
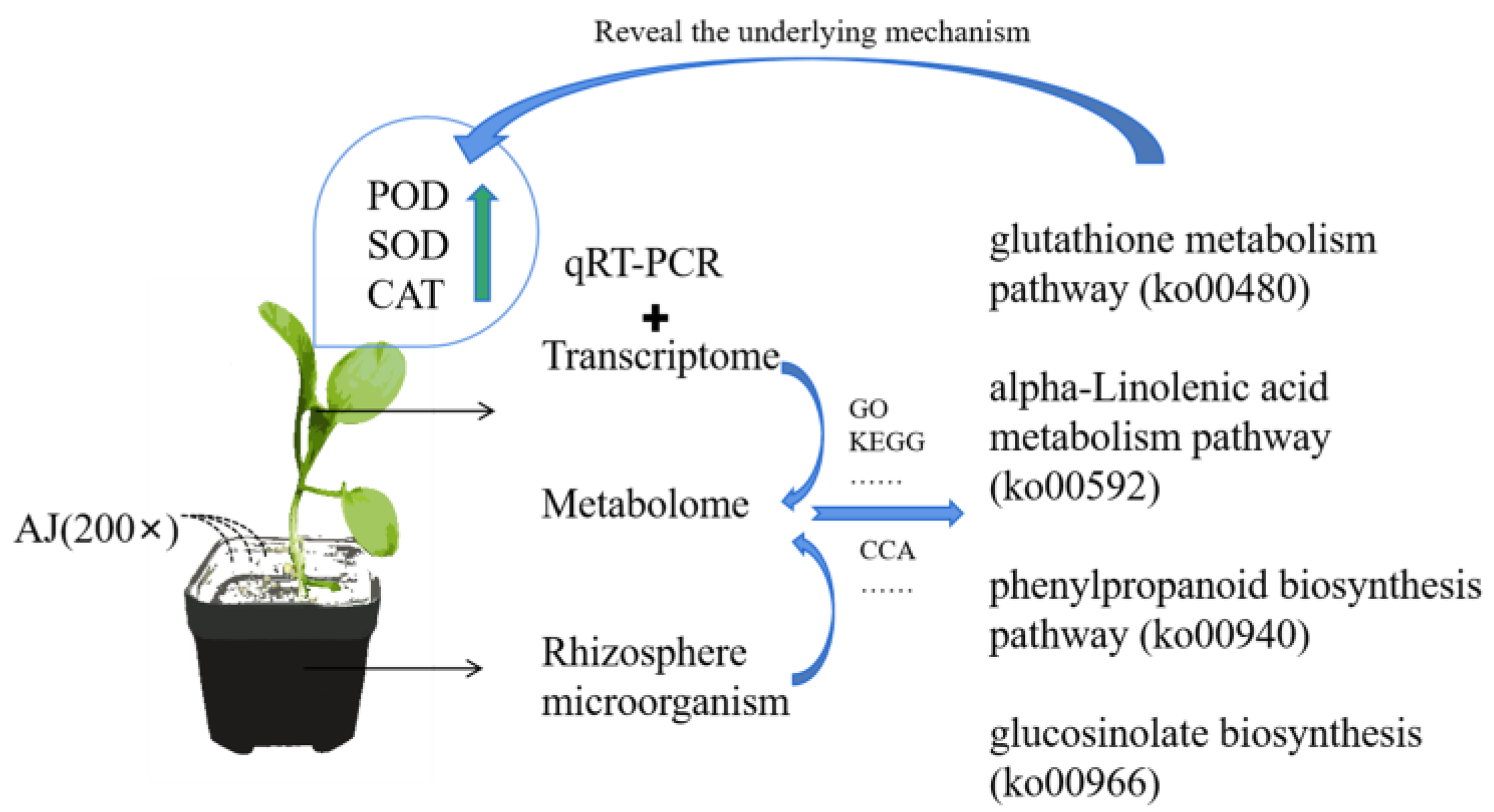
Agricultural Jiaosu Enhances the Stress Resistance of Pak Choi (Brassica rapa L. subsp. chinensis) by Recruiting Beneficial Rhizosphere Bacteria and Altering Metabolic Pathways



Abstract
:1. Introduction
2. Materials and Methods
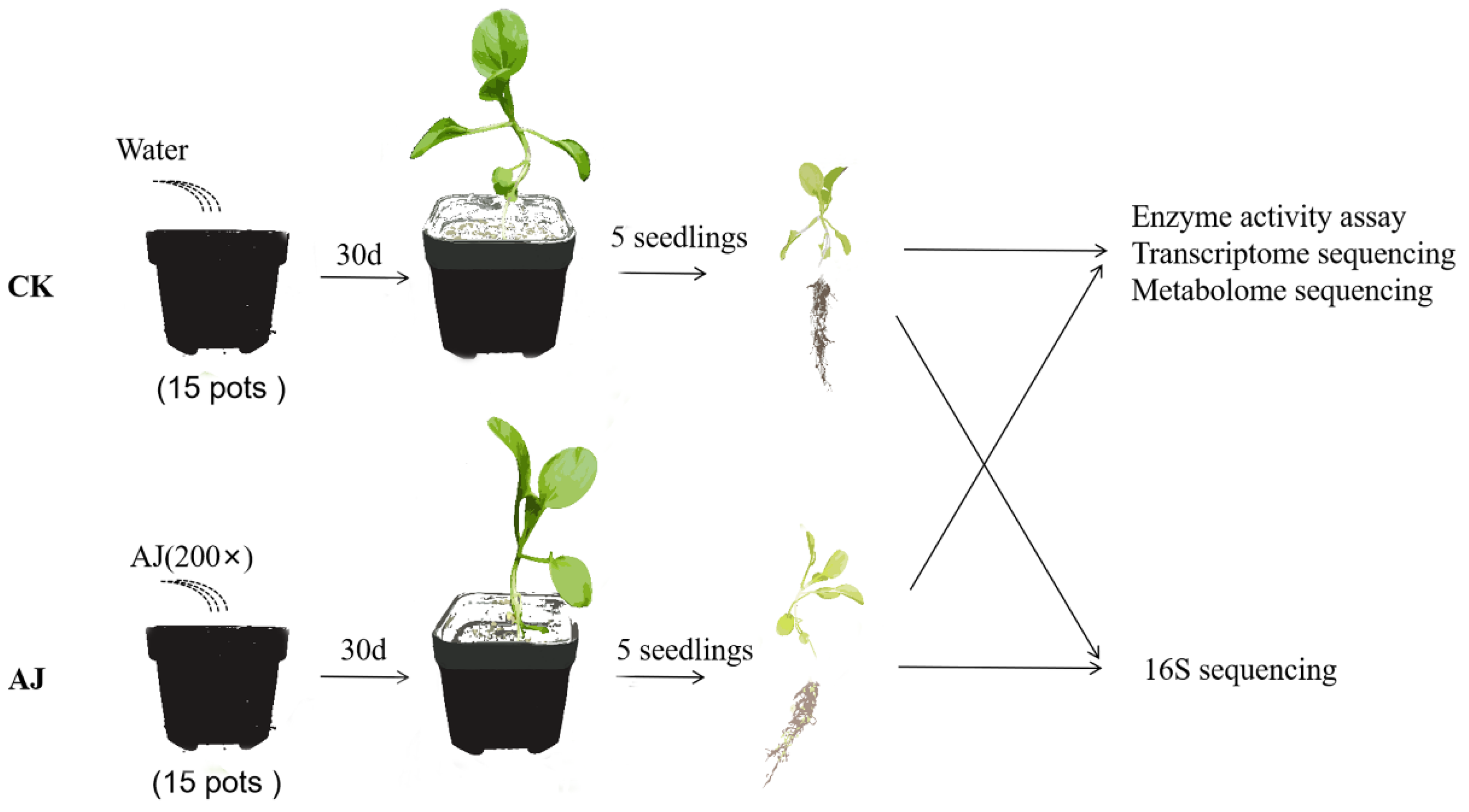
2.1. Plant Materials and Conditions of Planting
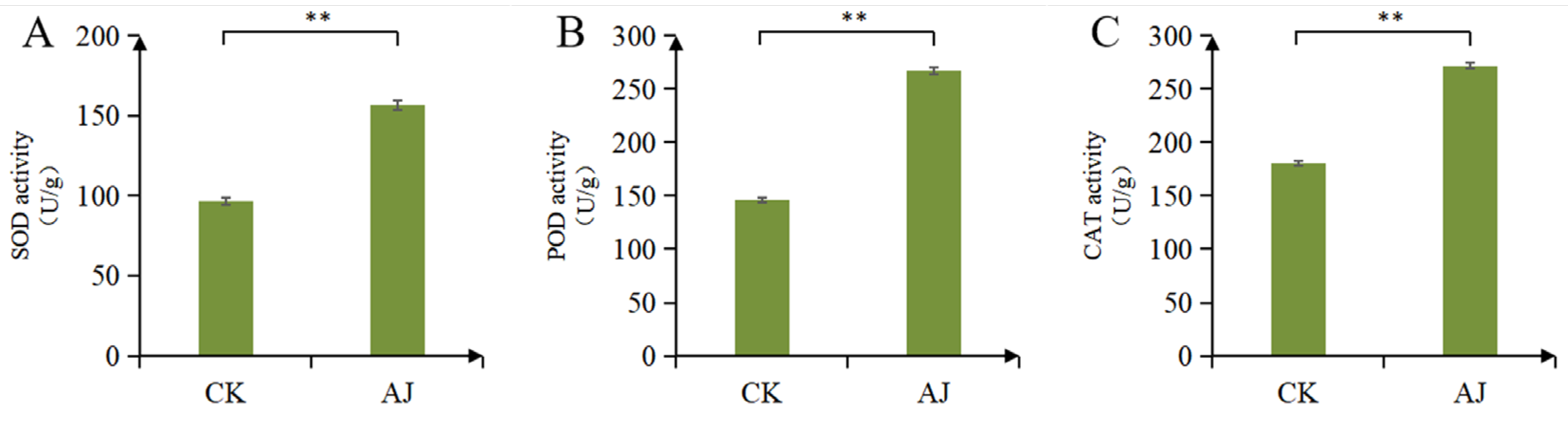
2.2. Measurement of Peroxidase, SOD, and CAT Activities
2.3. Transcriptome Analysis
2.4. Metabolome Analysis
2.5. Rhizosphere Microbial Analysis
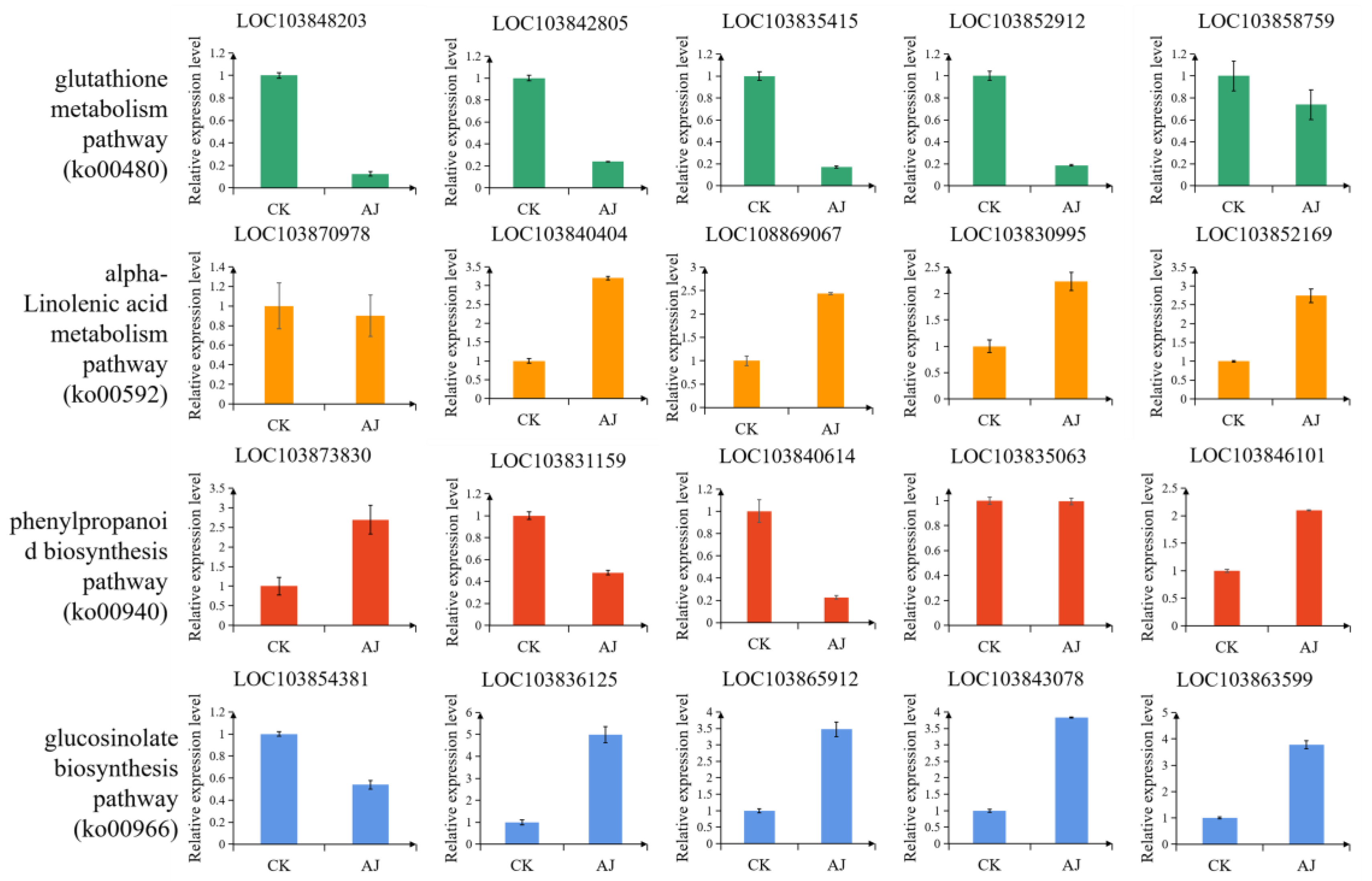
2.6. Quantitative Real-Time PCR
2.7. Statistical Analysis
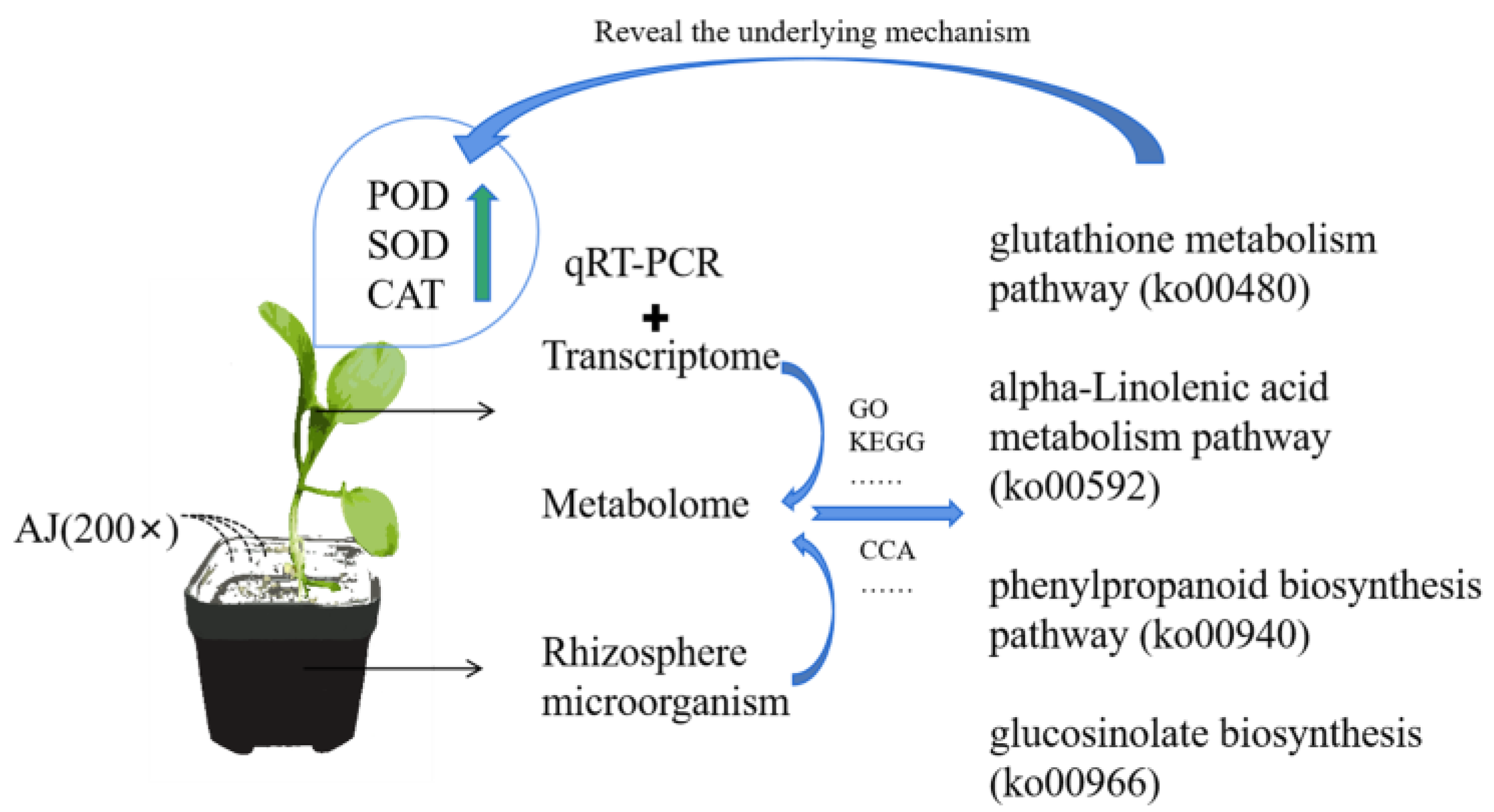
3. Results
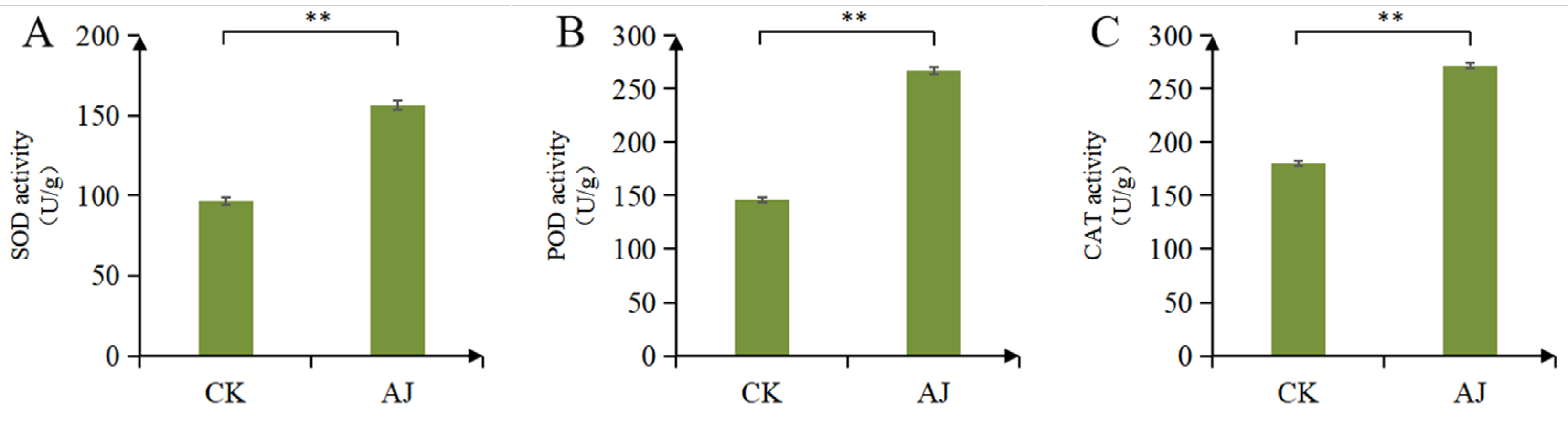
3.1. Effects of AJ on Pak Choi Stress Resistance
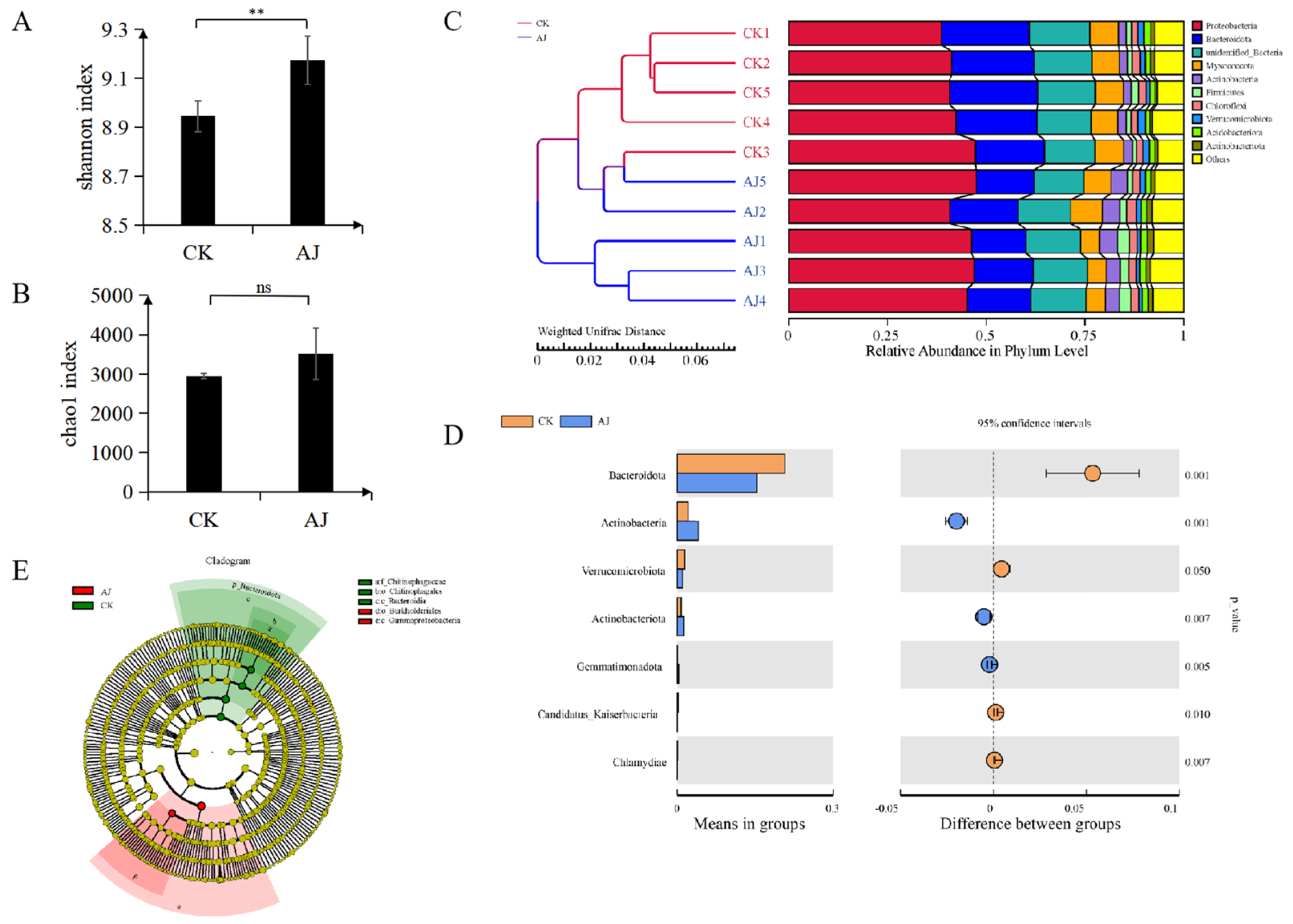
3.2. Rhizosphere Microbial Community Analysis
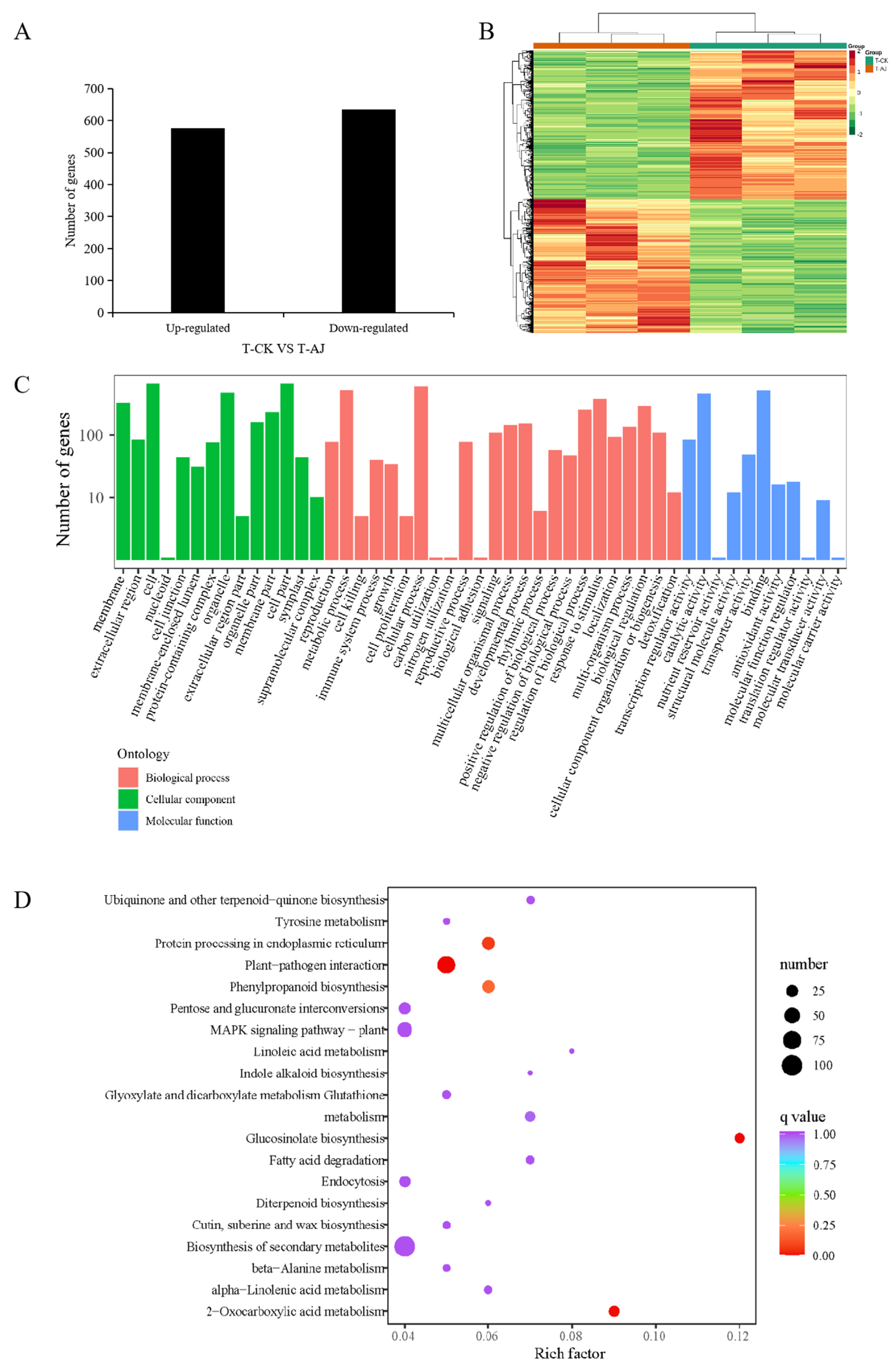
3.3. Transcriptome Analysis of AJ-Treated Pak Choi
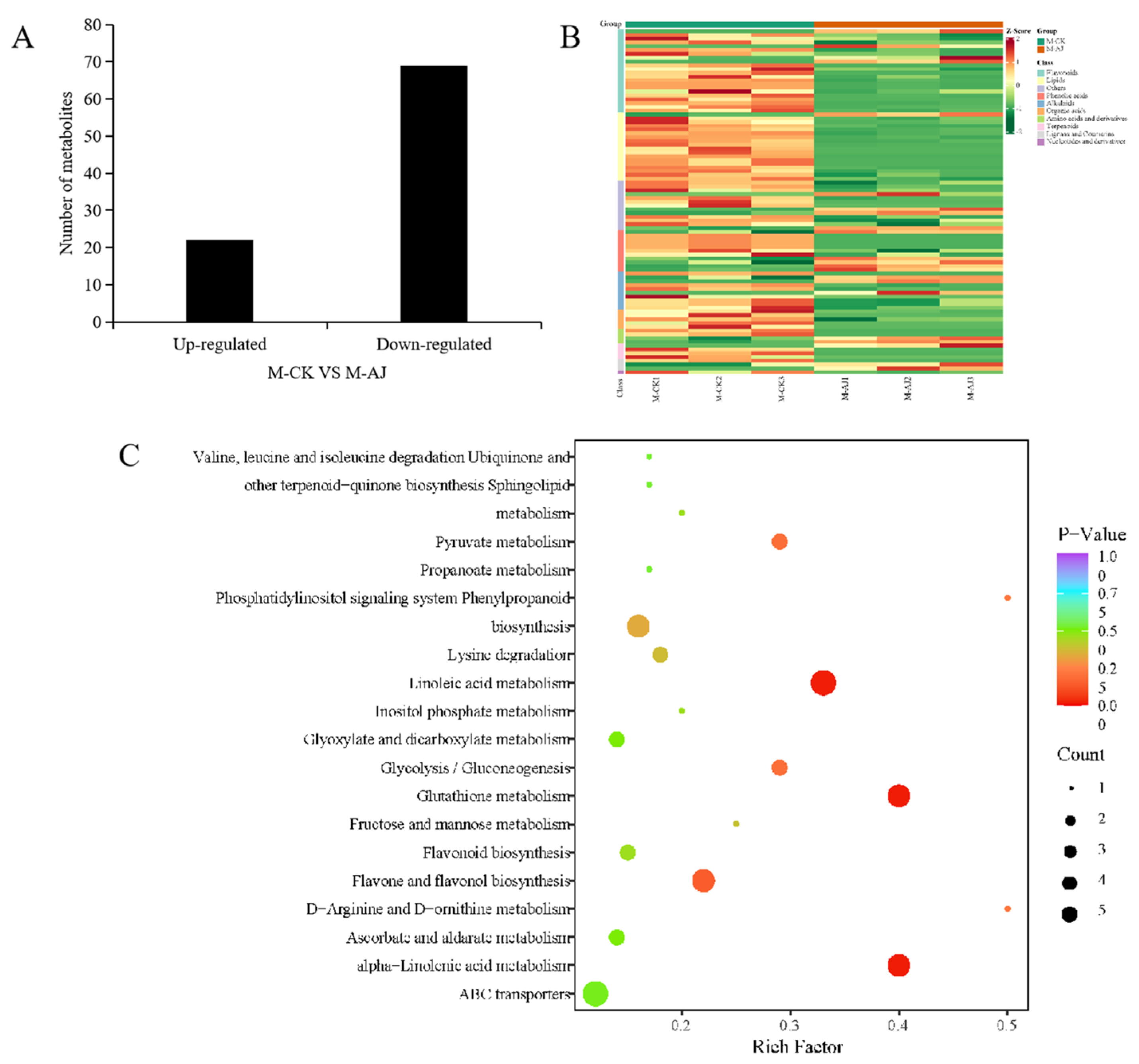
3.4. AJ Treatment Alters the Metabolic Pathway of Pak Choi
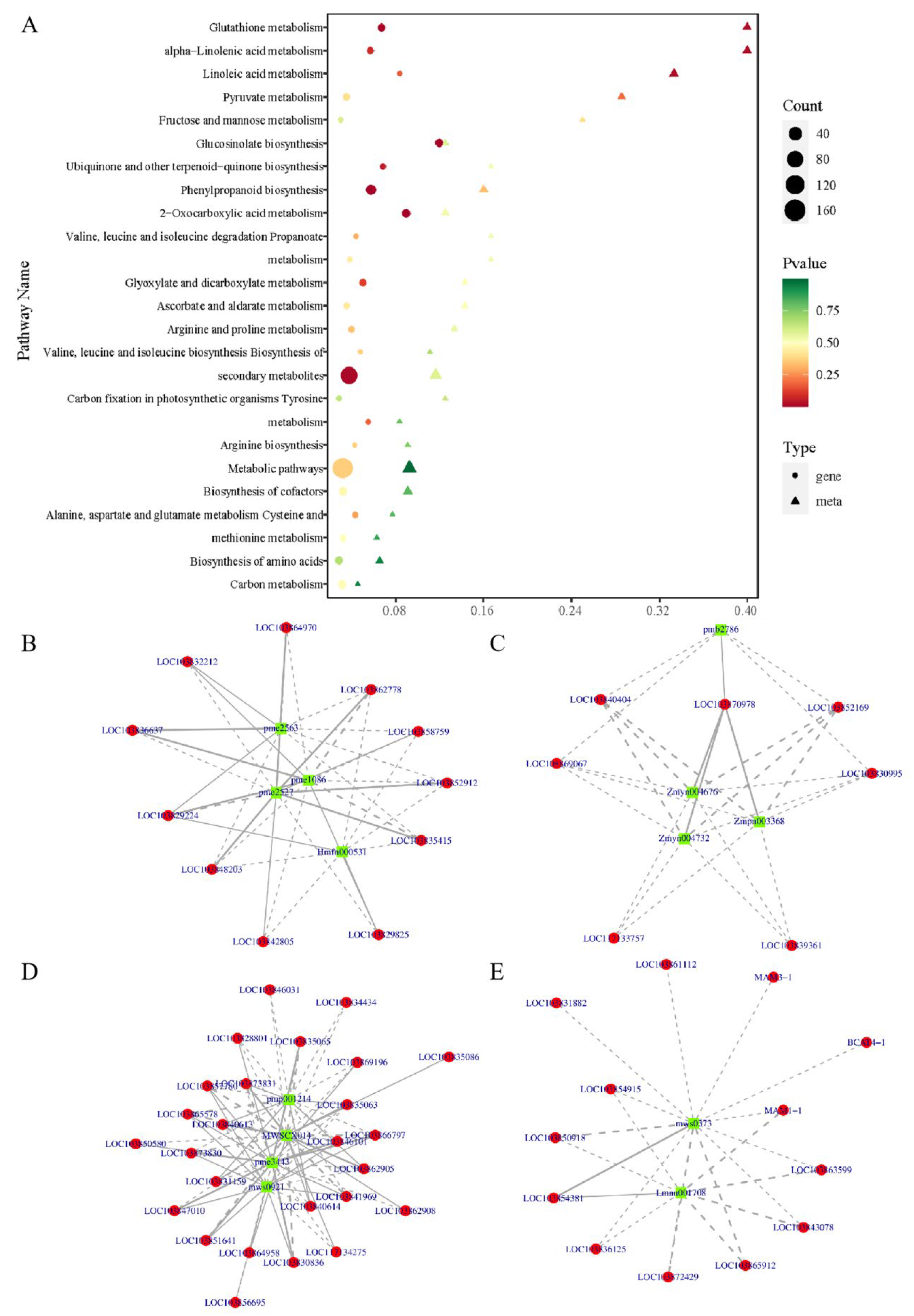
3.5. Integrated Transcriptome and Metabolome Analyses
3.6. Combined Microbiome and Metabolome Analysis
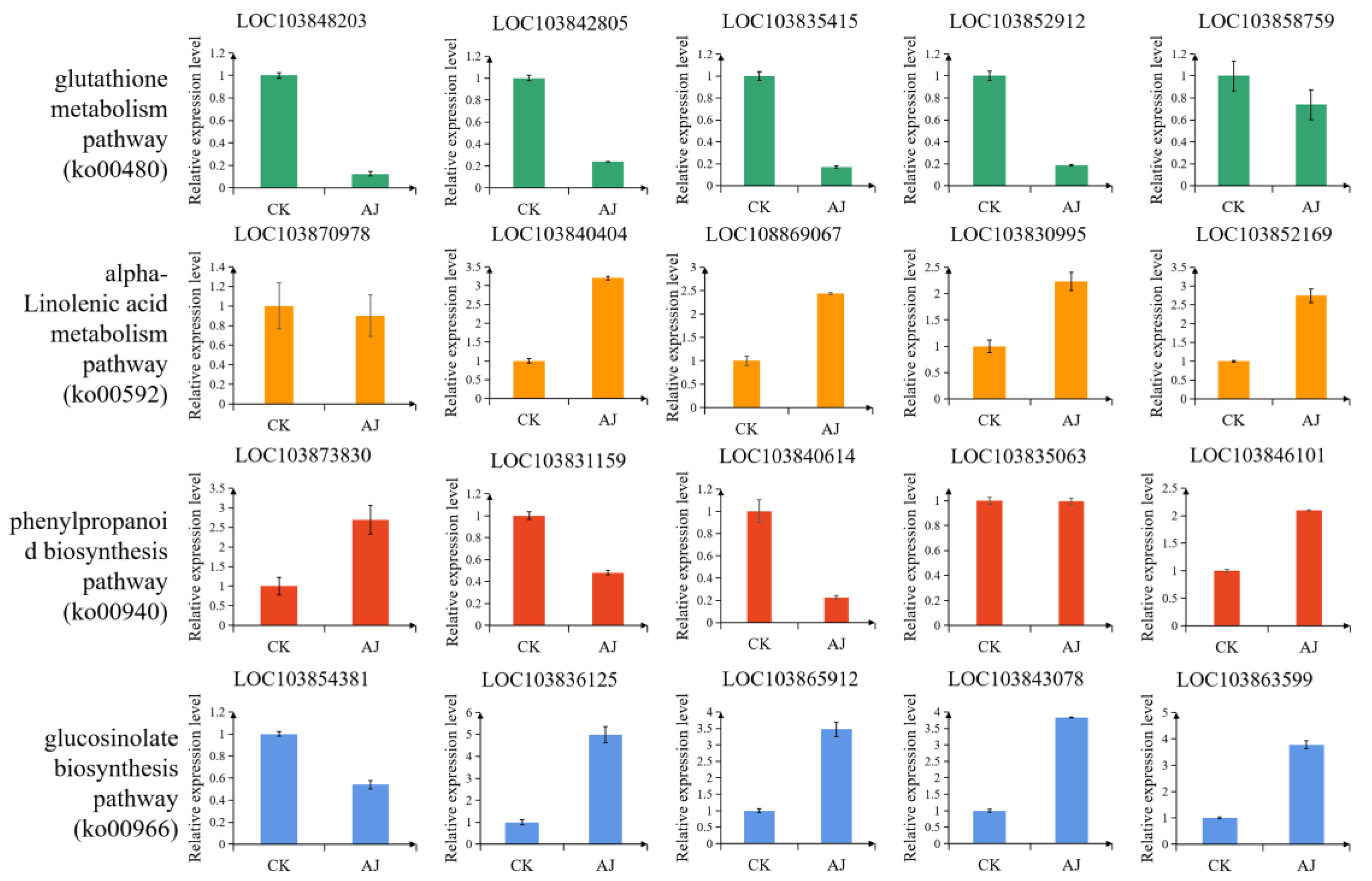
3.7. qRT-PCR
4. Discussion
4.1. Multiple Composition Guarantees Long-Term Effectiveness
4.2. AJ Drives Stress Resistance by Altering Transcriptional and Metabolic Pathways
4.3. AJ Drives Resistance by Altering the Rhizosphere Microbiome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, J.P.J.; Vadez, V. Review: An integrated framework for crop adaptation to dry environments: Responses to transient and terminal drought. Plant Sci. Int. J. Exp. Plan. Biol. 2016, 253, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Sun, H.; Yang, X.; Zhang, X. Drought coping strategies in cotton: Increased crop per drop. Plant Biotechnol. J. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lozano, J.M.; Porcel, R.; Azcón, C.; Aroca, R. Regulation by arbuscular mycorrhizae of the integrated physiological response to salinity in plants: New challenges in physiological and molecular studies. J. Exp. Bot. 2012, 63, 4033–4044. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Zheng, S.; Su, M.; Wang, L.; Zhang, T.; Wang, J.; Xie, H.; Wu, X.; Haq, S.I.U.; Qiu, Q.S. Small signaling molecules in plant response to cold stress. J. Plant Physiol. 2021, 266, 153534. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Y.; Jiang, Y.; Li, A.; Cheng, B.; Wu, J. OsASR6 Enhances Salt Stress Tolerance in Rice. Int. J. Mol. Sci. 2022, 23, 9340. [Google Scholar] [CrossRef]
