
Transcriptome Differential Expression Regulation Analysis of the Narrow-Leaf Mutant of Sorghum Bicolor

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Morphological Investigation of Sorghum Mutant and Analytical Stoma
2.2.2. RNA Extraction, cDNA Library Construction, and Sequencing
2.2.3. Alignment of Sequencing Data with the Reference Genome Sequence
2.2.4. Gene Expression Quantification and Differential Gene Screening
2.2.5. Real-Time PCR Analysis
2.2.6. Data Statistics and Analysis
3. Results and Analysis
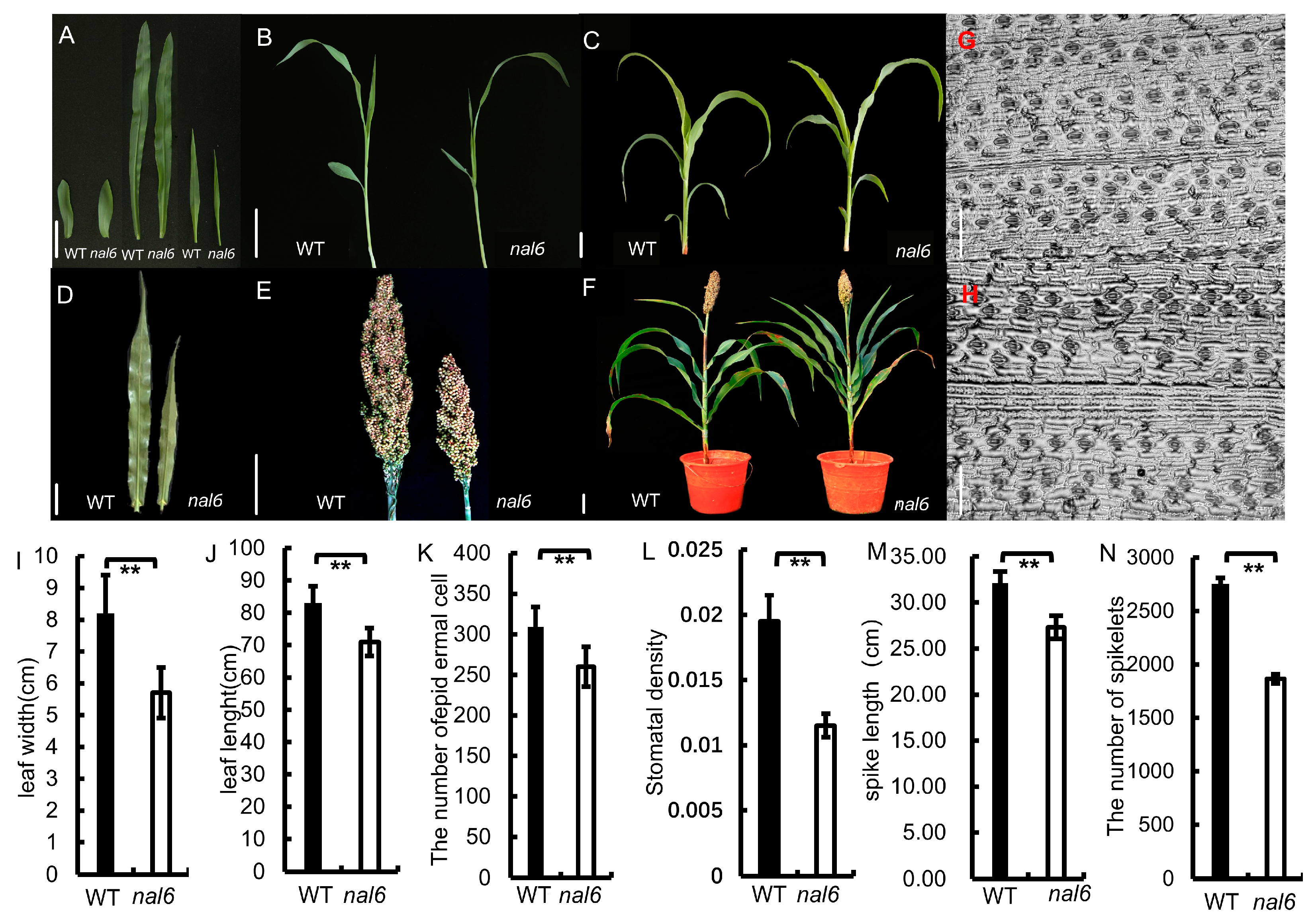
3.1. Phenotypic Characterization of the nal6 and Stomatal Density Analysis
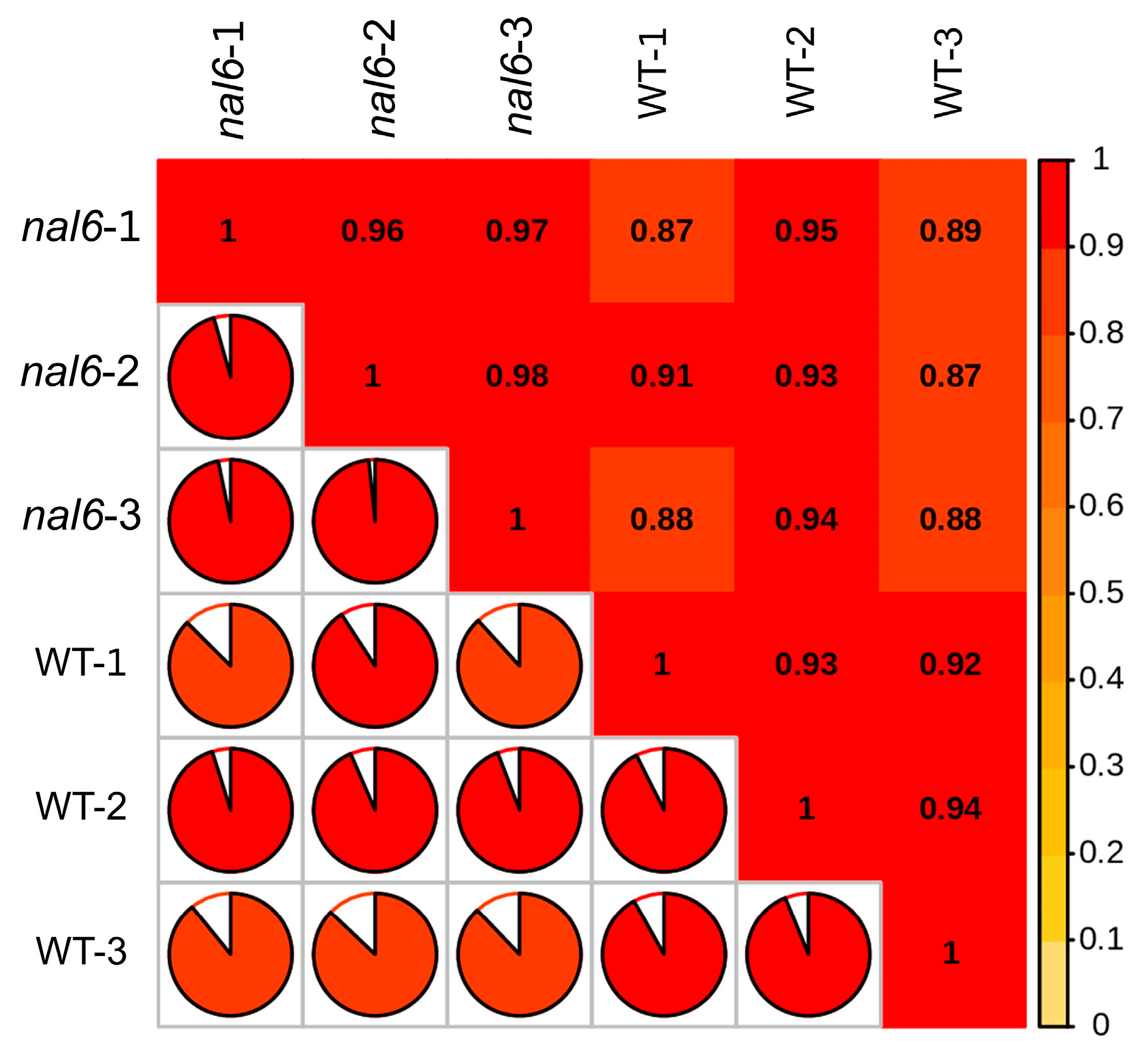
3.2. Comparison and Analysis of Transcriptome Data
3.3. DEG Selection, Functional Annotation, and Enrichment Analysis
3.3.1. DEG Statistical Screening
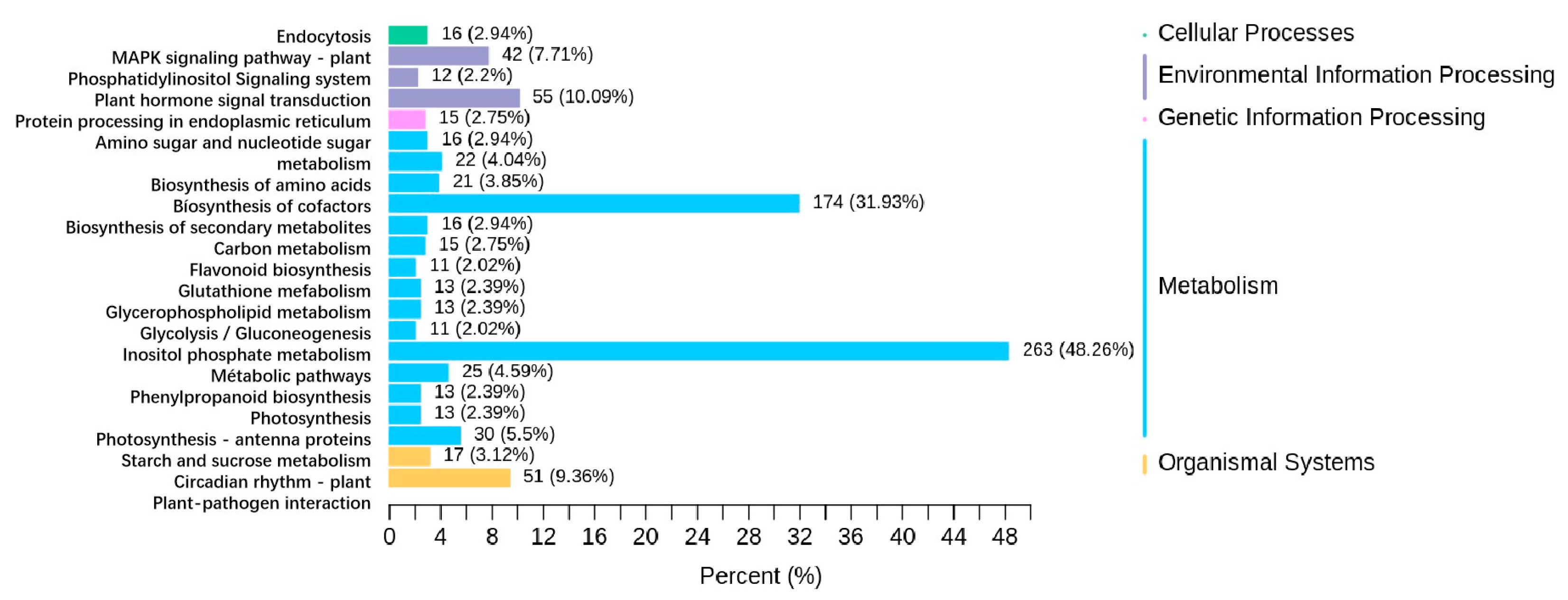
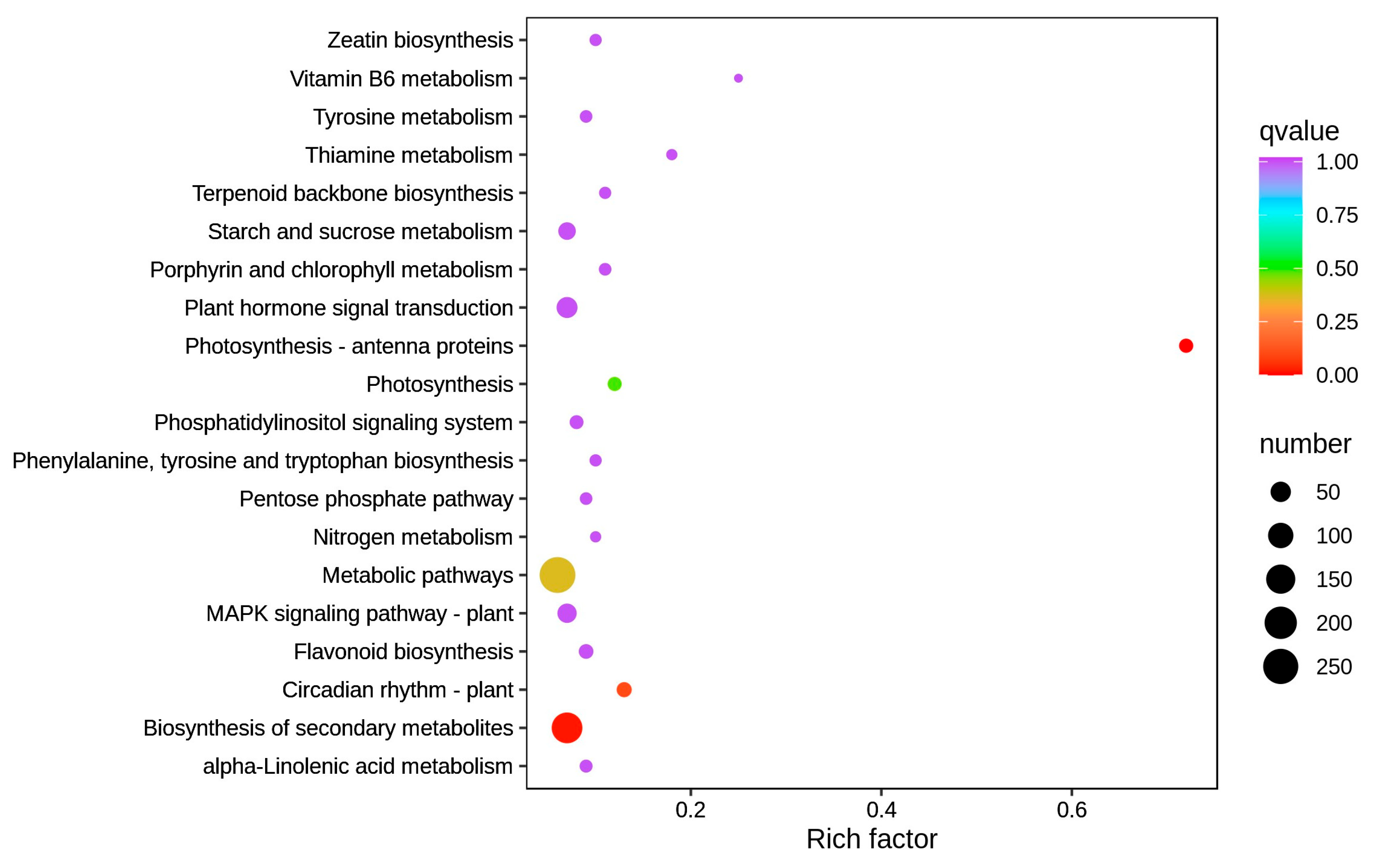
3.3.2. KEGG Annotation and Enrichment Analysis of DEGs
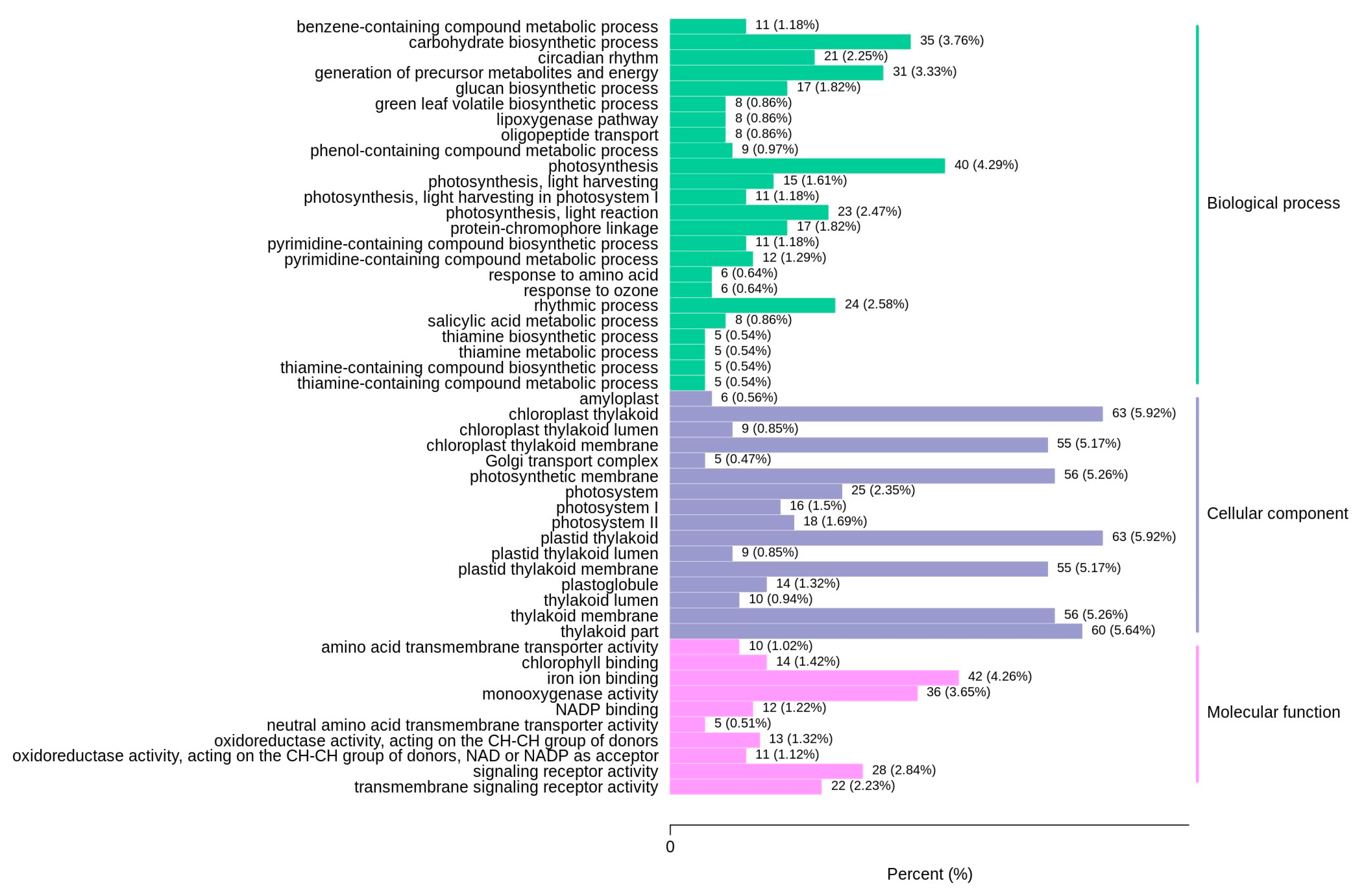
3.3.3. GO Classification and Enrichment Analysis of DEGs
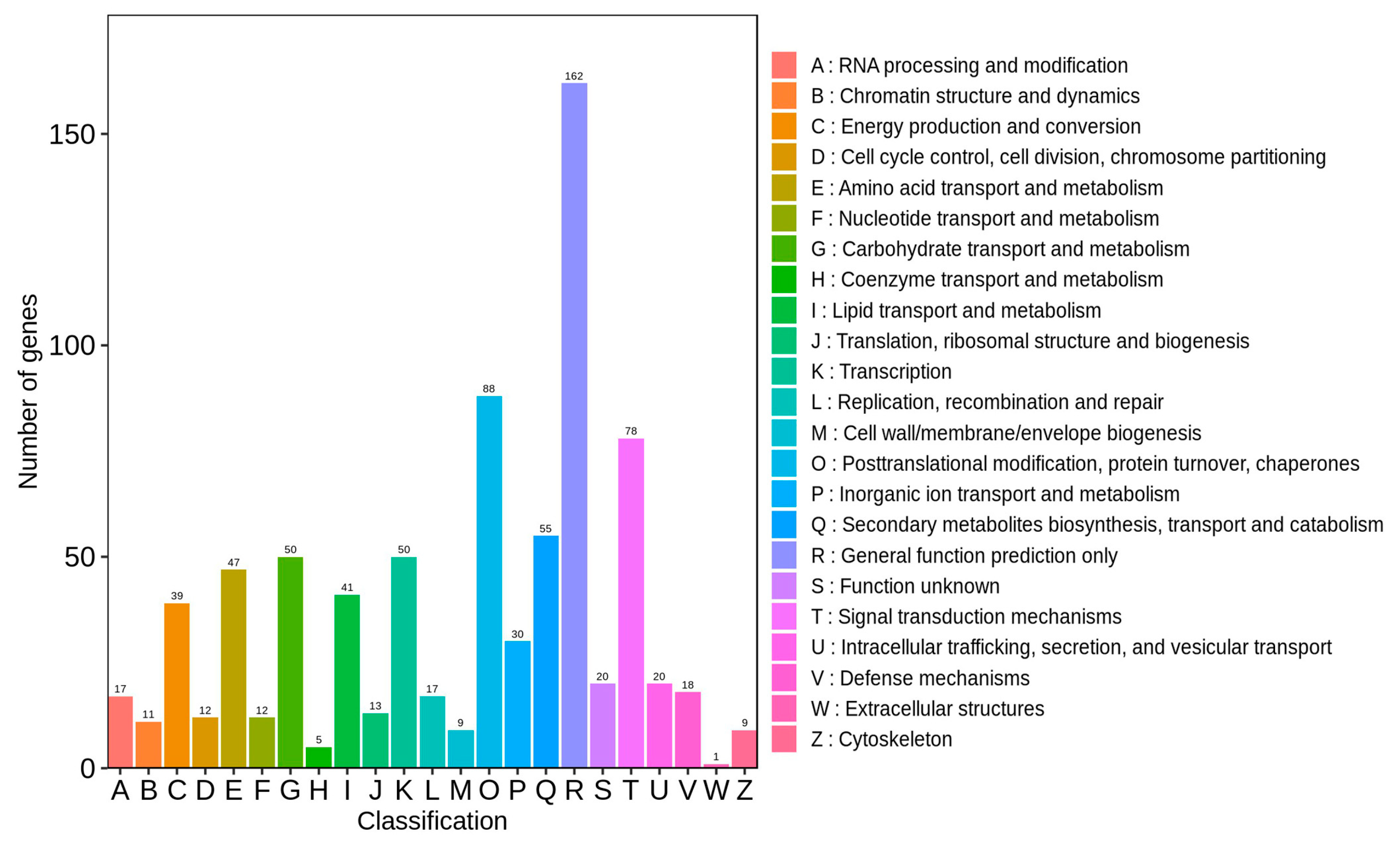
3.3.4. KOG Classification and Enrichment Analysis of DEGs
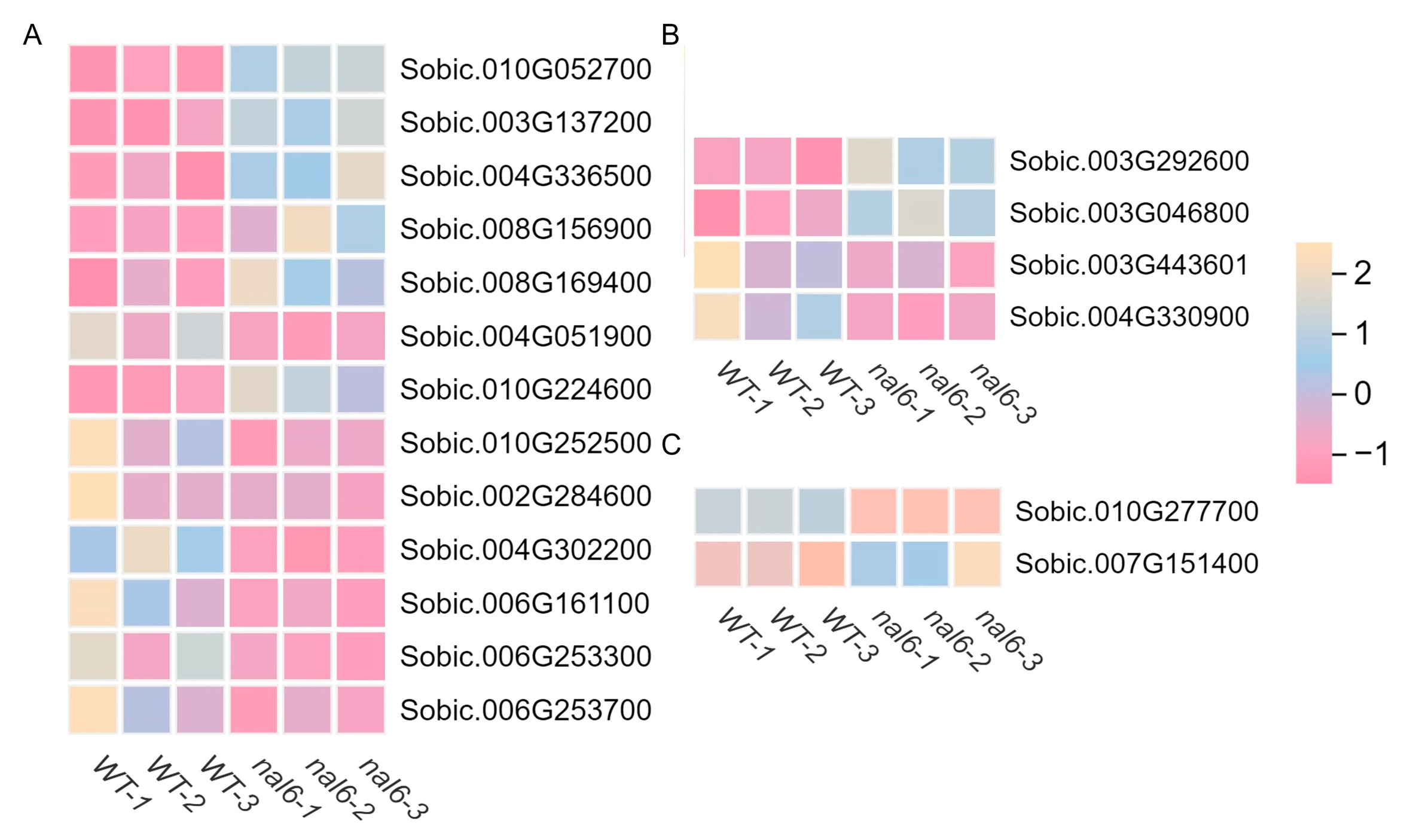
3.4. Mining and Analysis of Key Functional Genes
3.4.1. Mining of Genes Associated with Auxin Signaling and Zeatin Signaling
3.4.2. Mining of Genes Associated with Zeatin Biosynthesis
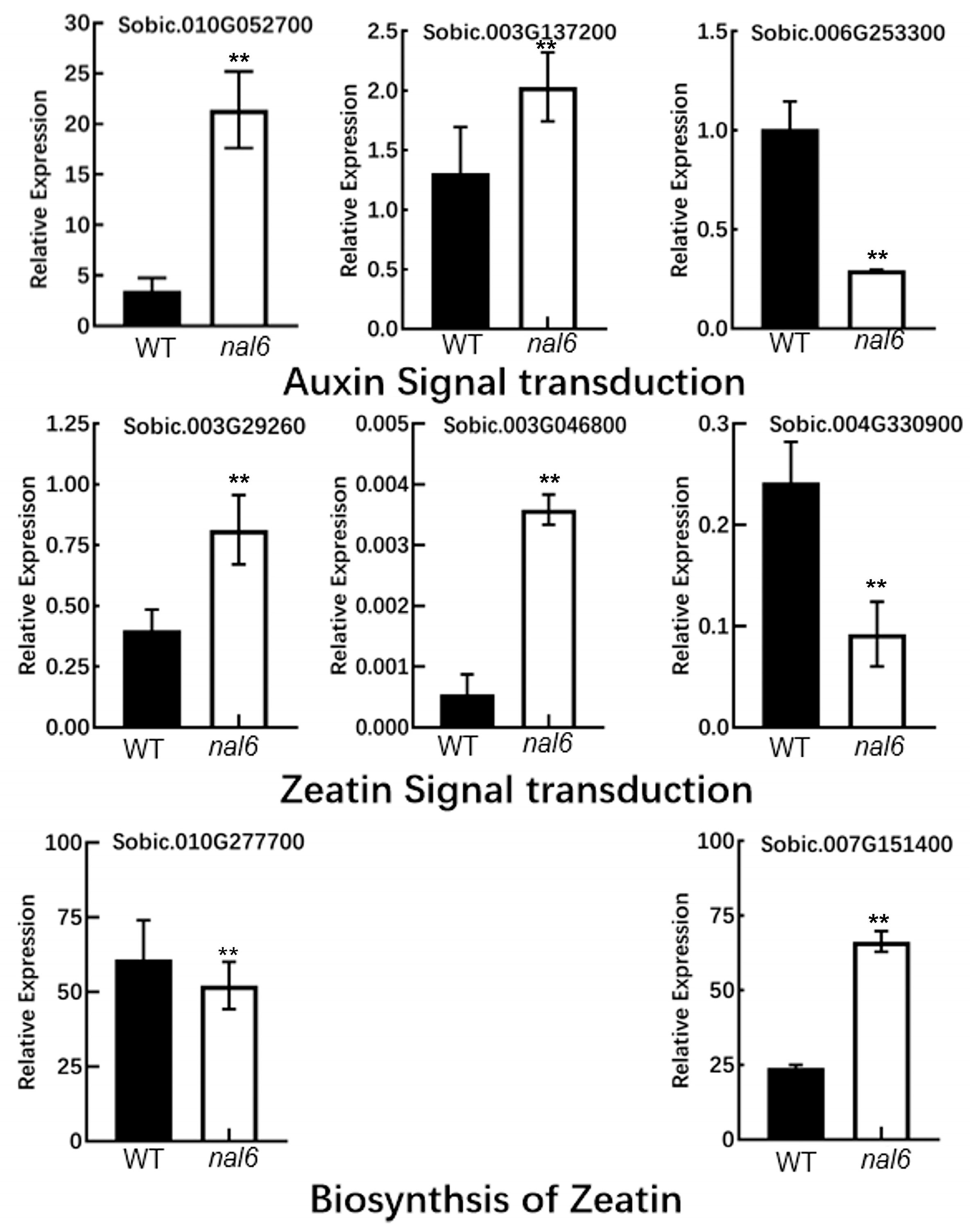
3.5. DEG Analysis by qRT-PCR
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
References
- Bai, S.; Yu, H.; Wang, B.; Li, J. Retrospective and perspective of rice breeding in China. J. Genet. Genom. 2018, 45, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Grown and development of the rice plant. In Fundamentals of Rice Crop Science; IRRI: Los Banos, Philippines, 1981; pp. 1–61. [Google Scholar]
- Zhang, L.Q.; Song, Z.F.; Zhang, Y.J.; Yang, Y.H.; Zhu, X.Y. Study on the relationship between stem tillering structure and leaf area index and yield of rice jointing stage. J. Yangzhou Univ. 2004, 25, 55–58. [Google Scholar] [CrossRef]
