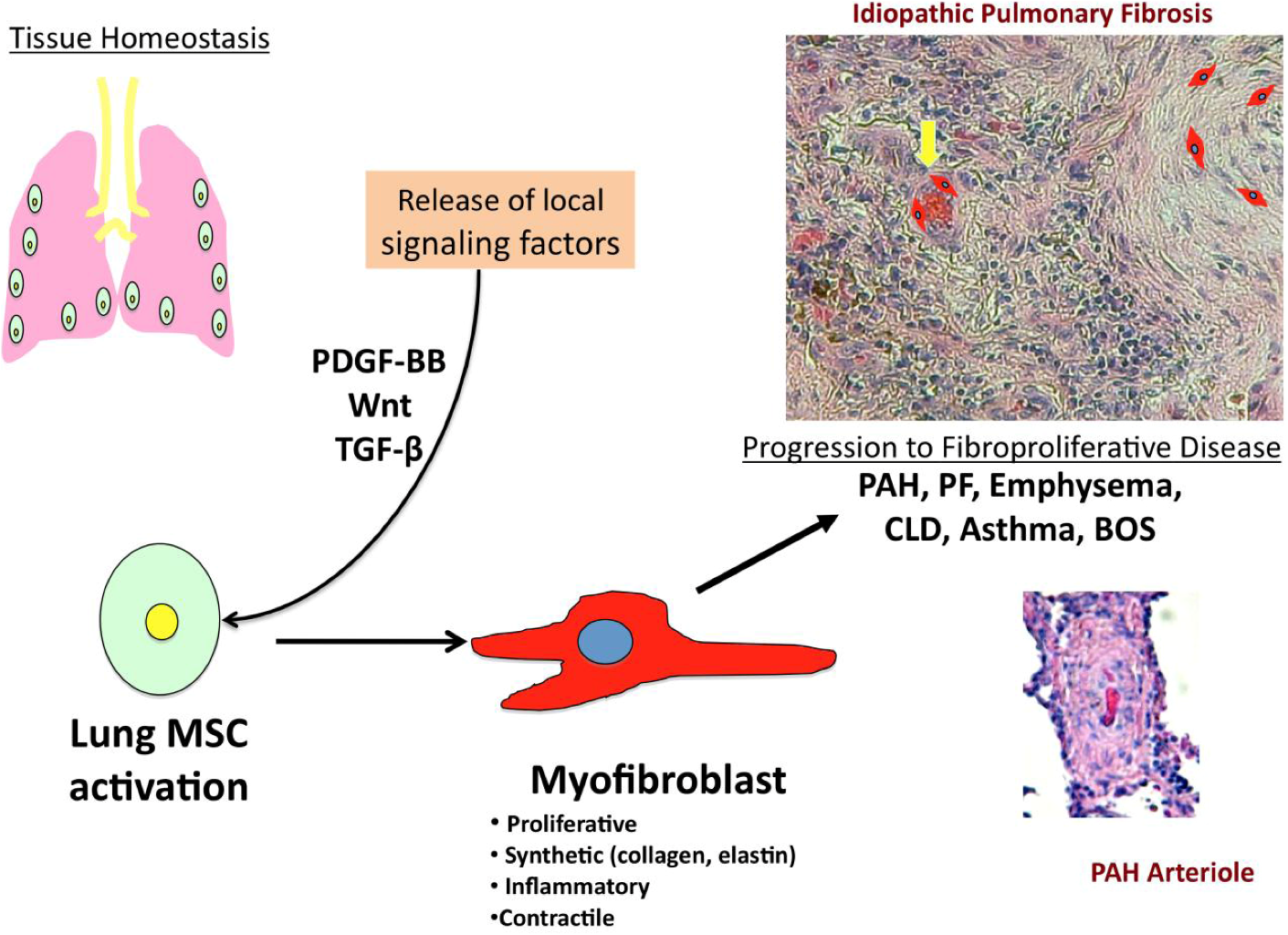
1.1. Stem Cell Therapy & Pulmonary Disease
1.2. Multipotent Resident Lung MSCs
2. PDGF-BB ad Wnt: Two Key Signaling Targets in Inflammatory Lung Disease and Vascular Remodeling
2.1. PDGF-BB Signaling & Fibro-Proliferative Lung Disorders
2.2. Wnt Signaling in Fibro-Proliferative Lung Disorders
3. Conclusions
Acknowledgments
Conflict of Interest
References
- Beers, M.F.; Morrissey, E.E. The three R's of lung health and disease: Repair, remodeling, and regeneration. J. Clin. Invest. 2011, 121, 2065–2073. [Google Scholar] [CrossRef]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; de Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Invest. 2011, 121, 4409–4419. [Google Scholar] [CrossRef]
- Martin, J.; Helm, K.; Ruegg, P.; Varella-Garcia, M.; Burnham, E.; Majka, S. Adult lung side population cells have mesenchymal stem cell potential. Cytotherapy 2008, 10, 140–151. [Google Scholar] [CrossRef]
- Summer, R.; Fitzsimmons, K.; Dwyer, D.; Murphy, J.; Fine, A. Isolation of an adult mouse lung mesenchymal progenitor cell population. Am. J. Respir. Cell Mol. Biol. 2007, 37, 152–159. [Google Scholar] [CrossRef]
- Lama, V.N.; Smith, L.; Badri, L.; Flint, A.; Andrei, A.-C.; Murray, S.; Wang, Z.; Liao, H.; Toews, G.B.; Krebsbach, P.H.; et al. Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J. Clin. Invest. 2007, 117, 989–996. [Google Scholar] [CrossRef]
- Jun, D.; Garat, C.; West, J.; Thorn, N.; Chow, K.; Cleaver, T.; Sullivan, T.; Torchia, E.C.; Childs, C.; Shade, T.; et al. The pathology of bleomycin-induced fibrosis is associated with loss of resident lung mesenchymal stem cells that regulate effector T-cell proliferation. Stem Cell. 2011, 29, 725–735. [Google Scholar] [CrossRef]
- Chateauvieux, S.; Ichanté, J.-L.; Delorme, B.; Frouin, V.; Piétu, G.; Langonné, A.; Gallay, N.; Sensebé, L.; Martin, M.T.; Moore, K.A.; et al. Molecular profile of mouse stromal mesenchymal stem cells. Physiol. Genom. 2007, 29, 128–138. [Google Scholar]
- McQualter, J.L.; Brouard, N.; Williams, B.; Baird, B.N.; Sims-Lucas, S.; Yuen, K.; Nilsson, S.K.; Simmons, P.J.; Bertoncello, I. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the Sca-1 positive cell fraction. Stem Cell. 2009, 27, 623–633. [Google Scholar] [CrossRef]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef]
- Lee, R.H.; Pulin, A.A.; Seo, M.J.; Kota, D.J.; Ylostalo, J.; Larson, B.L.; Semprun-Prieto, L.; Delafontaine, P.; Prockop, D.J. Intravenous hMSCs Improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009, 5, 54–63. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Hwang, S.-M.; Chen, K.-D.; Lee, Y.-S.; Hsu, L.-W.; Chang, Y.-J.; Wang, C.-N.; Peng, H.-H.; Chang, Y.-L.; Chao, A.-S.; et al. Functional network analysis of the transcriptomes of mesenchymal stem cells derived from amniotic fluid, amniotic membrane, cord blood, and bone marrow. Stem Cell. 2007, 25, 2511–2523. [Google Scholar] [CrossRef]
- Benvenuto, F.; Ferrari, S.; Gerdoni, E.; Gualandi, F.; Frassoni, F.; Pistoia, V.; Mancardi, G.; Uccelli, A. Human mesenchymal stem cells promote survival of T cells in a quiescent state. Stem Cell. 2007, 25, 1753–1760. [Google Scholar] [CrossRef]
