
Application of a High-Density Single Nucleotide Polymorphism Genetic Map in Mapping Quantitative Trait Loci of Early-Maturing Traits in Upland Cotton

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Mapping Populations
2.2. Field Experiments and Phenotypic Observations
2.3. DNA Extraction
2.4. GBS Library Construction and Illumina Sequencing
2.5. SNP Development
2.6. Genetic Map Construction
2.7. QTL Mapping
2.8. Comparison of the Physical Positions of SNP Markers Linked to QTLs
2.9. Candidate Gene Annotation
3. Results
3.1. Phenotypic Characteristics of Traits Related to Early Maturity in the Two Parents, the F2 Population, and the F2:3 Population
3.2. Genotyping by Sequencing (GBS)
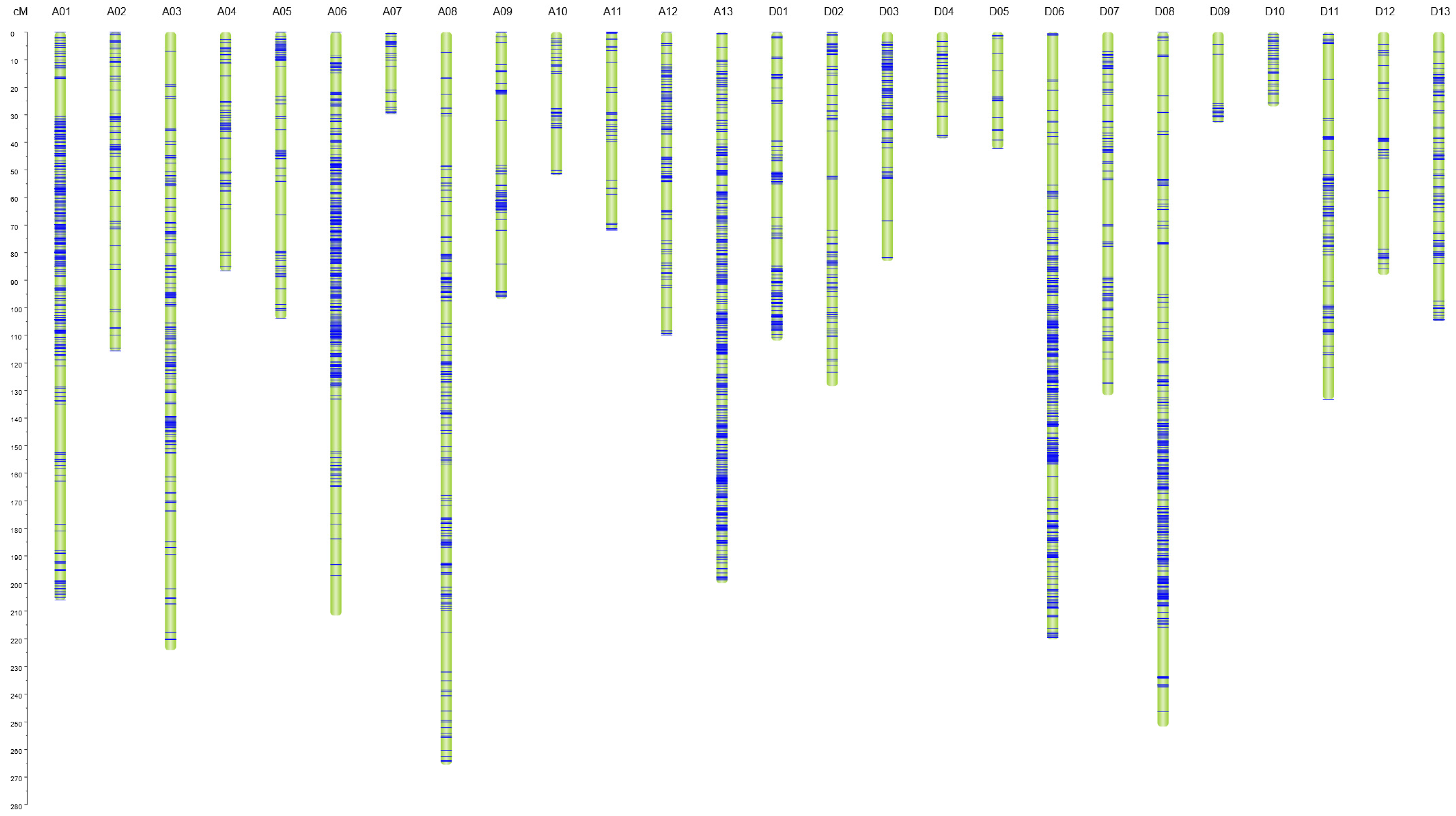
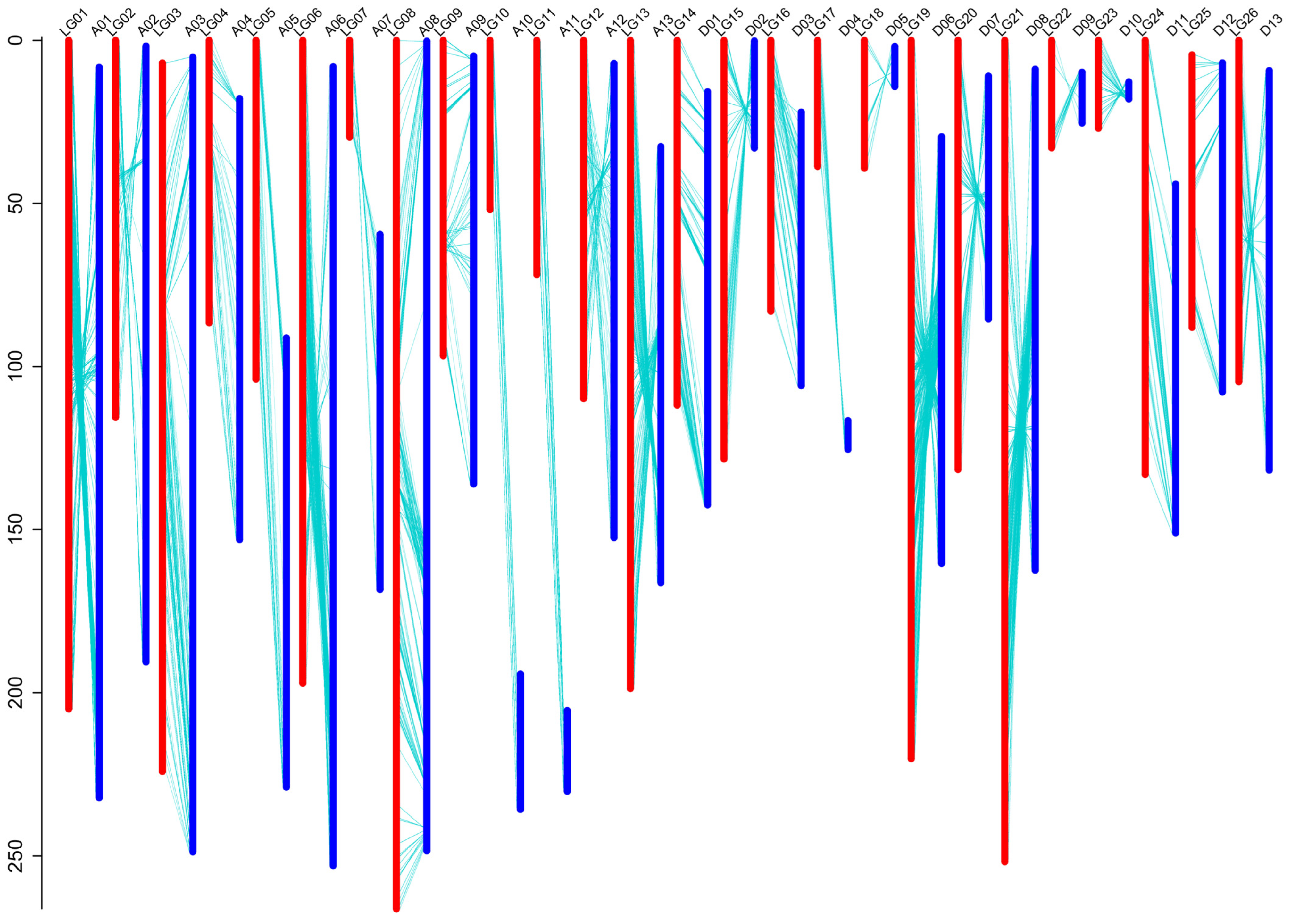
3.3. Construction of SNP-Based High Density Genetic Map
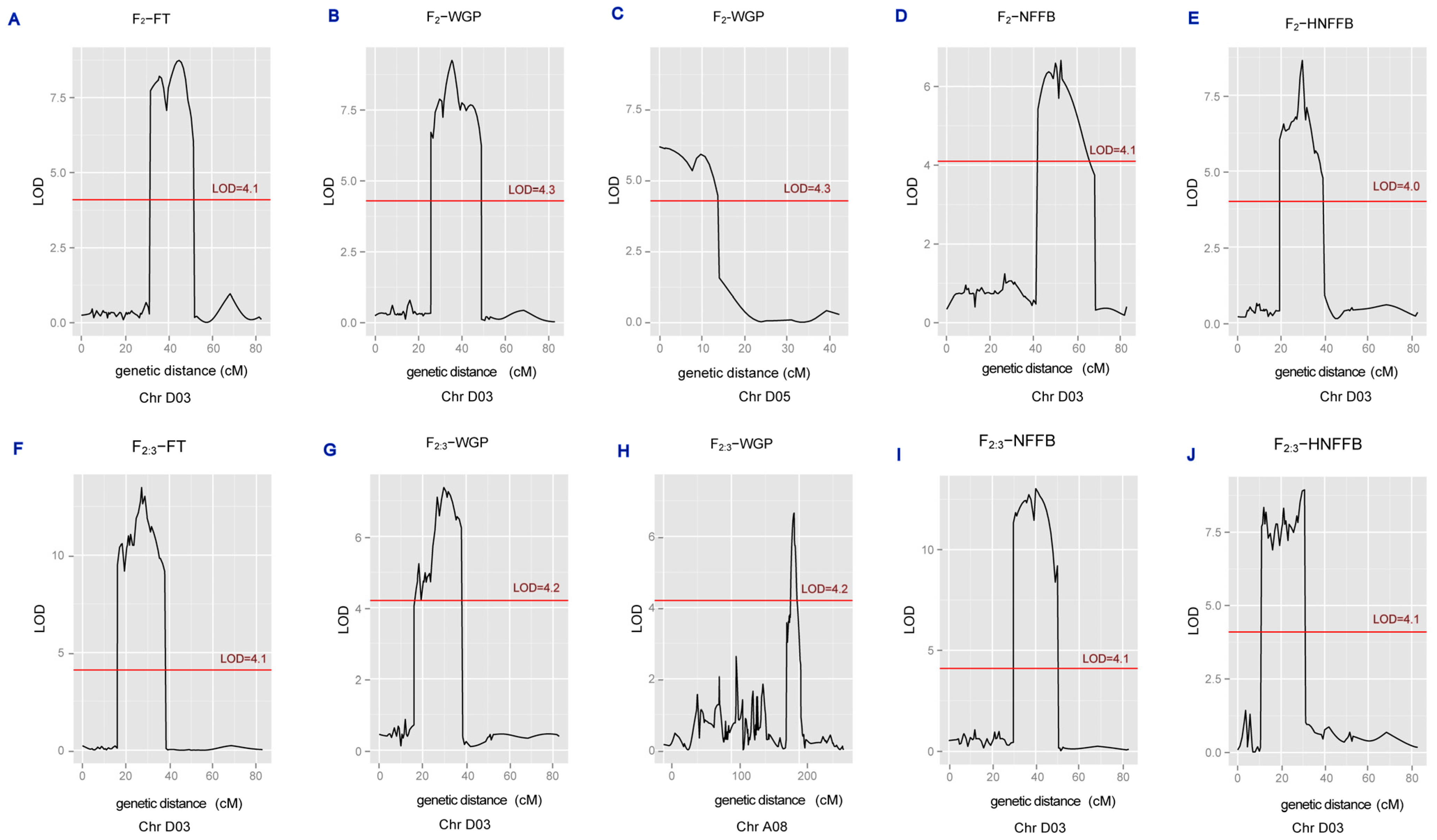
3.4. Mapping QTLs of Four Appearance Traits in the F2 and F2:3 Families
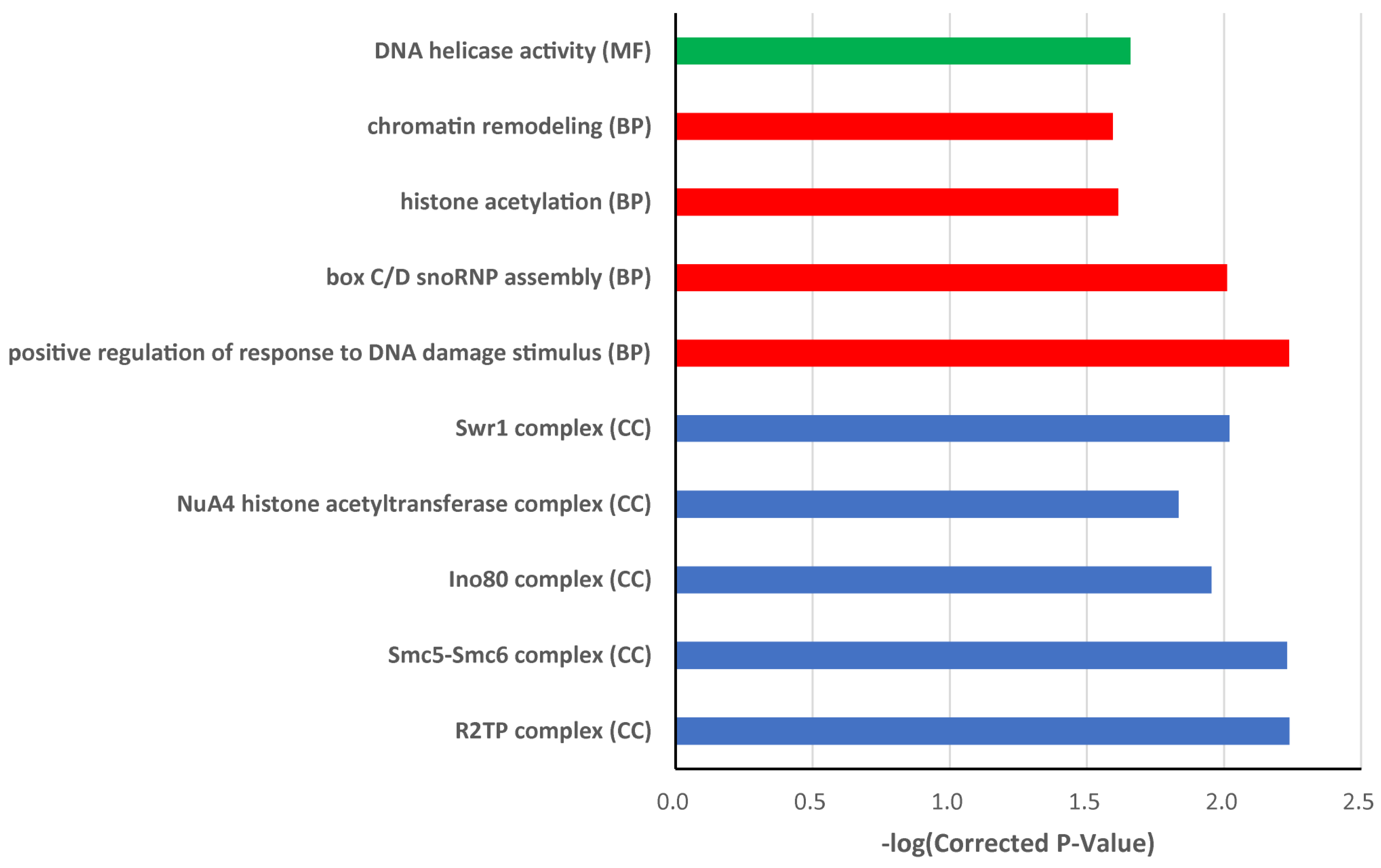
3.5. Candidate Gene Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Schefer, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, L.; Feng, W. Study on Strategy of Large Scale, Mechanization, Informationization, Intelligence and Social Services for Cotton Production. Eng. Sci. 2016, 18, 137–148. [Google Scholar]
- Du, X. Effects of Wheat-Cotton Cropping Systems on Cotton Yield Formation and Resource Use Efficiency. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2014. [Google Scholar]
- Shu, F. Prediction of Cotton Planting Area and Yield in China based on VAR. Cotton Sci. 2022, 44, 46–53. [Google Scholar]
- Yu, S.; Wang, H.; Wei, H.; Su, J. Research Progress and Application of Early Maturity in Upland Cotton. Cotton Sci. 2017, 29, 1–10. [Google Scholar]