- Chen, Z.X.; Pan, X.B.; H, J. Relationship between rice leaf curling traits and ideal plant shape. Jiangsu Agric. Res. 2001, 22, 88–91. [Google Scholar] [CrossRef]
- Yang, S.R.; Zhang, L.B.; Wang, J.M. Preliminary theory and method of ideal plant shape breeding in rice. Sci. Agric. Sin. 1984, 3, 6–13. [Google Scholar]
- Sazuka, T.; Kamiya, N.; Nishimura, T.; Ohmae, K.; Sato, Y.; Imamura, K.; Nagato, Y.; Koshiba, T.; Nagamura, Y.; Ashikari, M.; et al. A rice tryptophan deficient dwarf mutant, tdd1, contains a reduced level of indole acetic acid and develops abnormal flowers and organless embryos. Plant J. 2009, 60, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Fang, J.J.; Lou, L.; Zhao, J.F.; Yuan, S.J.; Yin, L.; Sun, W.; Peng, L.X.; Guo, B.T.; Li, X.Y. Characterization of a null allelic mutant of the rice NAL1 gene reveals its role in regulating cell division. PLoS ONE 2015, 10, e0118169. [Google Scholar] [CrossRef]
- Xu, Y.X.; Xiao, M.Z.; Liu, Y.; Fu, J.L.; He, Y.; Jiang, D.A. The small auxin-up RNA OsSAUR45 affects auxin synthesis and transport in rice. Plant Mol. Biol. 2017, 94, 97–107. [Google Scholar] [CrossRef]
- Qi, J.; Qian, Q.; Bu, Q.Y.; Li, S.Y.; Chen, Q.; Sun, J.Q.; Liang, W.X.; Zhou, Y.H.; Chu, C.C.; Li, X.G.; et al. Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol. 2008, 147, 1947–1959. [Google Scholar] [CrossRef]
- Fujino, K.; Matsuda, Y.; Ozawa, K.; Nishimura, T.; Koshiba, T.; Fraaije, M.W.; Sekiguchi, H. NARROW LEAF 7 controls leaf shape mediated by auxin in rice. Mol. Genet. Genom. 2008, 279, 499–507. [Google Scholar] [CrossRef]
- Ohmori, Y.; Tanaka, W.; Kojima, M.; Sakakibara, H.; Hirano, H.Y. WUSCHEL-RELATED HOMEOBOX4 is involved in meristem maintenance and is negatively regulated by the CLE gene FCP1 in rice. Plant Cell 2013, 25, 229–241. [Google Scholar] [CrossRef]