- Ao, H.; Xie, X.; Huang, M.; Zou, Y. Decreasing hill density combined with increasing nitrogen rate led to yield decline in hybrid rice under low-light conditions. Sci. Rep. 2019, 9, 15786. [Google Scholar] [CrossRef]
- Li, R.; Zhang, J.; Wei, J.; Wang, H.; Ma, R. Functions and mechanisms of the CBL–CIPK signaling system in plant response to abiotic stress. Prog. Nat. Sci. 2009, 19, 667–676. [Google Scholar] [CrossRef]
- Cooper, M.; Gho, C.; Leafgren, R.; Tang, T.; Messina, C. Breeding drought-tolerant maize hybrids for the US corn-belt: Discovery to product. J. Exp. Bot. 2014, 65, 6191–6204. [Google Scholar] [CrossRef]
- Tang, L.; Ma, W.; Noor, M.A.; Li, L.; Hou, H.; Zhang, X.; Zhao, M. Density resistance evaluation of maize varieties through new “Density-Yield Model” and quantification of varietal response to gradual planting density pressure. Sci. Rep. 2018, 8, 17281. [Google Scholar] [CrossRef]
- Jud, W.; Winkler, J.B.; Niederbacher, B.; Niederbacher, S.; Schnitzler, J.P. Volatilomics: A non-invasive technique for screening plant phenotypic traits. Plant Methods 2018, 14, 109. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, F.; Chen, L.; Pan, Y.; Sun, L.; Bao, N.; Zhang, T.; Cui, C.X.; Qiu, Z.; Zhang, Y.; et al. Jasmonate-mediated wound signalling promotes plant regeneration. Nat. Plants 2019, 5, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lozano-Torres, J.L.; Blilou, I.; Zhang, X.; Zhai, Q.; Smant, G.; Li, C.; Scheres, B. A Jasmonate Signaling Network Activates Root Stem Cells and Promotes Regeneration. Cell 2019, 177, 942–956.e14. [Google Scholar] [CrossRef] [PubMed]
- Gilliard, G.; Huby, E.; Cordelier, S.; Ongena, M.; Dhondt-Cordelier, S.; Deleu, M. Protoplast: A Valuable Toolbox to Investigate Plant Stress Perception and Response. Front. Plant Sci. 2021, 12, 749581. [Google Scholar] [CrossRef]
- Lu, J.; Du, J.; Tian, L.; Li, M.; Zhang, X.; Zhang, S.; Wan, X.; Chen, Q. Divergent Response Strategies of CsABF Facing Abiotic Stress in Tea Plant: Perspectives from Drought-Tolerance Studies. Front. Plant Sci. 2021, 12, 763843. [Google Scholar] [CrossRef]
