
Comparison of Single-Trait and Multi-Trait GBLUP Models for Genomic Prediction in Red Clover


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Planting, Sampling, and DNA Extraction
2.3. Genotyping-by-Sequencing (GBS) and Read Pre-Processing
2.4. Read Alignment and SNP Discovery
2.5. Field Trials and Phenotyping
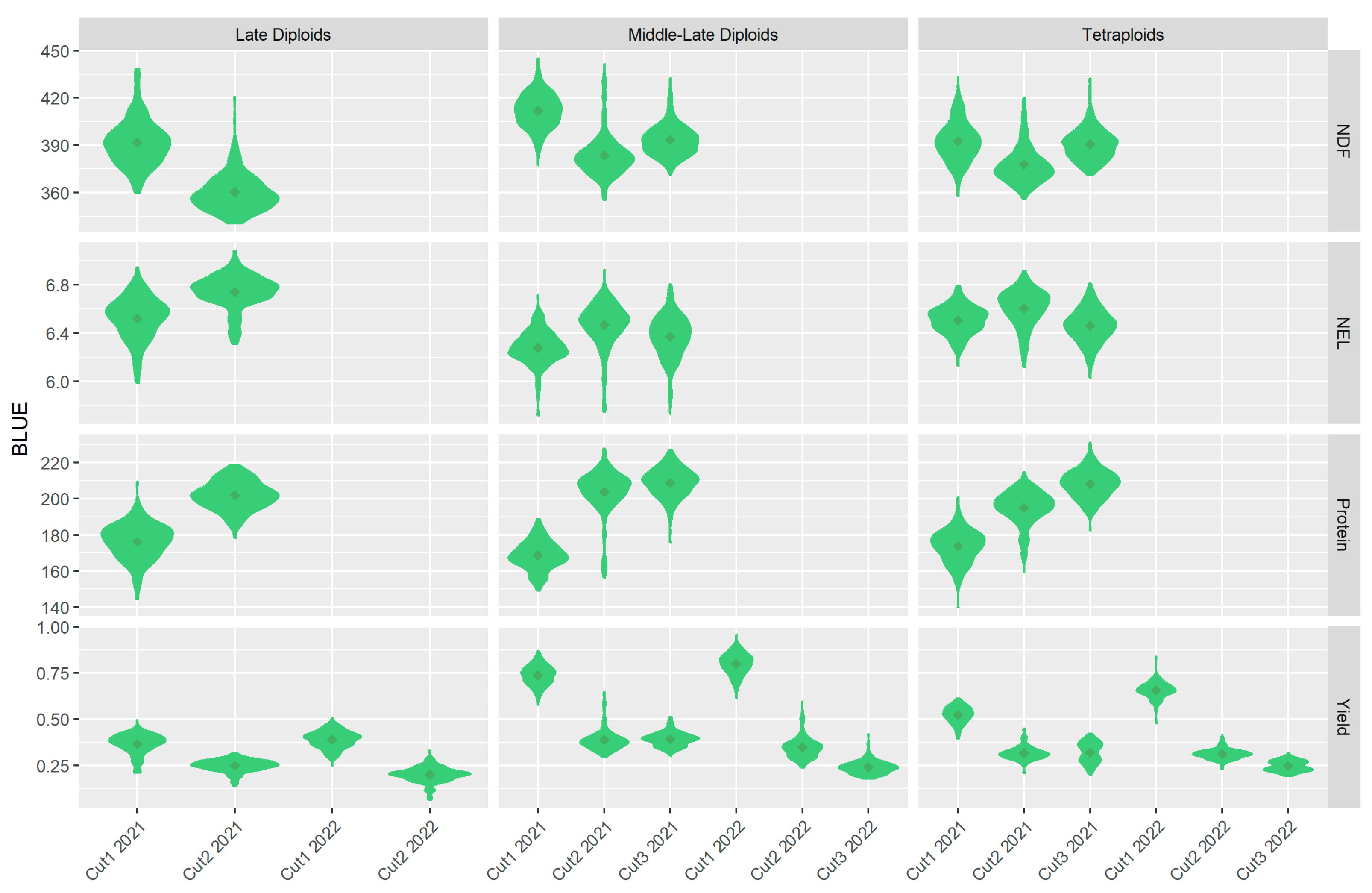
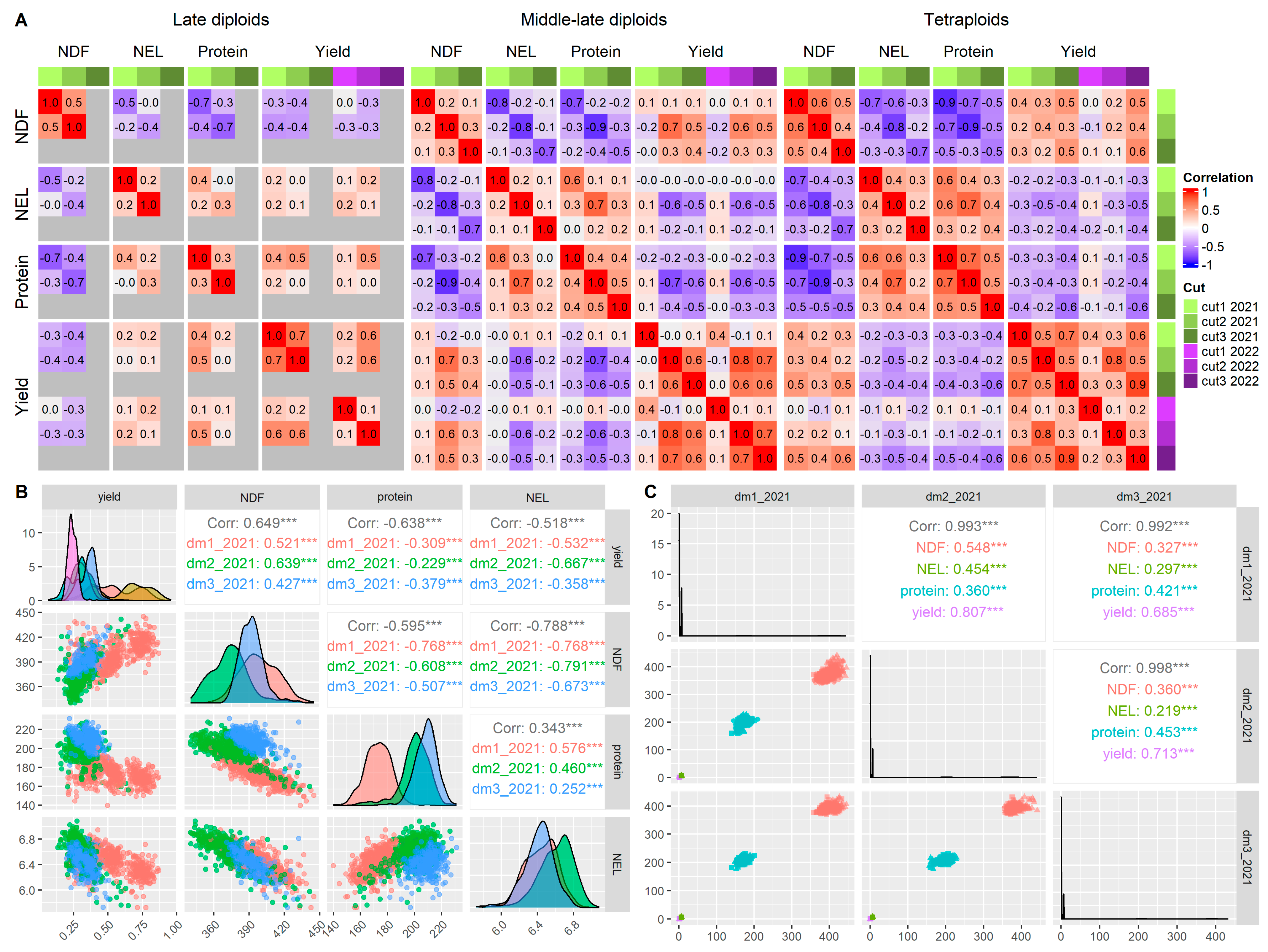
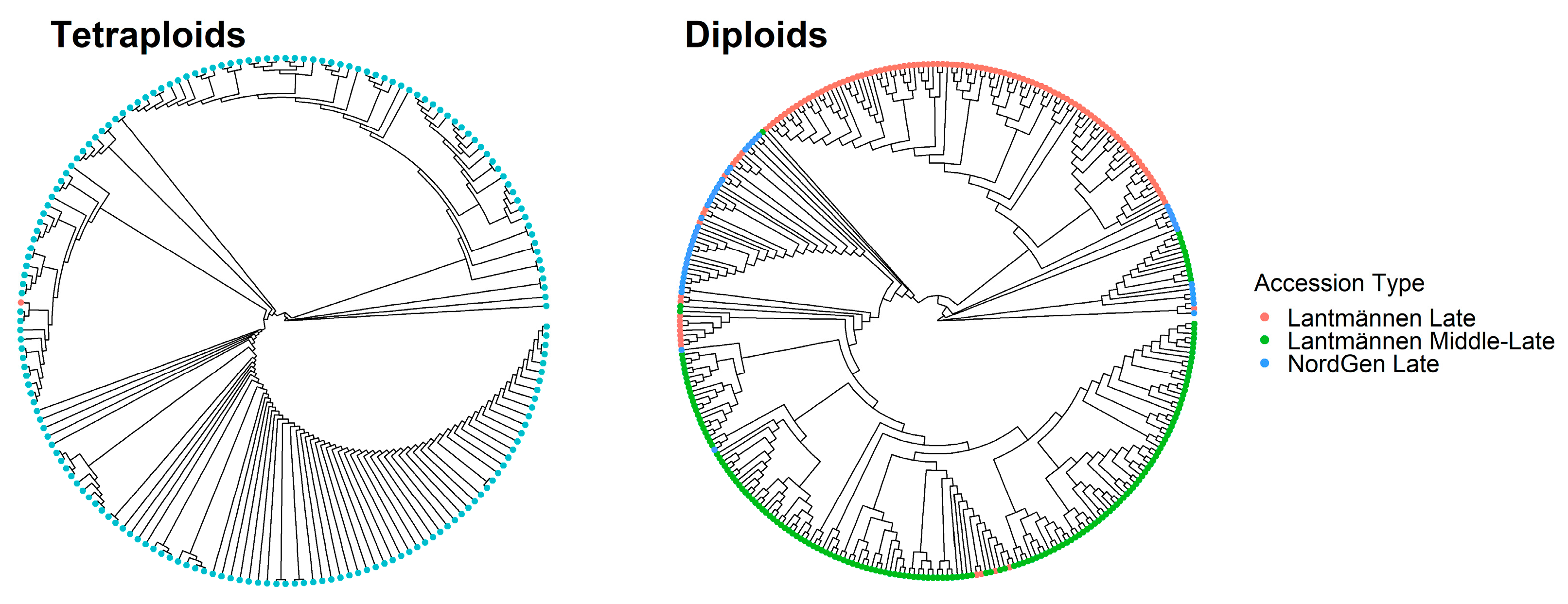
2.6. Population Structure and Phenotypic Data Evaluation
2.7. The Different Models
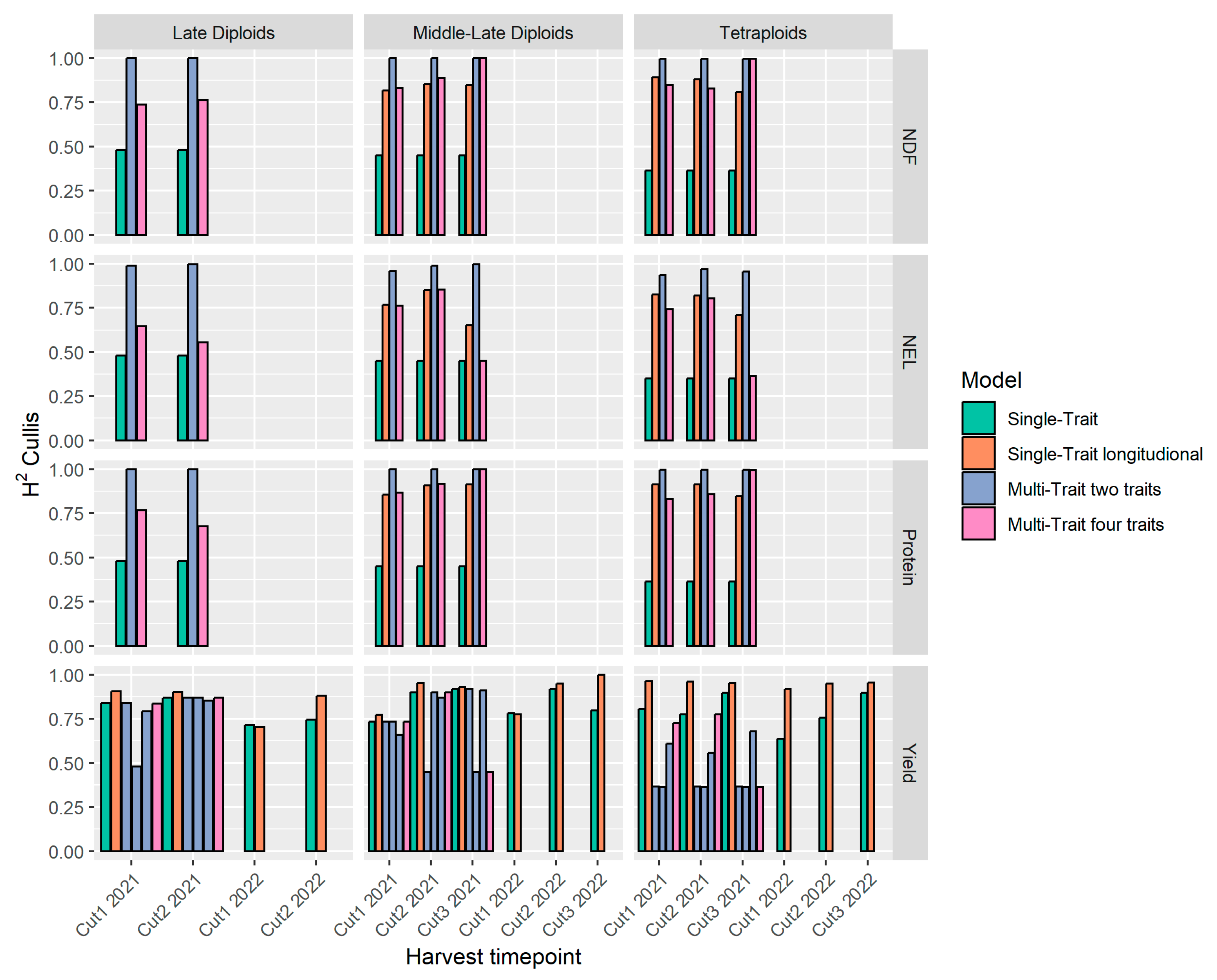