- Fatima, S.; Zhou, S.; Sorrentino, B.P. Abcg2 expression marks tissue-specific stem cells in multiple organs in a mouse progeny tracking model. Stem Cell. 2011, 30, 210–221. [Google Scholar]
- Hennrick, K.T.; Keeton, A.G.; Nanua, S.; Kijek, T.G.; Goldsmith, A.M.; Sajjan, U.S.; Bentley, J.K.; Lama, V.N.; Moore, B.B.; Schumacher, R.E.; et al. Lung cells from neonates show a mesenchymal stem cell phenotype. Am. J. Respir. Crit. Care Med. 2007, 175, 1158–1164. [Google Scholar] [CrossRef]
- Jarvinen, L.; Badri, L.; Wettlaufer, S.; Ohtsuka, T.; Standiford, T.J.; Toews, G.B.; Pinsky, D.J.; Peters-Golden, M.; Lama, V.N. Lung resident mesenchymal stem cells isolated from human lung allografts inhibit T cell proliferation via a soluble mediator. J. Immunol. 2008, 181, 4389–4396. [Google Scholar]
- Lee, J.W.; Gupta, N.; Serikov, V.; Matthay, M.A. Potential application of mesenchymal stem cells in acute lung injury. Expet. Opin. Biol. Ther. 2009, 9, 1259–1270. [Google Scholar] [CrossRef]
- Bonner, J.C. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair 2010, 3, 15. [Google Scholar] [CrossRef]
- Pierro, M.; Thebaud, B. Mesenchymal stem cells in chronic lung disease: Culprit or savior? Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L732–734. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Y.; Han, Q.; Jia, M.; Liao, L.; Qi, M.; Zhao, R.C. Injured microenvironment directly guides the differentiation of engrafted Flk-1(+) mesenchymal stem cell in lung. Exp. Hematol. 2007, 35, 1466–1475. [Google Scholar] [CrossRef]
- Popova, A.P.; Bozyk, P.D.; Bentley, J.K.; Linn, M.J.; Goldsmith, A.M.; Schumacher, R.E.; Weiner, G.M.; Filbrun, A.G.; Hershenson, M.B. Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics 2010, 126, e1127–1133. [Google Scholar] [CrossRef]
- Toti, P.; Buonocore, G.; Tanganelli, P.; Catella, A.M.; Palmeri, M.L.; Vatti, R.; Seemayer, T.A. Bronchopulmonary dysplasia of the premature baby: An immunohistochemical study. Pediatr. Pulmonol. 1997, 24, 22–28. [Google Scholar]
- Walker, N.; Badri, L.; Wettlaufer, S.; Flint, A.; Sajjan, U.; Krebsbach, P.H.; Keshamouni, V.G.; Peters-Golden, M.; Lama, V.N. Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am. J. Pathol. 2011, 178, 2461–2469. [Google Scholar] [CrossRef]
- Todd, J.L.; Palmer, S.M. Bronchiolitis obliterans syndrome: The final frontier for lung transplantation. Chest 2011, 140, 502–508. [Google Scholar] [CrossRef]
- Descalzi, D.; Folli, C.; Scordamaglia, F.; Riccio, A.M.; Gamalero, C.; Canonica, G.W. Importance of fibroblasts-myofibroblasts in asthma-induced airway remodeling. Recent Pat. Inflamm. Allergy Drug Discov. 2007, 1, 237–241. [Google Scholar] [CrossRef]
- Al-Muhsen, S.; Johnson, J.R.; Hamid, Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011, 128, 451–462, quiz 463- quiz 454. [Google Scholar] [CrossRef]
- Brewster, C.E.; Howarth, P.H.; Djukanovic, R.; Wilson, J.; Holgate, S.T.; Roche, W.R. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1990, 3, 507–511. [Google Scholar]