- Zhao, L.; Chen, S.; Xu, S. An estimation of heritability of the indicative characters of earliness in upland cotton. Acta Genet. Sin. 1974, 1, 107–116. [Google Scholar]
- Niu, Y. Research of precocious hybrid second generation genetic parameters of main characters in upland cotton. Shanxi Agric. Sci. 1981, 6, 8–12. [Google Scholar]
- Yu, S.; Huang, Z. Inheritance analysis on earliness components of short season cotton varieties in G. hirsutum. Sci. Agric. Sin. 1990, 23, 48–54. [Google Scholar]
- Godoy, A.; Palomo, G. Genetic analysis of earliness in upland cotton (Gossypium hirsutum L.) Ⅰ. morphological and phonological variables. Euphytica 1999, 105, 155–160. [Google Scholar] [CrossRef]
- Song, M.; Yu, S.; Fan, S.; Yuan, R.; Huang, Z. Genetic Analysis of Main Agronomic Traits in Short Season Upland Cotton (G. hirsutum L.). Cotton Sci. 2005, 17, 94–98. [Google Scholar]
- Fan, S. Study on Inheritance of Earliness and It’s Relative Traits of Short-Season Cotton and QTLs Mapping. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2004. [Google Scholar]
- Zhao, S.; Pang, C.; Wei, H.; Wang, H.; Li, L.; Su, J.; Fan, S.; Yu, S. Genetic Inheritance of Earliness Traits in Upland Cotton (Gossypium hirsutum L.) Inferred by Joint Analysis of Multiple Generations. Cotton Sci. 2017, 29, 119–127. [Google Scholar]
- Fan, S.; Yu, S.; Song, M.; Yuan, R. Construction of Molecular Linkage Map and QTL Mapping for Earliness in Short Season Cotton. Cotton Sci. 2006, 18, 135–139. [Google Scholar]
- Guo, Y.; McCarty, J.C.; Jenkins, J.N.; Saha, S. QTLs for node of first fruiting branch in a cross of an Upland cotton (Gossypium hirsutum L.) cultivar with primitive accession Texas 701. Euphytica 2008, 163, 113–122. [Google Scholar] [CrossRef]
- Guo, Y.; McCarty, J.; Jenkins, J.; An, C.; Saha, S. Genetic detection of node of first fruiting branch in crosses of a cultivar with two exotic accessions of upland cotton. Euphytica 2009, 166, 317–329. [Google Scholar] [CrossRef]