- Chen, K.; Guo, T.; Li, X.M.; Yang, Y.B.; Dong, N.Q.; Shi, C.L.; Ye, W.W.; Shan, J.X.; Lin, H.X. NAL8 encodes a prohibitin that contributes to leaf and spikelet development by regulating mitochondria and chloroplasts stability in rice. BMC Plant Biol. 2019, 19, 395. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.C.; Yan, S.Y.; Jiang, S.; Bai, L.; Liu, Y.C.; Peng, S.S.; Chen, R.B.; Liu, Q.; Xiao, Y.H.; Kang, H.X. Identification of a Rice Leaf Width Gene Narrow Leaf 22 (NAL22) through Genome-Wide Association Study and Gene Editing Technology. Int. J. Mol. Sci. 2023, 24, 4073. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Ohmori, Y.; Takebayashi, Y.; Sakakibara, H.; Hirano, H.Y. WUSCHEL-RELATED HOMEOBOX4 acts as a key regulator in early leaf development in rice. PLoS Genet. 2018, 14, e1007365. [Google Scholar] [CrossRef] [PubMed]
- Honda, E.; Yew, C.L.; Yoshikawa, T.; Sato, Y.; Hibara, K.I.; Itoh, J.I. LEAF LATERAL SYMMETRY1, a Member of the WUSCHEL-RELATED HOMEOBOX3 Gene Family, Regulates Lateral Organ Development Differentially from Other Paralogs, NARROW LEAF2 and NARROW LEAF3 in Rice. Plant Cell Physiol. 2018, 59, 376–391. [Google Scholar] [CrossRef]
- Zheng, X.L.; Zhang, S.; Liang, Y.L.; Zhang, R.; Liu, L.; Qin, P.C.; Zhang, Z.; Wang, Y.; Zhou, J.P.; Tang, X.; et al. Loss-function mutants of OsCKX gene family based on CRISPR-Cas systems revealed their diversified roles in rice. Plant Genome 2023, 16, e20283. [Google Scholar] [CrossRef]
- Guo, T.T.; Wang, D.F.; Fang, J.J.; Zhao, J.F.; Yuan, S.J.; Xiao, L.T.; Li, X.Y. Mutations in the rice OSCHR4 gene, encoding a CHD3 family chromatin remodeler, induce narrow and rolled leaves with increased cuticular wax. Int. J. Mol. Sci. 2019, 20, 2567. [Google Scholar] [CrossRef]
- Ma, L.; Sang, X.C.; Zhang, T.; Yu, Z.Y.; Li, Y.F.; Zhao, F.M.; Wang, Z.W.; Wang, Y.T.; Yu, P.; Wang, N.; et al. Abnormal VASCULAR Bundles regulates cell proliferation and procambium cell establishment during aerial organ development in rice. New Phytol. 2017, 213, 275–286. [Google Scholar] [CrossRef]
- Wang, H.M.; Shi, Y.F.; Zhang, X.B.; Xu, X.; Wu, J.L. Characterization of a novel rice dynamic narrow-rolled leaf mutant with deficiencies in aromatic amino acids. Int. J. Mol. Sci. 2020, 21, 1521. [Google Scholar] [CrossRef]