- Bentley, J.K.; Popova, A.P.; Bozyk, P.D.; Linn, M.J.; Baek, A.E.; Lei, J.; Goldsmith, A.M.; Hershenson, M.B. Ovalbumin sensitization and challenge increases the number of lung cells possessing a mesenchymal stromal cell phenotype. Respir. Res. 2010, 11, 127. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar]
- Barst, R.J. PDGF signaling in pulmonary arterial hypertension. J. Clin. Invest. 2005, 115, 2691–2694. [Google Scholar] [CrossRef]
- Berg, J.Ä.; Breen, E.Ä.; Fu, Z.; Mathieu-Costello, O.; West, J.Ä. Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor-B in lung parenchyma. Am. J. Respir. Crit. Care Med. 1998, 158, 1920–1928. [Google Scholar]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 2005, 115, 2811–2821. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Seeger, W.; Grimminger, F. Imatinib for the treatment of pulmonary arterial hypertension. New Engl. J. Med. 2005, 353, 1412–1413. [Google Scholar] [CrossRef]
- Lamé, M.W.; Jones, A.D.; Wilson, D.W.; Segall, H.J. Monocrotaline pyrrole targets proteins with and without cysteine residues in the cytosol and membranes of human pulmonary artery endothelial cells. Proteomics 2005, 5, 4398–4413. [Google Scholar] [CrossRef]
- Adachi, M.; Osawa, Y.; Uchinami, H.; Kitamura, T.; Accili, D.; Brenner, D.A. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology 2007, 132, 1434–1446. [Google Scholar] [CrossRef]
- Desfaits, A.-C.C.; Raymond, J. Growth factors stimulate neointimal cells in vitro and increase the thickness of the neointima formed at the neck of porcine aneurysms treated by embolization. Stroke 2000, 31, 498–507. [Google Scholar] [CrossRef]
- Fishbein, I.; Waltenberger, J.; Banai, S.; Rabinovich, L.; Chorny, M.; Levitzki, A.; Gazit, A.; Huber, R.; Mayr, U.; Gertz, S.D.; et al. Local delivery of platelet-derived growth factor receptor-specific tyrphostin inhibits neointimal formation in rats. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 667–676. [Google Scholar] [CrossRef]
- Sirois, M.G.; Simons, M.; Edelman, E.R. Antisense oligonucleotide inhibition of PDGFR-beta receptor subunit expression directs suppression of intimal thickening. Circulation 1997, 95, 669–676. [Google Scholar] [CrossRef]
- Sheu, J.-R.; Wu, C.-H.; Chen, Y.-C.; Hsiao, G.; Lin, C.-H. Mechanisms in the inhibition of neointimal hyperplasia with triflavin in a rat model of balloon angioplasty. J. Lab. Clin. Med. 2001, 137, 270–278. [Google Scholar] [CrossRef]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef]
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Gröne, H.-J.; Lipson, K.E.; et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–935. [Google Scholar] [CrossRef]
- Wang, S.; Wilkes, M.C.; Leof, E.B.; Hirschberg, R. Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005, 19, 1–11. [Google Scholar] [CrossRef]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.R.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. DGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar] [CrossRef]