- Liang, Y. Construction of Chromosome Segment Substitution Lines and Primary QTL mapping in Early-Maturing Upland Cotton. Master’s Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2010. [Google Scholar]
- Cui, Q. QTL Location of Earliness and Fiber Quality in Cotton Using RILs. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2011. [Google Scholar]
- Li, C.; Wang, C.; Dong, N.; Wang, X.; Zhao, H.; Converse, R.; Xia, Z.; Wang, R.; Wang, Q. QTL detection for node of first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 2012, 188, 441–451. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Dong, N.; Zhao, H.; Xia, Z.; Wang, R.; Converse, R.; Wang, Q. QTL analysis for early-maturing traits in cotton using two upland cotton (Gossypium hirsutum L.) crosses. Breed. Sci. 2013, 63, 154–163. [Google Scholar] [CrossRef]
- Li, C.; Song, L.; Zhao, H.; Xia, Z.; Jia, Z.; Wang, X.; Dong, N.; Wang, Q. Quantitative trait loci mapping for plant architecture traits across two upland cotton populations using SSR markers. J. Agric. Sci. 2014, 152, 275–287. [Google Scholar] [CrossRef]
- Liu, R.; Ai, N.; Zhu, X.; Liu, F.; Guo, W.; Zhang, T. Genetic analysis of plant height using two immortalized populations of “CRI12 × J8891” in Gossypium hirsutum L. Euphytica 2014, 196, 51–61. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, C.; Zhang, Y.; Yan, Q.; Hu, W.; Yang, L.; Wang, Z.; Li, F. Recent progression and future perspectives in cotton genomic breeding. J. Integr. Plant Biol. 2023, 65, 548–569. [Google Scholar]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Legare, G.; Boyle, B.; Normandeau, E.; Laroche, J.; Larose, S.; Jean, M.; et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef]