- Huo, T.; Wang, C.T.; Yu, T.F.; Wang, D.M.; Li, M.; Zhao, D.; Li, X.T.; Fu, J.D.; Xu, Z.S.; Song, X.Y. Overexpression of ZmWRKY65 transcription factor from maize confers stress resistances in transgenic Arabidopsis. Sci. Rep. 2021, 11, 4024. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.R.; Baker, B.J. The evolution of resistance genes in multi-protein plant resistance systems. Curr. Opin. Genet. Dev. 2007, 17, 493–499. [Google Scholar] [CrossRef]
- Yoshida, K. Plant biotechnology—Genetic engineering to enhance plant salt tolerance. J. Biosci. Bioeng. 2002, 94, 585–590. [Google Scholar] [CrossRef]
- Fan, R.; Su, X.; Guo, Y.; Sun, F.; Qu, Y.; Chen, Q. Cotton seedling drought tolerance is improved via salt preconditioning. Protoplasma 2021, 258, 263–277. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Liu, Y.; Zhang, Q.; Wei, Q.; Zhang, W. Nitric oxide enhances salt tolerance in maize seedlings through increasing activities of proton-pump and Na+/H+ antiport in the tonoplast. Planta 2006, 224, 545–555. [Google Scholar] [CrossRef]
- Doan, T.T.; Henry-des-Tureaux, T.; Rumpel, C.; Janeau, J.L.; Jouquet, P. Impact of compost, vermicompost and biochar on soil fertility, maize yield and soil erosion in Northern Vietnam: A three years mesocosm experiment. Sci. Total Environ. 2015, 514, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Dring, M.J. Stress Resistance and Disease Resistance in Seaweeds: The Role of Reactive Oxygen Metabolism. Adv. Bot. Res. 2005, 43, 175–207. [Google Scholar]
- Ruzzi, M.; Aroca, R. Plant growth-promoting rhizobacteria act as biostimulants in horticulture. Sci. Hortic. 2015, 196, 124–134. [Google Scholar] [CrossRef]
- Vitale, L.; Vitale, E.; Francesca, S.; Lorenz, C.; Arena, C. Plant-Growth Promoting Microbes Change the Photosynthetic Response to Light Quality in Spinach. Plants 2023, 12, 1149. [Google Scholar] [CrossRef] [PubMed]
- Vitale, L.; Vitale, E.; Guercia, G.; Turano, M.; Arena, C. Effects of different light quality and biofertilizers on structural and physiological traits of spinach plants. Photosynthetica 2020, 58, 932–943. [Google Scholar] [CrossRef]
- Evseeva, N.V.; Tkachenko, O.V.; Denisova, A.Y.; Burygin, G.L.; Veselov, D.S.; Matora, L.Y.; Shchyogolev, S.Y. Functioning of plant-bacterial associations under osmotic stress in vitro. World J. Microbiol. Biotechnol. 2019, 35, 195. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Zheng, Z.; Meng, X.; Cai, Y.; Liu, J.; Hu, Y.; Yan, S.; Wang, X. A microbial ecosystem: Agricultural Jiaosu achieves effective and lasting antifungal activity against Botrytis cinerea. AMB Express 2020, 10, 216. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Cheng, X.; Zheng, Z.; Wu, X.; Dong, X.; Hu, Y.; Wang, X. Agricultural Jiaosu: An Eco-Friendly and Cost-Effective Control Strategy for Suppressing Fusarium Root Rot Disease in Astragalus membranaceus. Front. Microbiol. 2022, 13, 823704. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Zheng, Y. Review on drought response in plants from phenotype to molecular. Curr. Biotechnol. 2017, 7, 169–176. [Google Scholar]
- Leksungnoen, N. The Relationship between Salinity and Drought Tolerance in Turfgrasses and Woody Species; Utah State University: Logan, UT, USA, 2012. [Google Scholar]
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of Plant Responses to Salt Stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef]
- Vitale, L.; Vitale, E.; Bianchi, A.R.; Maio, A.D.; Arena, C. Role of Poly (ADP-Ribose) Polymerase (PARP) Enzyme in the Systemic Acquired Acclimation Induced by Light Stress in Phaseolus vulgaris L. Plants. Plants 2022, 11, 1870. [Google Scholar] [CrossRef]
- Gohari, G.; Panahirad, S.; Sepehri, N.; Akbari, A.; Zahedi, S.M.; Jafari, H.; Dadpour, M.R.; Fotopoulos, V. Enhanced tolerance to salinity stress in grapevine plants through application of carbon quantum dots functionalized by proline. Environ. Sci. Pollut. Res. Int. 2021, 28, 42877–42890. [Google Scholar] [CrossRef]
- Shao, H.B.; Chu, L.Y.; Shao, M.A.; Jaleel, C.A.; Mi, H.M. Higher plant antioxidants and redox signaling under environmental stresses. Comptes Rendus Biol. 2008, 331, 433–441. [Google Scholar] [CrossRef]
- Taj, Z.; Challabathula, D. Protection of Photosynthesis by Halotolerant Staphylococcus sciuri ET101 in Tomato (Lycoperiscon esculentum) and Rice (Oryza sativa) Plants During Salinity Stress: Possible Interplay between Carboxylation and Oxygenation in Stress Mitigation. Front. Microbiol. 2020, 11, 547750. [Google Scholar] [CrossRef] [PubMed]
- Sewelam, N.; Kazan, K.; Thomas-Hall, S.R.; Kidd, B.N.; Manners, J.M.; Schenk, P.M. Ethylene response factor 6 is a regulator of reactive oxygen species signaling in Arabidopsis. PLoS ONE 2013, 8, e70289. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Sax, M.; Chakrabartty, A.; Hew, C.L. Crystal structure of an antifreeze polypeptide and its mechanistic implications. Nature 1988, 333, 232–237. [Google Scholar] [CrossRef]
- Kenward, K.D.; Altschuler, M.; Hildebrand, D.; Davies, P.L. Accumulation of type I fish antifreeze protein in transgenic tobacco is cold-specific. Plant Mol. Biol. 1993, 23, 377–385. [Google Scholar] [CrossRef]