2.8. Predictive Ability and Reliability
3. Results
3.1. Genotyping
3.2. Genetic Diversity of Accessions
3.3. Model Reliability and Predictive Ability
4. Discussion
4.1. Genotype and Phenotype Data
4.2. Trial Design and BLUEs
4.3. Correlations Between Traits and Their Effect on Predictability and Reliability
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, R.R.; Taylor, N.L.; Bowley, S.R. Red Clover. In Clover Science and Technology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1985; pp. 457–470. ISBN 978-0-89118-218-4. [Google Scholar]
- Taylor, N.L.; Quesenberry, K.H. Historical Perspectives. In Red Clover Science; Taylor, N.L., Quesenberry, K.H., Eds.; Current Plant Science and Biotechnology in Agriculture; Springer: Dordrecht, The Netherlands, 1996; pp. 1–10. ISBN 978-94-015-8692-4. [Google Scholar]
- Sato, S.; Isobe, S.; Asamizu, E.; Ohmido, N.; Kataoka, R.; Nakamura, Y.; Kaneko, T.; Sakurai, N.; Okumura, K.; Klimenko, I.; et al. Comprehensive Structural Analysis of the Genome of Red Clover (Trifolium pratense L.). DNA Res. 2005, 12, 301–364. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.L.; Quesenberry, K.H. Tetraploid Red Clover. In Red Clover Science; Taylor, N.L., Quesenberry, K.H., Eds.; Current Plant Science and Biotechnology in Agriculture; Springer: Dordrecht, The Netherlands, 1996; pp. 161–169. ISBN 978-94-015-8692-4. [Google Scholar]
- Taylor, N.L.; Giri, N. Frequency and Stability of Tetraploids from 2X–4X Crosses in Red Clover1. Crop Sci. 1983, 23, 1191–1194. [Google Scholar] [CrossRef]
- Öhberg, H. Studies of the Persistence of Red Clover Cultivars in Sweden. 2008. Available online: https://pub.epsilon.slu.se/1741/ (accessed on 7 July 2021).