- Liu, X.F.; Li, M.; Liu, K.; Tang, D.; Sun, M.F.; Li, Y.F.; Shen, Y.; Du, G.J.; Cheng, Z.K. Semi-rolled Leaf2 modulates rice leaf rolling by regulating abaxial side cell differentiation. J. Exp. Bot. 2016, 67, 2139–2150. [Google Scholar] [CrossRef]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Lanctot, A.; Nemhauser, J.L. It’s morphin’ time: How multiple signals converge on ARF transcription factors to direct development. Curr. Opin. Plant Biol. 2020, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Miotk, A.; Šutiković, Z.; Ermakova, O.; Wenzl, C.; Medzihradszky, A.; Gaillochet, C.; Forner, J.; Utan, G.; Brackmann, K.; et al. WUSCHEL acts as an auxin response rheostat to maintain apical stem cells in arabidopsis. Nat. Commun. 2019, 10, 5093. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Eiguchi, M.; Hibara, K.-I.; Ito, J.-I.; Nagato, Y. Rice slender Leaf 1 gene encodes cellulose synthase-like D4 and is specifically expressed in M-phase cells to regulate cell proliferation. J. Exp. Bot. 2013, 64, 2049–2061. [Google Scholar] [CrossRef]
- Zhao, S.S.; Zhao, L.; Liu, F.X.; Wu, Y.Z.; Zhu, Z.F.; Sun, C.Q.; Tan, L.B. Narrow and rolled Leaf 2 regulates leaf shape, male fertility, and seed size in rice. J. Integr. Plant Biol. 2016, 58, 983–996. [Google Scholar] [CrossRef]
- Li, J.H.; Dong, S.; Li, W.H.; Zhang, L.X.; Lu, X.C. Phenotypic Identification and Genetic Analysis of the Leaf Angle Mutant LA1 in Sorghum. J. Shenyang Agric. Univ. 2021, 52, 460–466. [Google Scholar]
- Dong, S.; Lu, X.C.; Li, J.H.; Han, J.Y. Phenotypic Identification and Genetic Analysis of an Upright Leaf Mutant in Sorghum. J. Plant Genet. Resour. 2022, 23, 177–182. [Google Scholar] [CrossRef]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of Rna-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef]
- Gao, Z.X.; Gang, S.; Lu, X.C.; Wang, P.; Li, Z.J.; Wang, Y.W.; Li, J.H. Mapping and Candidate Gene Analysis of the Yellow Seed 2 Gene for Yellow Grains in Sorghum. J. Plant Genet. Resour. 2024, 25, 1565–1572. [Google Scholar] [CrossRef]