- Veevers-Lowe, J.; Ball, S.G.; Shuttleworth, A.; Kielty, C.M. Mesenchymal stem cell migration is regulated by fibronectin through α5β1-integrin-mediated activation of PDGFR-β and potentiation of growth factor signals. J. Cell Sci. 2011, 124, 1288–1300. [Google Scholar] [CrossRef]
- Dhar, K.; Dhar, G.; Majumder, M.; Haque, I.; Mehta, S.; van Veldhuizen, P.J.; Banerjee, S.K.; Banerjee, S. Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1. Mol. Cancer 2010, 9, 209. [Google Scholar] [CrossRef]
- Baksh, D.; Boland, G.M.; Tuan, R.S. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J. Cell. Biochem. 2007, 101, 1109–1124. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Spencer, G.J.; Heath, D.J.; Genever, P.G. Expression profiling and functional analysis of Wnt signaling mechanisms in mesenchymal stem cells. Stem Cell. 2004, 22, 849–860. [Google Scholar] [CrossRef]
- Kalani, M.Y.S.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L.; Palmer, T.D.; Nusse, R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16970–16975. [Google Scholar]
- Kirton, J.P.; Crofts, N.J.; George, S.J.; Brennan, K.; Canfield, A.E. Wnt/β-catenin signaling stimulates chondrogenic and inhibits adipogenic differentiation of pericytes. Circ. Res. 2007, 101, 581–589. [Google Scholar] [CrossRef]
- Ling, L.; Nurcombe, V.; Cool, S.M. Wnt signaling controls the fate of mesenchymal stem cells. Gene 2009, 433, 1–7. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Berg, T.; DeLanghe, S.; Al Alam, D.; Utley, S.; Estrada, J.; Wang, K.S. β-catenin regulates mesenchymal progenitor cell differentiation during hepatogenesis. J. Surg. Res. 2010, 164, 276–285. [Google Scholar] [CrossRef]
- Kuhl, M.; Sheldahl, L.C.; Malbon, C.C.; Moon, R.T. Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J. Biol. Chem. 2000, 275, 12701–12711. [Google Scholar]
- Boutros, M.; Paricio, N.; Strutt, D.I.; Mlodzik, M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 1998, 94, 109–118. [Google Scholar] [CrossRef]
- Cohen, E.D.; Ihida-Stansbury, K.; Lu, M.M.; Panettieri, R.A.; Jones, P.L.; Morrisey, E.E. Wnt signaling regulates smooth muscle precursor development in the mouse lung via a tenascin C/PDGFR pathway. J. Clin. Invest. 2009, 119, 2538–2549. [Google Scholar] [CrossRef]
- Badri, L.; Lama, V.N. Lysophosphatidic acid induces migration of human lung-resident mesenchymal stem cells through the beta-catenin pathway. Stem Cell. 2012, 30, 2010–2019. [Google Scholar] [CrossRef]
- Deng, L.; Hu, S.; Baydoun, A.R.; Chen, J.; Chen, X.; Cong, X. Aspirin induces apoptosis in mesenchymal stem cells requiring Wnt/beta-catenin pathway. Cell Prolif. 2009, 42, 721–730. [Google Scholar] [CrossRef]
- De Boer, J.; Wang, H.J.; van Blitterswijk, C. Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 2004, 10, 393–401. [Google Scholar] [CrossRef]
- Uren, A.; Reichsman, F.; Anest, V.; Taylor, W.G.; Muraiso, K.; Bottaro, D.P.; Cumberledge, S.; Rubin, J.S. Secreted frizzled-related protein-1 binds directly to Wingless and is a biphasic modulator of Wnt signaling. J. Biol. Chem. 2000, 275, 4374–4382. [Google Scholar]