- Jia, X.; Pang, C.; Wei, H.; Wang, H.; Ma, Q.; Yang, J.; Cheng, S.; Su, J.; Fan, S.; Wusiman, N.; et al. High-density linkage map construction and QTL analysis for earliness-related traits in Gossypium hirsutum L. BMC Genom. 2016, 17, 909. [Google Scholar] [CrossRef]
- Li, L.; Zhao, S.; Su, J.; Fan, S.; Pang, C.; Wei, H.; Wang, H.; Gu, L.; Zhang, C.; Liu, G.; et al. High-density genetic linkage map construction by F2 populations and QTL analysis of early-maturity traits in upland cotton (Gossypium hirsutum L.). PLoS ONE 2017, 12, e0182918. [Google Scholar] [CrossRef]
- Qi, H.; Wang, N.; Qiao, W.; Xu, Q.; Zhou, H.; Shi, J.; Yan, G.; Huang, Q. Construction of a high-density genetic map using genotyping by sequencing (GBS) for quantitative trait loci (QTL) analysis of three plant morphological traits in upland cotton (Gossypium hirsutum L.). Euphytica 2017, 213, 83. [Google Scholar] [CrossRef]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP array for cotton and high-density mapping of intraspecific and Interspecific populations of Gossypium spp. G3 Genes Genome Genet. 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Pang, C.; Wei, H.; Li, L.; Liang, B.; Wang, C.; Song, M.; Wang, H.; Zhao, S.; Jia, X.; et al. Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genom. 2016, 17, 687. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Ai, N.; Li, Y.; Song, J. A genome-wide association study of early-maturation traits in upland cotton based on the CottonSNP8oK array. J. Integr. Plant Biol. 2018, 60, 970–985. [Google Scholar] [CrossRef]
- Li, L.; Zhang, C.; Huang, J.; Liu, Q.; Wei, H.; Wang, H.; Liu, G.; Gu, L.; Yu, S. Genomic analyses reveal the genetic basis of early maturity and identification of loci and candidate genes in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 109–123. [Google Scholar] [CrossRef]