- Meng, B.X.; Wang, T.; Luo, Y.; Xu, D.Z.; Li, L.Z.; Diao, Y.; Gao, Z.Y.; Hu, Z.L.; Zheng, X.F. Genome-wide association study identified novel candidate loci/genes affecting lodging resistance in rice. Genes 2021, 12, 718. [Google Scholar] [CrossRef]
- Ran, X.J.; Zhao, F.; Wang, Y.J.; Liu, J.; Zhuang, Y.L.; Ye, L.H.; Qi, M.F.; Cheng, J.F.; Zhang, Y.J. Plant regulomics: A data-driven interface for retrieving upstream regulators from plant multi-omics data. Plant J. 2020, 101, 237–248. [Google Scholar] [CrossRef]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of differential gene expression results of Rna-seq data: Review and integration. Brief. Bioinf. 2019, 20, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. Rna-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for Rna-seq data with Deseq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Han, L.L.; Jiang, C.G.; Zhang, W.; Wang, H.W.; Li, K.; Liu, X.G.; Liu, Z.F.; Wu, Y.J.; Huang, C.L.; Hu, X.J. Morphological characterization and transcriptome analysis of new dwarf and narrow-leaf (dnl2) mutant in maize. Int. J. Mol. Sci. 2022, 23, 795. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.F.; Gu, J.Y.; Zhao, M.H.; Zhao, L.S.; Guo, H.J.; Xiong, H.C.; Xie, Y.D.; Zhao, S.R.; Ding, Y.P.; Qiao, W.C.; et al. Transcriptome analysis of wheat erect leaf mutant Mths29. J. Plant Genet. Resour. 2022, 23, 871–880. [Google Scholar] [CrossRef]
- Guo, S.L.; Lu, X.M.; Qi, J.S.; Wei, L.M.; Zhang, X.; Han, X.H.; Yue, R.Q.; Wang, Z.H.; Tie, S.G.; Chen, Y.H. Exploration of regulatory genes related to morphogenesis in maize leaves using Rna-seq. Chin. J. Agric. Sci. 2020, 53, 1–17. [Google Scholar] [CrossRef]
- Israeli, A.; Capua, Y.; Shwartz, I.; Tal, L.; Meir, Z.; Levy, M.; Bar, M.; Efroni, I.; Ori, N. Multiple auxin-response regulators enable stability and variability in leaf development. Curr. Biol. 2019, 29, 1746–1759. [Google Scholar] [CrossRef]
- Satterlee, J.W.; Scanlon, M.J. Coordination of leaf development across developmental axes. Plants 2019, 8, 433. [Google Scholar] [CrossRef]
- Lawson, T.; Matthews, J. Guard cell metabolism and stomatal function. Annu. Rev. Plant Biol. 2020, 71, 273–302. [Google Scholar] [CrossRef]