- Kovtun, Y.; Chiu, W.L.; Tena, G.; Sheen, J. Functional analysis of oxidative stress-activated mitogen-activated protein kinase cascade in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 2940–2945. [Google Scholar] [CrossRef]
- Shou, H.; Bordallo, P.; Fan, J.B.; Yeakley, J.M.; Bibikova, M.; Sheen, J.; Wang, K. Expression of an active tobacco mitogen-activated protein kinase enhances freezing tolerance in transgenic maize. Proc. Natl. Acad. Sci. USA 2004, 101, 3298–3303. [Google Scholar] [CrossRef]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Fang, X.; Wang, Z.; Shangguan, L.; Liu, T.; Chen, C.; Liu, Z.; Ge, M.; Zhang, C.; Zheng, T.; et al. Multi-omics analyses on the response mechanisms of ‘Shine Muscat’ grapevine to low degree of excess copper stress (Low-ECS). Environ. Pollut. 2021, 286, 117278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhang, S.; Yang, W.; Li, B.; Lan, C.; Zhang, J.; Yuan, L.; Wang, Y.; Xie, Q.; Han, J.; et al. Multi-omic dissection of the drought resistance traits of soybean landrace LX. Plant Cell Environ. 2021, 44, 1379–1398. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Zhang, J.; Li, D.; Yan, L.; You, M.; Zhang, J.; Lei, X.; Chang, D.; Ji, X.; et al. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to High Temperature Stress. Front. Plant Sci. 2021, 12, 753011. [Google Scholar] [CrossRef]
- Hübner, S.; Korol, A.B.; Schmid, K.J. RNA-Seq analysis identifies genes associated with differential reproductive success under drought-stress in accessions of wild barley Hordeum spontaneum. BMC Plant Biol. 2015, 15, 134. [Google Scholar] [CrossRef]
- Fracasso, A.; Trindade, L.M.; Amaducci, S. Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 2016, 16, 115. [Google Scholar] [CrossRef]
- Dong, B.; Wu, B.; Hong, W.; Li, X.; Li, Z.; Xue, L.; Huang, Y. Transcriptome analysis of the tea oil camellia (Camellia oleifera) reveals candidate drought stress genes. PLoS ONE 2017, 12, e0181835. [Google Scholar] [CrossRef]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, W.; Zhao, Y.; Xu, Y.; Song, S.; Chong, K. Comparative metabolomic analysis reveals a reactive oxygen species-dominated dynamic model underlying chilling environment adaptation and tolerance in rice. New Phytol. 2016, 211, 1295–1310. [Google Scholar] [CrossRef]