- Kohel, R.J.; Richmond, T.R.; Lewis, C.F. Texas Marker1. Description of a Genetic Standard for Gossypium hirsutum L. Crop Sci. 1970, 10, 670–671. [Google Scholar] [CrossRef]
- Song, M.; Yang, Z.; Fan, S.; Zhu, H.; Pang, C.; Tian, M.; Yu, S. Physiological and Biochemical Analysis and Identification of A Short Season Cotton Virescent Mutant. Sci. Agric. Sin. 2011, 44, 3709–3720. [Google Scholar]
- Yu, S.; Fan, S.; Song, M.; Huang, Z.; Yuan, R. A new short-season cotton variety-Zhongmian Suo 58. China Cotton 2007, 34, 16–19. [Google Scholar]
- Mao, G.; Wei, H.; Hu, W.; Ma, Q.; Zhang, M.; Wang, H.; Yu, S. Fine mapping and molecular characterization of the virescent gene vsp in Upland cotton (Gossypium hirsutum). Theor. Appl. Genet. 2019, 132, 2069–2086. [Google Scholar] [CrossRef]
- Paterson, A.H.; Brubaker, C.L.; Wendel, J.F. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Rep. 1993, 11, 122–127. [Google Scholar] [CrossRef]
- Song, M. Genetic Analysis and Comparative Study on the Main Characteristics of Virescent Mutants in Upland Cotton (Gossypium hirsutum L.). Master’s Thesis, Graduate School of Chinese Academy of Agricultural Sciences, Beijing, China, 2014. [Google Scholar]
- Dong, W.; Wu, D.; Li, G.; Wu, D.; Wang, Z. Next-generation sequencing from bulked segregant analysis identifies a dwarfism gene in watermelon. Sci. Rep. 2018, 8, 2908. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, C.; Zhou, Y.; Hao, Z.; Wang, Z.; Zeng, X.; Di, H.; Li, M.; Zhang, D.; Yong, H.; et al. Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genom. 2016, 17, 178. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, T.; Zhong, Y.; Li, X.; Huang, J. Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genet. Genomes 2016, 12, 76. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Ooijen, J.W.V. JoinMap® 4.0, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherland, 2006.