- Qin, W.T.; Yang, C.Q.; Deng, J.C.; Iqbal, N.; Yang, F.; Wang, X.C.; Yang, W.Y.; Liu, J. Transcriptomic analysis of soybean seed aging. J. Northeast Agric. Univ. 2018, 49, 1–9. [Google Scholar] [CrossRef]
- Saha, A.; Das, S.; Moin, M.; Dutta, M.; Bakshi, A.; Madhav, M.S.; Kirti, P.B. Genome-wide identification and comprehensive expression profiling of ribosomal protein small subunit (RPS) genes and their comparative analysis with the large subunit (RPL) genes in rice. Front. Plant Sci. 2017, 8, 1553. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.X.; Wang, X.F.; Liu, R.H.; Du, W.; Ma, M.C.; Han, Y.H.; Li, H.Y.; Liu, L.L.; Hou, S.Y. Comparative transcriptomic analysis reveals the regulatory mechanism of the gibberellic acid pathway of tartary buckwheat (Fagopyrum tataricum (L.) Gaertn.) dwarf mutants. BMC Plant Biol. 2021, 21, 206. [Google Scholar] [CrossRef]
- Wang, C.Y.; Zhu, Z.X.; Li, D.; Cong, L.; Zhang, L.X. Screening and identification of EMS mutagenesis and mutants in sorghum. Biotechnol. Bull. 2014, 9, 78–83. [Google Scholar] [CrossRef]
- Yang, Q.; Yin, J.J.; Li, G.; Qi, L.; Yang, F.; Wang, R.; Li, G.J. Reference gene selection for Qrt-pcr in Caragana korshinskii Kom. under different stress conditions. Mol. Biol. Rep. 2014, 41, 2325–2334. [Google Scholar] [CrossRef]
- Bae, K.-D.; Um, T.-Y.; Yang, W.-T.; Park, T.-H.; Hong, S.-Y.; Kim, K.-M.; Chung, Y.-S.; Yun, D.-J.; Kim, D.-H. Characterization of dwarf and narrow leaf (dnl-4) mutant in rice. Plant Signal. Behav. 2021, 16, 1849490. [Google Scholar] [CrossRef]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inzé, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Gajdosová, S.; Spíchal, L.; Kamínek, M.; Hoyerová, K.; Novák, O.; Dobrev, P.I.; Galuszka, P.; Klíma, P.; Gaudinová, A.; Zizková, E.; et al. Distribution, biological activities, metabolism, and the conceivable function of cis-zeatin-type cytokinins in plants. J. Exp. Bot. 2011, 62, 2827–2840. [Google Scholar] [CrossRef]
- Thilakarathne, A.S.; Liu, F.; Zou, Z. Plant Signaling Hormones and Transcription Factors: Key Regulators of Plant Responses to Growth, Development, and Stress. Plants 2025, 14, 1070. [Google Scholar] [CrossRef]