- Wang, Y.; Lysøe, E.; Armarego-Marriott, T.; Erban, A.; Paruch, L.; van Eerde, A.; Bock, R.; Liu-Clarke, J. Transcriptome and metabolome analyses provide insights into root and root-released organic anion responses to phosphorus deficiency in oat. J. Exp. Bot. 2018, 69, 3759–3771. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Chiniquy, D.; Pierroz, G.; Deng, S.; Gao, C.; Diamond, S.; Simmons, T.; Wipf, H.M.; Caddell, D.; et al. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nat. Commun. 2021, 12, 3209. [Google Scholar] [CrossRef] [PubMed]
- de Vries, F.T.; Griffiths, R.I.; Knight, C.G.; Nicolitch, O.; Williams, A. Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 2020, 368, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Ma, L.; Qi, W.; Bai, J.; Li, H.; Fang, Y.; Xu, J.; Xu, Y.; Zeng, X.; Pu, Y.; Wang, W.; et al. Genome-Wide Identification and Analysis of the Ascorbate Peroxidase (APX) Gene Family of Winter Rapeseed (Brassica rapa L.) Under Abiotic Stress. Front. Genet. 2021, 12, 753624. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef]
- Naservafaei, S.; Sohrabi, Y.; Moradi, P.; Mac Sweeney, E.; Mastinu, A. Biological Response of Lallemantia iberica to Brassinolide Treatment under Different Watering Conditions. Plants 2021, 10, 496. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, N.; Li, Y.; Zhu, C.; Qu, B.; Liu, H.; Li, R.; Bai, Y.; Shen, Q.; Salles, J.F. Bio-organic soil amendment promotes the suppression of Ralstonia solanacearum by inducing changes in the functionality and composition of rhizosphere bacterial communities. New Phytol. 2022, 235, 1558–1574. [Google Scholar] [CrossRef]
- Lourenço, K.S.; Suleiman, A.K.A.; Pijl, A.; van Veen, J.A.; Cantarella, H.; Kuramae, E.E. Resilience of the resident soil microbiome to organic and inorganic amendment disturbances and to temporary bacterial invasion. Microbiome 2018, 6, 142. [Google Scholar] [CrossRef]
- Arun, C.; Sivashanmugam, P. Solubilization of waste activated sludge using a garbage enzyme produced from different pre-consumer organic waste. RSC Adv. 2015, 5, 51421–51427. [Google Scholar] [CrossRef]
- Mittler, R.; Zilinskas, B.A. Purification and characterization of pea cytosolic ascorbate peroxidase. Plant Physiol. 1991, 97, 962–968. [Google Scholar] [CrossRef]
- Mittler, R.; Zilinskas, B.A. Regulation of pea cytosolic ascorbate peroxidase and other antioxidant enzymes during the progression of drought stress and following recovery from drought. Plant J. 1994, 5, 397–405. [Google Scholar] [CrossRef]