- Wang, S.; Basten, C.J.; Zeng, Z.B. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2011. [Google Scholar]
- Zeng, Z.B. Theoretical basis for separation of multipli linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 1993, 90, 10972–10976. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Fan, S.; Pang, C.; Wei, H.; Yu, S.; Song, M. Research on the Main Photosynthetic Characteristics and Agronomic Traits in Virescent Cotton Materials. Cotton Sci. 2014, 26, 531–538. [Google Scholar]
- Wang, J.; Lu, X.; Wang, Y.; Wang, S.; Yin, Z.; Fu, X.; Wang, D.; Chen, X.; Guo, L.; Chen, C.; et al. Characteristics and Cold Tolerance of Upland Cotton Genetic Standard Line TM-1. Sci. Agric. Sin. 2022, 58, 1503–1517. [Google Scholar]
- Li, H.; Zhang, L.; Wang, J. Analysis and Answers to Frequently Asked Questions in Quantitative Trait Locus Mapping. Acta Agron. Sin. 2010, 36, 918–931. [Google Scholar] [CrossRef]
- Mao, G. Fine Mapping and Candidate Genes Analysis of Two Virescent Genes (v1 and vsp) in Upland Cotton. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2018. [Google Scholar]
- Jessberger, R. The many functions of SMC proteins in chromosome dynamics. Nat. Rev. Mol. Cell Biol. 2002, 3, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.C. Chromatin modifiers that control plant development. Curr. Opin. Plant Biol. 2006, 9, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Park, C.; Lee, J.; Oh, M.; Noh, B.; Lee, I. Arabidopsis homologs of components of the SWR1 complex regulate flowering and plant development. Development 2007, 134, 1931–1941. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | In the Year 2015 | In the Year 2016 | ||||
|---|---|---|---|---|---|---|
| TM-1 | vsp | CCRI58 | TM-1 | Vsp | CCRI58 | |
| FT (d) | 63.09 ± 2.84 | 53.45 ± 1.48 | 54.12 ± 1.99 | 65.85 ± 0.28 | 60.96 ± 0.28 | 60.10 ± 0.26 |
| WGP(d) | 119.2 ± 2.57 | 104.9 ± 5.97 | 100.7 ± 3.79 | 123.57 ± 0.76 | 105.70 ± 0.20 | 105.31 ± 0.85 |
| NFFB (num) | 7.71 ± 1.14 | 4.83 ± 0.47 | 4.71 ± 0.55 | 7.85 ± 0.22 | 6.44 ± 0.23 | 6.14 ± 0.17 |
| HNFFB (cm) | -- | -- | -- | 18.64 ± 1.24 | 12.2 ± 1.06 | 12.67 ± 0.49 |
| Traits | Population | Range | Mean ± SD | K | SE | |K|/SE | S | SE | |S|/SE | K-S Test |
|---|---|---|---|---|---|---|---|---|---|---|
| FT (d) | F2 | 51.0–67.0 | 60.42 ± 3.03 | −0.130 | 0.485 | 0.27 | −0.065 | 0.245 | 0.27 | 0.038 |
| WGP (d) | F2 | 98.0–131.0 | 112.32 ± 7.15 | 0.172 | 0.488 | 0.35 | 0.639 | 0.246 | 2.60 | 0.000 |
| NFFB (num) | F2 | 4.0–10.0 | 6.40 ± 1.13 | 1.207 | 0.481 | 2.51 | 0.737 | 0.243 | 3.03 | 0.000 |
| HNFFB (cm) | F2 | 11.0–32.0 | 19.36 ± 4.63 | −0.176 | 0.483 | 0.36 | 0.466 | 0.244 | 1.91 | 0.200 |
| FT (d) | F2:3 | 62–69 | 64.79 ± 1.48 | −0.392 | 0.481 | 0.81 | 0.189 | 0.243 | 0.78 | 0.000 |
| WGP (d) | F2:3 | 105–124 | 115.24 ± 4.22 | −0.716 | 0.481 | 1.49 | −0.161 | 0.243 | 0.66 | 0.009 |