- Korasick, D.A.; Jez, J.M.; Strader, L.C. Refining the nuclear auxin response pathway through structural biology. Curr. Opin. Plant Biol. 2015, 27, 22–28. [Google Scholar] [CrossRef]
- Zhou, D.; Li, Y.J.; Xie, X.R.; Ding, W.Y.; Chen, L.B.; Li, T.; Tang, J.N.; Tan, X.Y.; Liu, W.Z.; Heng, Y.Q. Copy number variation of NAL23 causes narrow-leaf development in rice. J. Genet. Genom. 2024, 51, 880–883. [Google Scholar] [CrossRef]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family in rice (Oryza sativa). Genomics 2006, 88, 360–371. [Google Scholar] [CrossRef]
- Du, M.Y.; Li, R.; Ruan, G.X.; Li, Y.S.; Wan, P.; Yang, K. Transcriptome analysis and hormone response of adzuki bean dwarf stalk narrow leaf mutant. J. Beijing Univ. Agric. 2020, 35, 18–24. [Google Scholar] [CrossRef]
- Wang, B.; Smith, S.M.; Li, J.Y. Genetic regulation of shoot architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.Q.; Liu, Q.Y.; Wang, B.S.; Yuan, F. Roles of phytohormones and their signaling pathways in leaf development and stress responses. J. Agric. Food Chem. 2021, 69, 3566–3584. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, D.; Yan, S.; Liu, S.M.; Liu, B.; Kang, H.X.; Wang, G.-L. Dissection of the genetic architecture of rice tillering using a genome-wide association study. Rice 2019, 12, 43. [Google Scholar] [CrossRef]
- Roy, N.; Kabir, A.H.; Zahan, N.; Mouna, S.T.; Chakravarty, S.; Rahman, A.H.; Bayzid, M.S. Genome wide association studies on seven yield-related traits of 183 rice varieties in Bangladesh. Plant Direct 2024, 8, e593. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Pathway Name/Function | Sequence |
|---|---|---|
| Sobic.010G052700F | Auxin signal transduction | AAGGTCAAGATGGAAGGGGTG |
| Sobic.010G052700R | Auxin signal transduction | TTCTTCTGCATGACCTGCATC |
| Sobic.003G137200F | Auxin signal transduction | TCGTCCATGGATAGCAGCAC |
| Sobic.003G137200R | Auxin signal transduction | GCGTAGAAGGGATGCTCCTC |
| Sobic.006G253300F | Auxin signal transduction | AGGAGGAGTACGGCTTCCC |