- Amdahl, H.; Aamlid, T.S.; Ergon, Å.; Kovi, M.R.; Marum, P.; Alsheikh, M.; Rognli, O.A. Seed Yield of Norwegian and Swedish Tetraploid Red Clover (Trifolium pratense L.) Populations. Crop Sci. 2016, 56, 603–612. [Google Scholar] [CrossRef]
- Zanotto, S.; Palmé, A.; Helgadóttir, Á.; Daugstad, K.; Isolahti, M.; Öhlund, L.; Marum, P.; Moen, M.A.; Veteläinen, M.; Rognli, O.A.; et al. Trait Characterization of Genetic Resources Reveals Useful Variation for the Improvement of Cultivated Nordic Red Clover. J. Agron. Crop Sci. 2021, 207, 492–503. [Google Scholar] [CrossRef]
- Osterman, J.; Hammenhag, C.; Ortiz, R.; Geleta, M. Insights into the Genetic Diversity of Nordic Red Clover (Trifolium pratense) Revealed by SeqSNP-Based Genic Markers. Front. Plant Sci. 2021, 12, 2402. [Google Scholar] [CrossRef]
- Osterman, J.; Hammenhag, C.; Ortiz, R.; Geleta, M. Discovering Candidate SNPs for Resilience Breeding of Red Clover. Front. Plant Sci. 2022, 13, 997860. [Google Scholar] [CrossRef] [PubMed]
- Jordbruksaktuellt 23 år innan ny sort når Marknaden. Available online: https://www.ja.se/artikel/2226512/23-r-innan-ny-sort-nr-marknaden.html (accessed on 2 November 2020).
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Hayes, B.J.; Cogan, N.O.I.; Pembleton, L.W.; Goddard, M.E.; Wang, J.; Spangenberg, G.C.; Forster, J.W. Prospects for Genomic Selection in Forage Plant Species. Plant Breed. 2013, 132, 133–143. [Google Scholar] [CrossRef]
- Taylor, N.L.; Quesenberry, K.H. Reproductive Biology, Genetics and Evolution. In Red Clover Science; Taylor, N.L., Quesenberry, K.H., Eds.; Current Plant Science and Biotechnology in Agriculture; Springer: Dordrecht, The Netherlands, 1996; pp. 25–43. ISBN 978-94-015-8692-4. [Google Scholar]
- Futschik, A.; Schlötterer, C. The Next Generation of Molecular Markers from Massively Parallel Sequencing of Pooled DNA Samples. Genetics 2010, 186, 207–218. [Google Scholar] [CrossRef]
- Frey, L.A.; Vleugels, T.; Ruttink, T.; Schubiger, F.X.; Pegard, M.; Skøt, L.; Grieder, C.; Studer, B.; Roldán-Ruiz, I.; Kölliker, R. Phenotypic Variation and Quantitative Trait Loci for Resistance to Southern Anthracnose and Clover Rot in Red Clover. Theor. Appl. Genet. 2022, 135, 4337–4349. [Google Scholar] [CrossRef]
- Nay, M.M.; Grieder, C.; Frey, L.A.; Amdahl, H.; Radovic, J.; Jaluvka, L.; Palmé, A.; Skøt, L.; Ruttink, T.; Kölliker, R. Multi-Location Trials and Population-Based Genotyping Reveal High Diversity and Adaptation to Breeding Environments in a Large Collection of Red Clover. Front. Plant Sci. 2023, 14, 1128823. [Google Scholar] [CrossRef] [PubMed]
- Fè, D.; Cericola, F.; Byrne, S.; Lenk, I.; Ashraf, B.H.; Pedersen, M.G.; Roulund, N.; Asp, T.; Janss, L.; Jensen, C.S.; et al. Genomic Dissection and Prediction of Heading Date in Perennial Ryegrass. BMC Genom. 2015, 16, 921. [Google Scholar] [CrossRef] [PubMed]
- Skøt, L.; Nay, M.M.; Grieder, C.; Frey, L.A.; Pégard, M.; Öhlund, L.; Amdahl, H.; Radovic, J.; Jaluvka, L.; Palmé, A.; et al. Including marker x environment interactions improves genomic prediction in red clover (Trifolium pratense L.). Front. Plant Sci. 2024, 15, 1407609. [Google Scholar] [CrossRef] [PubMed]
- Tucak, M.; Popović, S.; Čupić, T.; Španić, V.; Meglič, V. Variation in Yield, Forage Quality and Morphological Traits of Red Clover (Trifolium pratense L.) Breeding Populations and Cultivars. Zemdirb.-Agric. 2013, 100, 63–70. [Google Scholar] [CrossRef]
- Abd El Moneim, A.M.; Khair, M.A.; Rihawi, S. Effect of Genotypes and Plant Maturity on Forage Quality of Certain Forage Legume Species Under Rainfed Conditions. J. Agron. Crop Sci. 1990, 164, 85–92. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics, 4th ed.; Addison Wesley Longman: Harlow, UK, 1996. [Google Scholar]