- Foyer, C.H.; Souriau, N.; Perret, S.; Lelandais, M.; Kunert, K.J.; Pruvost, C.; Jouanin, L. Overexpression of glutathione reductase but not glutathione synthetase leads to increases in antioxidant capacity and resistance to photoinhibition in poplar trees. Plant Physiol. 1995, 109, 1047–1057. [Google Scholar] [CrossRef]
- Samis, K.; Bowley, S.; McKersie, B. Pyramiding Mn-superoxide dismutase transgenes to improve persistence and biomass production in alfalfa. J. Exp. Bot. 2002, 53, 1343–1350. [Google Scholar]
- Li, C.; Qi, Y.; Zhao, C.; Wang, X.; Zhang, Q. Transcriptome Profiling of the Salt Stress Response in the Leaves and Roots of Halophytic Eutrema salsugineum. Front. Genet. 2021, 12, 770742. [Google Scholar] [CrossRef]
- Zhao, C.; Yang, M.; Wu, X.; Wang, Y.; Zhang, R. Physiological and transcriptomic analyses of the effects of exogenous melatonin on drought tolerance in maize (Zea mays L.). Plant Physiol. Biochem. 2021, 168, 128–142. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Yang, K.; Han, H.; Li, Y.; Ye, J.; Xu, F. Significance of miRNA in enhancement of flavonoid biosynthesis. Plant Biol. 2022, 24, 217–226. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, C.; Chao, H.; Long, Y.; Wu, J.; Li, Z.; Ge, X.; Xia, H.; Yin, Y.; Batley, J.; et al. Integration of metabolome and transcriptome reveals flavonoid accumulation in the intergeneric hybrid between Brassica rapa and Raphanus sativus. Sci. Rep. 2019, 9, 18368. [Google Scholar] [CrossRef]
- Muthusamy, M.; Kim, J.H.; Kim, S.H.; Park, S.Y.; Lee, S.I. BrPP5.2 Overexpression Confers Heat Shock Tolerance in Transgenic Brassica rapa through Inherent Chaperone Activity, Induced Glucosinolate Biosynthesis, and Differential Regulation of Abiotic Stress Response Genes. Int. J. Mol. Sci. 2021, 22, 6437. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, C.; Zhu, F.; Li, Y. Mild osmotic stress promotes 4-methoxy indolyl-3-methyl glucosinolate biosynthesis mediated by the MKK9-MPK3/MPK6 cascade in Arabidopsis. Plant Cell Rep. 2017, 36, 543–555. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Deng, J.; Dai, L.; Liu, W.; Pan, B.; Wang, C.; Zhang, D.; Li, Z. Molecular mechanism of the response of Zygosaccharomyces rouxii to D-fructose stress by the glutathione metabolism pathway. FEMS Yeast Res. 2020, 20, 5. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, R.; Xiang, C.; Zhang, R.; Wang, Q.; Wang, T.; Li, X.; Lu, X.; Gao, S.; Liu, Z.; et al. Transcriptomic and Physiological Analysis Reveal That α-Linolenic Acid Biosynthesis Responds to Early Chilling Tolerance in Pumpkin Rootstock Varieties. Front. Plant Sci. 2021, 12, 669565. [Google Scholar] [CrossRef]