| NFFB (num) | F2:3 | 5.9–8.2 | 6.94 ± 0.52 | −0.416 | 0.481 | 0.86 | 0.037 | 0.243 | 0.15 | 0.000 |
| HNFFB (cm) | F2:3 | 12.3–20.7 | 15.36 ± 1.71 | 0.343 | 0.481 | 0.71 | 0.651 | 0.243 | 2.68 | 0.098 |
| Traits | F2:3-NFFB | F2:3-FT | F2:3-WGP | F2:3-HNNFB | F2-NFFB | F2-FT | F2-WGP | F2-HNFFB |
|---|---|---|---|---|---|---|---|---|
| F2:3-NFFB | 1 | |||||||
| F2:3-FT | 0.57 ** | 1 | ||||||
| F2:3-WGP | 0.46 ** | 0.70 ** | 1 | |||||
| F2:3-HNNFB | 0.63 ** | 0.40 ** | 0.43 ** | 1 | ||||
| F2-NFFB | 0.51 ** | 0.38 ** | 0.36 ** | 0.36 ** | 1 | |||
| F2-FT | 0.30 ** | 0.45 ** | 0.37 ** | 0.24 * | 0.39 ** | 1 | ||
| F2-WGP | 0.32 ** | 0.45 ** | 0.42 ** | 0.18 | 0.39 ** | 0.81 ** | 1 | |
| F2-HNFFB | 0.41 ** | 0.37 ** | 0.41 ** | 0.53 ** | 0.66 ** | 0.43 ** | 0.30 ** | 1 |
| Marker Type | Numbers |
|---|---|
| ef × eg | 116 |
| nn × np | 8253 |
| ab × cc | 8 |
| aa × bb | 28,684 |
| ab × cd | 0 |
| lm × ll | 27,866 |
| hk × hk | 29,825 |
| cc × ab | 6 |
| Total markers | 94,758 |
| Linkage Group | Chromosome | SNP Number | Physical Distance /Mb | Genetic Distance /cM | Average Gap /cM | Maximal Gap /cM |
|---|---|---|---|---|---|---|
| LG01 | A01 | 537 | 99.88 | 205.97 | 0.38 | 17.46 |
| LG02 | A02 | 132 | 83.45 | 115.67 | 0.88 | 14.38 |
| LG03 | A03 | 294 | 100.26 | 224.17 | 0.76 | 12.44 |
| LG04 | A04 | 99 | 62.91 | 86.65 | 0.88 | 15.72 |
| LG05 | A05 | 120 | 92.05 | 103.91 | 0.87 | 13.31 |
| LG06 | A06 | 617 | 103.17 | 211.61 | 0.34 | 19.05 |
| LG07 | A07 | 49 | 78.25 | 29.70 | 0.61 | 8.64 |
| LG08 | A08 | 328 | 103.63 | 265.47 | 0.81 | 18.14 |
| LG09 | A09 | 108 | 75.00 | 96.79 | 0.90 | 16.25 |
| LG10 | A10 | 57 | 100.87 | 51.91 | 0.91 | 15.47 |
| LG11 | A11 | 68 | 93.32 | 71.85 | 1.06 | 14.16 |
| LG12 | A12 | 157 | 87.48 | 109.87 | 0.70 | 10.44 |
| LG13 | A13 | 667 | 79.96 | 199.90 | 0.30 | 5.01 |
| LG14 | D01 | 217 | 61.46 | 111.93 | 0.52 | 13.50 |
| LG15 | D02 | 120 | 67.28 | 128.41 | 1.07 | 18.63 |
| LG16 | D03 | 147 | 46.69 | 83.05 | 0.56 | 15.38 |
| LG17 | D04 | 47 | 51.45 | 38.63 | 0.82 | 6.96 |
| LG18 | D05 | 31 | 61.93 | 42.26 | 1.36 | 9.32 |
| LG19 | D06 | 572 | 64.29 | 220.24 | 0.39 | 16.38 |
| LG20 | D07 | 145 | 55.31 | 131.66 | 0.91 | 16.39 |
| LG21 | D08 | 459 | 65.89 | 251.66 | 0.55 | 18.50 |
| LG22 | D09 | 46 | 51.00 | 32.98 | 0.72 | 17.93 |
| LG23 | D10 | 50 | 63.37 | 26.97 | 0.54 | 2.98 |
| LG24 | D11 | 162 | 66.09 | 133.15 | 0.82 | 14.24 |
| LG25 | D12 | 89 | 59.11 | 88.05 | 0.99 | 18.65 |
| LG26 | D13 | 136 | 60.53 | 104.69 | 0.77 | 13.72 |
| Total | 5454 | 1934.63 | 3167.14 | 0.58 | 19.05 |
| Trait | QTL | Generation | Chr | Position (cM) | LOD | R2 (%) | 0.95% CI /cM | Physical Interval/Mb | Left Marker | Right Marker |
|---|---|---|---|---|---|---|---|---|---|---|
| NFFB | qNFFB-D03-1 | F2:3 | D03 | 40.01 | 13.03 | 43.1 | 39.5–44.7 | 35.29–38.17 | mk3284 | mk3293 |