- Leyns, L.; Bouwmeester, T.; Kim, S.H.; Piccolo, S.; de Robertis, E.M. Frzb-1 is a secreted antagonist of Wnt signaling expressed in the Spemann organizer. Cell 1997, 88, 747–756. [Google Scholar] [CrossRef]
- Semenov, M.V.; Tamai, K.; Brott, B.K.; Kuhl, M.; Sokol, S.; He, X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 2001, 11, 951–961. [Google Scholar] [CrossRef]
- Cho, S.W.; Her, S.J.; Sun, H.J.; Choi, O.K.; Yang, J.Y.; Kim, S.W.; Kim, S.Y.; Shin, C.S. Differential effects of secreted frizzled-related proteins (sFRPs) on osteoblastic differentiation of mouse mesenchymal cells and apoptosis of osteoblasts. Biochem. Biophys. Res. Commun. 2008, 367, 399–405. [Google Scholar] [CrossRef]
- Bodine, P.V.; Billiard, J.; Moran, R.A.; Ponce-de-Leon, H.; McLarney, S.; Mangine, A.; Scrimo, M.J.; Bhat, R.A.; Stauffer, B.; Green, J.; et al. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J. Cell. Biochem. 2005, 96, 1212–1230. [Google Scholar] [CrossRef]
- Surmann-Schmitt, C.; Widmann, N.; Dietz, U.; Saeger, B.; Eitzinger, N.; Nakamura, Y.; Rattel, M.; Latham, R.; Hartmann, C.; von der Mark, H.; et al. Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J. Cell Sci. 2009, 122, 3627–3637. [Google Scholar] [CrossRef]
- Horwitz, E.M. Dkk-1-mediated expansion of adult stem cells. Trends Biotechnol. 2004, 22, 386–388. [Google Scholar] [CrossRef]
- Kneidinger, N.; Yildirim, A.O.; Callegari, L.; Takenaka, S.; Stein, M.M.; Dumitrascu, R.; Bohla, A.; Bracke, K.R.; Morty, R.E.; Brusselle, G.G.; et al. Activation of the WNT/beta-catenin pathway attenuates experimental emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 723–733. [Google Scholar] [CrossRef]
- Foronjy, R.; Imai, K.; Shiomi, T.; Mercer, B.; Sklepkiewicz, P.; Thankachen, J.; Bodine, P.; D'Armiento, J. The divergent roles of secreted frizzled related protein-1 (SFRP1) in lung morphogenesis and emphysema. Am. J. Pathol. 2010, 177, 598–607. [Google Scholar] [CrossRef]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Invest. 2009, 119, 772–787. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Foronjy, R.F.; Majka, S.M. The Potential for Resident Lung Mesenchymal Stem Cells to Promote Functional Tissue Regeneration: Understanding Microenvironmental Cues. Cells 2012, 1, 874-885. https://doi.org/10.3390/cells1040874
Foronjy RF, Majka SM. The Potential for Resident Lung Mesenchymal Stem Cells to Promote Functional Tissue Regeneration: Understanding Microenvironmental Cues. Cells. 2012; 1(4):874-885. https://doi.org/10.3390/cells1040874
Chicago/Turabian StyleForonjy, Robert F., and Susan M. Majka. 2012. "The Potential for Resident Lung Mesenchymal Stem Cells to Promote Functional Tissue Regeneration: Understanding Microenvironmental Cues" Cells 1, no. 4: 874-885. https://doi.org/10.3390/cells1040874
APA StyleForonjy, R. F., & Majka, S. M. (2012). The Potential for Resident Lung Mesenchymal Stem Cells to Promote Functional Tissue Regeneration: Understanding Microenvironmental Cues. Cells, 1(4), 874-885. https://doi.org/10.3390/cells1040874