- Alam, S.I.; Kim, M.W.; Shah, F.A.; Saeed, K.; Ullah, R.; Kim, M.O. Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain. Cells 2021, 10, 2274. [Google Scholar] [CrossRef]
- Wu, L.; Chen, J.; Wu, H.; Qin, X.; Wang, J.; Wu, Y.; Khan, M.U.; Lin, S.; Xiao, Z.; Luo, X.; et al. Insights into the Regulation of Rhizosphere Bacterial Communities by Application of Bio-organic Fertilizer in Pseudostellaria heterophylla Monoculture Regime. Front. Microbiol. 2016, 7, 1788. [Google Scholar] [CrossRef]
- Lee, S.M.; Kong, H.G.; Song, G.C.; Ryu, C.M. Disruption of Firmicutes and Actinobacteria abundance in tomato rhizosphere causes the incidence of bacterial wilt disease. ISME J. 2021, 15, 330–347. [Google Scholar] [CrossRef]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 2022, 13, 836. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Name | Plant Height (cm) | Fresh Weight (g) | Root Length (cm) | SPAD |
|---|---|---|---|---|
| CK | 12.40a | 3.16a | 4.80a | 27.60b |
| AJ | 12.80a | 3.62a | 7.60a | 28.40a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Gao, Y.; Wang, Z.; Cai, Y.; Wang, X. Agricultural Jiaosu Enhances the Stress Resistance of Pak Choi (Brassica rapa L. subsp. chinensis) by Recruiting Beneficial Rhizosphere Bacteria and Altering Metabolic Pathways. Agronomy 2023, 13, 2310. https://doi.org/10.3390/agronomy13092310
Cheng X, Gao Y, Wang Z, Cai Y, Wang X. Agricultural Jiaosu Enhances the Stress Resistance of Pak Choi (Brassica rapa L. subsp. chinensis) by Recruiting Beneficial Rhizosphere Bacteria and Altering Metabolic Pathways. Agronomy. 2023; 13(9):2310. https://doi.org/10.3390/agronomy13092310
Chicago/Turabian StyleCheng, Xiaoqian, Youhui Gao, Ziyu Wang, Yafan Cai, and Xiaofen Wang. 2023. "Agricultural Jiaosu Enhances the Stress Resistance of Pak Choi (Brassica rapa L. subsp. chinensis) by Recruiting Beneficial Rhizosphere Bacteria and Altering Metabolic Pathways" Agronomy 13, no. 9: 2310. https://doi.org/10.3390/agronomy13092310
APA StyleCheng, X., Gao, Y., Wang, Z., Cai, Y., & Wang, X. (2023). Agricultural Jiaosu Enhances the Stress Resistance of Pak Choi (Brassica rapa L. subsp. chinensis) by Recruiting Beneficial Rhizosphere Bacteria and Altering Metabolic Pathways. Agronomy, 13(9), 2310. https://doi.org/10.3390/agronomy13092310