- Semagn, K.; Crossa, J.; Cuevas, J.; Iqbal, M.; Ciechanowska, I.; Henriquez, M.A.; Randhawa, H.; Beres, B.L.; Aboukhaddour, R.; McCallum, B.D.; et al. Comparison of Single-Trait and Multi-Trait Genomic Predictions on Agronomic and Disease Resistance Traits in Spring Wheat. Theor. Appl. Genet. 2022, 135, 2747–2767. [Google Scholar] [CrossRef]
- Cuevas, J.; Reslow, F.; Crossa, J.; Ortiz, R. Modeling Genotype × Environment Interaction for Single and Multitrait Genomic Prediction in Potato (Solanum tuberosum L.). G3 Genes Genomes Genet. 2023, 13, jkac322. [Google Scholar] [CrossRef]
- De Vega, J.J.; Ayling, S.; Hegarty, M.; Kudrna, D.; Goicoechea, J.L.; Ergon, Å.; Rognli, O.A.; Jones, C.; Swain, M.; Geurts, R.; et al. Red Clover (Trifolium pratense L.) Draft Genome Provides a Platform for Trait Improvement. Sci. Rep. 2015, 5, 17394. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Empfehlungen zur Energie und Nährstoffversorgung der Milchkühe und Aufzuchtrinder; DLG Verlag: Frankfurt, Germany, 2001; Available online: https://www.dlg-verlag.de/shop/empfehlungen-zur-energie-und-nahrstoffversorgung-von-milchkuhen.html (accessed on 30 March 2024).
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Moehring, J.; Williams, E.R.; Piepho, H.-P. Efficiency of Augmented P-Rep Designs in Multi-Environmental Trials. Theor. Appl. Genet. 2014, 127, 1049–1060. [Google Scholar] [CrossRef]
- Leto, J.; Knežević, M.; Bošnjak, K.; Maćešić, D.; Štafa, Z.; Kozumplik, V. Yield and Forage Quality of Red Clover (Trifolium pratense L.) Cultivars in the Lowland and the Mountain Regions. Plant Soil Environ. 2004, 50, 391–396. [Google Scholar] [CrossRef]
- Jambagi, S.; Hodén, K.P.; Öhlund, L.; Dixelius, C. Red Clover Root-Associated Microbiota Is Shaped by Geographic Location and Choice of Farming System. J. Appl. Microbiol. 2023, 134, lxad067. [Google Scholar] [CrossRef]
- Bohmanova, J.; Miglior, F.; Jamrozik, J.; Misztal, I.; Sullivan, P.G. Comparison of Random Regression Models with Legendre Polynomials and Linear Splines for Production Traits and Somatic Cell Score of Canadian Holstein Cows. J. Dairy Sci. 2008, 91, 3627–3638. [Google Scholar] [CrossRef]
- Campbell, M.; Momen, M.; Walia, H.; Morota, G. Leveraging Breeding Values Obtained from Random Regression Models for Genetic Inference of Longitudinal Traits. Plant Genome 2019, 12, 180075. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osterman, J.; Gutiérrez, L.; Öhlund, L.; Ortiz, R.; Hammenhag, C.; Parsons, D.; Geleta, M. Comparison of Single-Trait and Multi-Trait GBLUP Models for Genomic Prediction in Red Clover. Agronomy 2024, 14, 2445. https://doi.org/10.3390/agronomy14102445
Osterman J, Gutiérrez L, Öhlund L, Ortiz R, Hammenhag C, Parsons D, Geleta M. Comparison of Single-Trait and Multi-Trait GBLUP Models for Genomic Prediction in Red Clover. Agronomy. 2024; 14(10):2445. https://doi.org/10.3390/agronomy14102445
Chicago/Turabian StyleOsterman, Johanna, Lucia Gutiérrez, Linda Öhlund, Rodomiro Ortiz, Cecilia Hammenhag, David Parsons, and Mulatu Geleta. 2024. "Comparison of Single-Trait and Multi-Trait GBLUP Models for Genomic Prediction in Red Clover" Agronomy 14, no. 10: 2445. https://doi.org/10.3390/agronomy14102445
APA StyleOsterman, J., Gutiérrez, L., Öhlund, L., Ortiz, R., Hammenhag, C., Parsons, D., & Geleta, M. (2024). Comparison of Single-Trait and Multi-Trait GBLUP Models for Genomic Prediction in Red Clover. Agronomy, 14(10), 2445. https://doi.org/10.3390/agronomy14102445