| Sobic.006G253300R | Auxin signal transduction | GTTGTGATCCACGGCCATTC |
| Sobic.003G292600F | Zeatin signal transduction | GTGGAAGACTTGCAGGACGA |
| Sobic.003G292600R | Zeatin signal transduction | CCTTGAAGCACCAATGCTGG |
| Sobic.003G046800F | Zeatin signal transduction | CGTAGTACAGAACGGCGTCA |
| Sobic.003G046800R | Zeatin signal transduction | ATCCACCTTCATCAGCTCGC |
| Sobic.004G330900F | Zeatin signal transduction | GTCCAGCTGCCACAAAAGTG |
| Sobic.004G330900R | Zeatin signal transduction | ACATTCTGCTGGGATGCACA |
| Sobic.010G277700F | Biosynthesis of zeatin | AGTGCCATCGGTTACAAGCA |
| Sobic.010G277700R | Biosynthesis of zeatin | TGCAGCCATGGTAAGCATCA |
| Sobic.007G151400F | Biosynthesis of zeatin | CGGAGGGCGAGATCTTCTAC |
| Sobic.007G151400R | Biosynthesis of zeatin | ATCGCCAGCGGGTCGTA |
| Sobic.001G112600F | internal standard | ATGGCTGACGCCGAGGATATCCA |
| Sobic.001G112600R | internal standard | GAGCCACACGGAGCTCGTTGTAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Y.; Zhao, Y.; Zhang, S.; Wang, C.; Cong, L.; Zhang, Y.; Gang, S.; Lu, X. Transcriptome Differential Expression Regulation Analysis of the Narrow-Leaf Mutant of Sorghum Bicolor. Agronomy 2025, 15, 1432. https://doi.org/10.3390/agronomy15061432
Li J, Wang Y, Zhao Y, Zhang S, Wang C, Cong L, Zhang Y, Gang S, Lu X. Transcriptome Differential Expression Regulation Analysis of the Narrow-Leaf Mutant of Sorghum Bicolor. Agronomy. 2025; 15(6):1432. https://doi.org/10.3390/agronomy15061432
Chicago/Turabian StyleLi, Jinhong, Yiwei Wang, Yuche Zhao, Shirui Zhang, Chunyu Wang, Ling Cong, Yanpeng Zhang, Shuang Gang, and Xiaochun Lu. 2025. "Transcriptome Differential Expression Regulation Analysis of the Narrow-Leaf Mutant of Sorghum Bicolor" Agronomy 15, no. 6: 1432. https://doi.org/10.3390/agronomy15061432
APA StyleLi, J., Wang, Y., Zhao, Y., Zhang, S., Wang, C., Cong, L., Zhang, Y., Gang, S., & Lu, X. (2025). Transcriptome Differential Expression Regulation Analysis of the Narrow-Leaf Mutant of Sorghum Bicolor. Agronomy, 15(6), 1432. https://doi.org/10.3390/agronomy15061432