| NFFB | qNFFB-D03-2 | F2:3 | D03 | 50.01 | 9.19 | 29.9 | 49.0–50.2 | 38.17–38.62 | mk3293 | mk3294 |
| NFFB | qNFFB-D03-3 | F2 | D03 | 52.51 | 6.66 | 21.4 | 51.5–57.6 | 38.66–38.79 | mk3295 | mk3299 |
| FT | qFT-D03-1 | F2:3 | D03 | 21.11 | 10.99 | 35.3 | 20.6–21.6 | 13.27–14.44 | mk3199 | mk3209 |
| FT | qFT-D03-2 | F2:3 | D03 | 27.21 | 13.47 | 46.4 | 26.2–27.7 | 25.70–30.27 | mk3250 | mk3259 |
| FT | qFT-D03-3 | F2 | D03 | 35.81 | 8.21 | 30.7 | 35.5–38.5 | 34.28–35.29 | mk3272 | mk3284 |
| FT | qFT-D03-4 | F2 | D03 | 45.01 | 8.74 | 37.2 | 44.2–48.5 | 35.87–38.17 | mk3292 | mk3293 |
| WGP | qWGP-A08-1 | F2:3 | A08 | 192.61 | 6.66 | 15.9 | 188.8–194.4 | 85.10–89.08 | mk1822 | mk1825 |
| WGP | qWGP-D03-1 | F2:3 | D03 | 18.21 | 5.23 | 11.6 | 16.4–19.2 | 8.79–14.43 | mk3191 | mk3208 |
| WGP | qWGP-D03-2 | F2:3 | D03 | 26.71 | 7.10 | 19.8 | 25.5–27.7 | 25.57–27.06 | mk3248 | mk3258 |
| WGP | qWGP-D03-3 | F2 | D03 | 29.71 | 7.88 | 20.6 | 27.2–31.3 | 26.39–30.63 | mk3257 | mk3268 |
| WGP | qWGP-D03-4 | F2 | D03 | 35.41 | 9.25 | 27.9 | 32.5–37.8 | 30.60–35.01 | mk3262 | mk3273 |
| WGP | qWGP-D05-5 | F2 | D05 | 0.01 | 6.21 | 23.1 | 0–7.7 | 5.50–5.68 | mk3402 | mk3398 |
| WGP | qWGP-D05-6 | F2 | D05 | 9.71 | 5.94 | 26.5 | 7.7–13 | 5.68–39.48 | mk3398 | mk6383 |
| HNFFB | qHNFFB-D03-1 | F2:3 | D03 | 12.01 | 8.34 | 28.4 | 11–12.5 | 11.90–23.28 | mk3194 | mk3247 |
| HNFFB | qHNFFB-D03-2 | F2:3 | D03 | 21.11 | 8.31 | 28.6 | 20.1–21.6 | 14.43–14.44 | mk3208 | mk3209 |
| HNFFB | qHNFFB-D03-3 | F2:3 | D03 | 30.71 | 8.94 | 33.5 | 28.2–31.8 | 27.06–30.60 | mk3258 | mk3266 |
| HNFFB | qHNFFB-D03-4 | F2 | D03 | 29.71 | 8.64 | 38.9 | 28.3–30.8 | 27.06–30.63 | mk3258 | mk3267 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, G.; Wei, H.; Chen, P.; Xing, F.; Wang, H. Application of a High-Density Single Nucleotide Polymorphism Genetic Map in Mapping Quantitative Trait Loci of Early-Maturing Traits in Upland Cotton. Agronomy 2023, 13, 2716. https://doi.org/10.3390/agronomy13112716
Mao G, Wei H, Chen P, Xing F, Wang H. Application of a High-Density Single Nucleotide Polymorphism Genetic Map in Mapping Quantitative Trait Loci of Early-Maturing Traits in Upland Cotton. Agronomy. 2023; 13(11):2716. https://doi.org/10.3390/agronomy13112716
Chicago/Turabian StyleMao, Guangzhi, Hengling Wei, Pengyun Chen, Feng Xing, and Hantao Wang. 2023. "Application of a High-Density Single Nucleotide Polymorphism Genetic Map in Mapping Quantitative Trait Loci of Early-Maturing Traits in Upland Cotton" Agronomy 13, no. 11: 2716. https://doi.org/10.3390/agronomy13112716
APA StyleMao, G., Wei, H., Chen, P., Xing, F., & Wang, H. (2023). Application of a High-Density Single Nucleotide Polymorphism Genetic Map in Mapping Quantitative Trait Loci of Early-Maturing Traits in Upland Cotton. Agronomy, 13(11), 2716. https://doi.org/10.3390/agronomy13112716