
Identification and Analysis of MADS-box, WRKY, NAC, and SBP-box Transcription Factor Families in Diospyros oleifera Cheng and Their Associations with Sex Differentiation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Gene Family Members and Protein Physicochemical Property Analysis
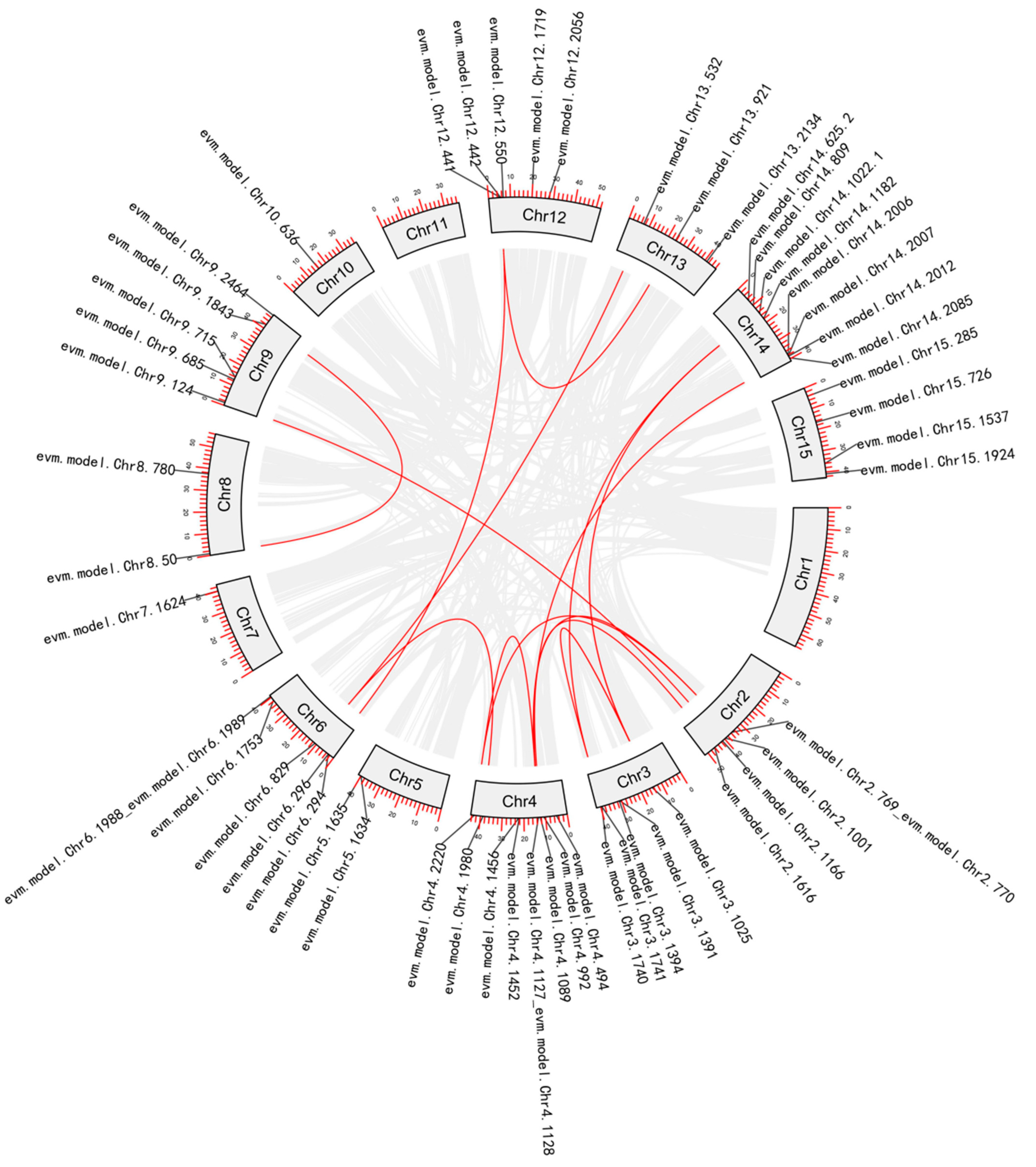
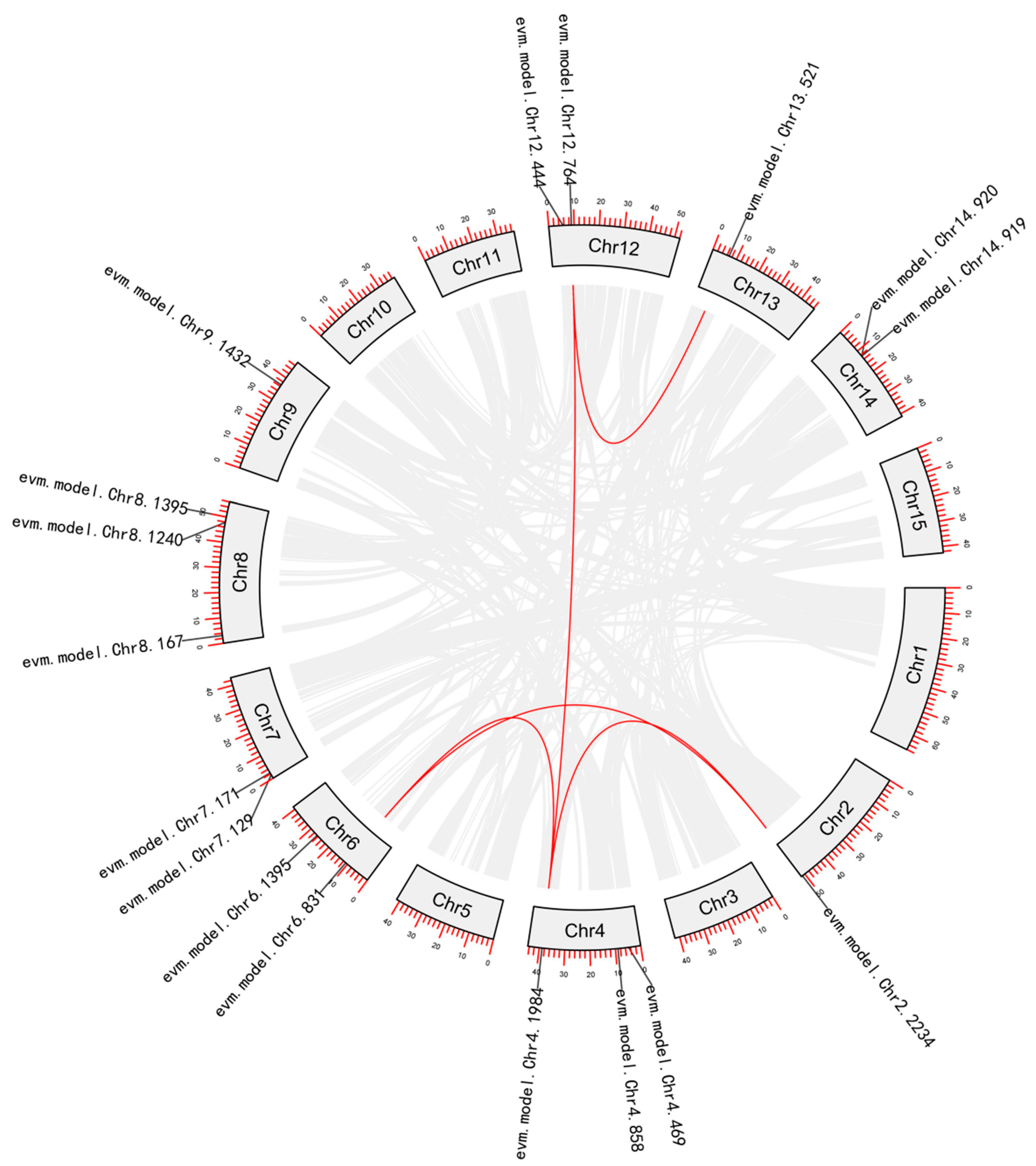
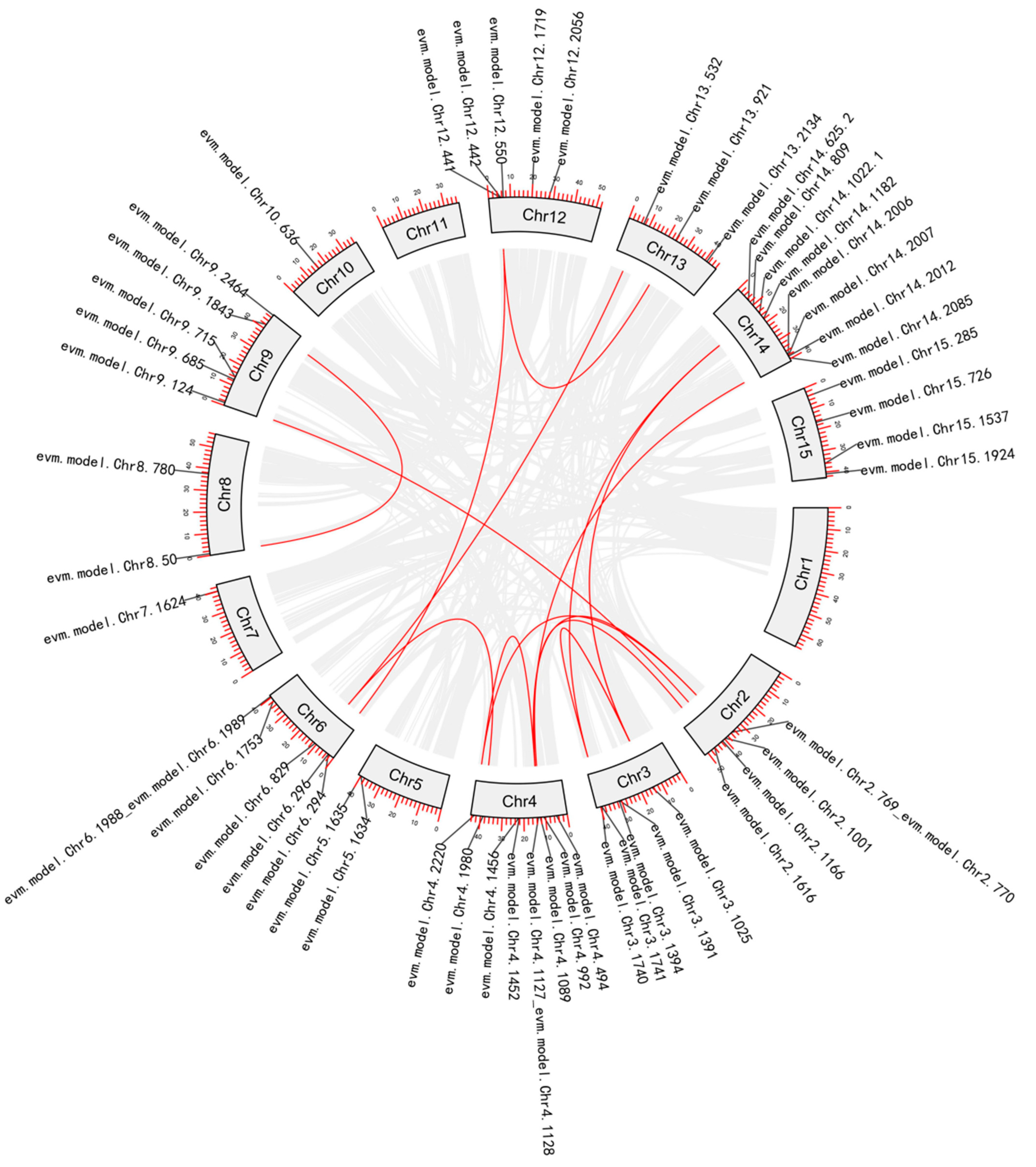
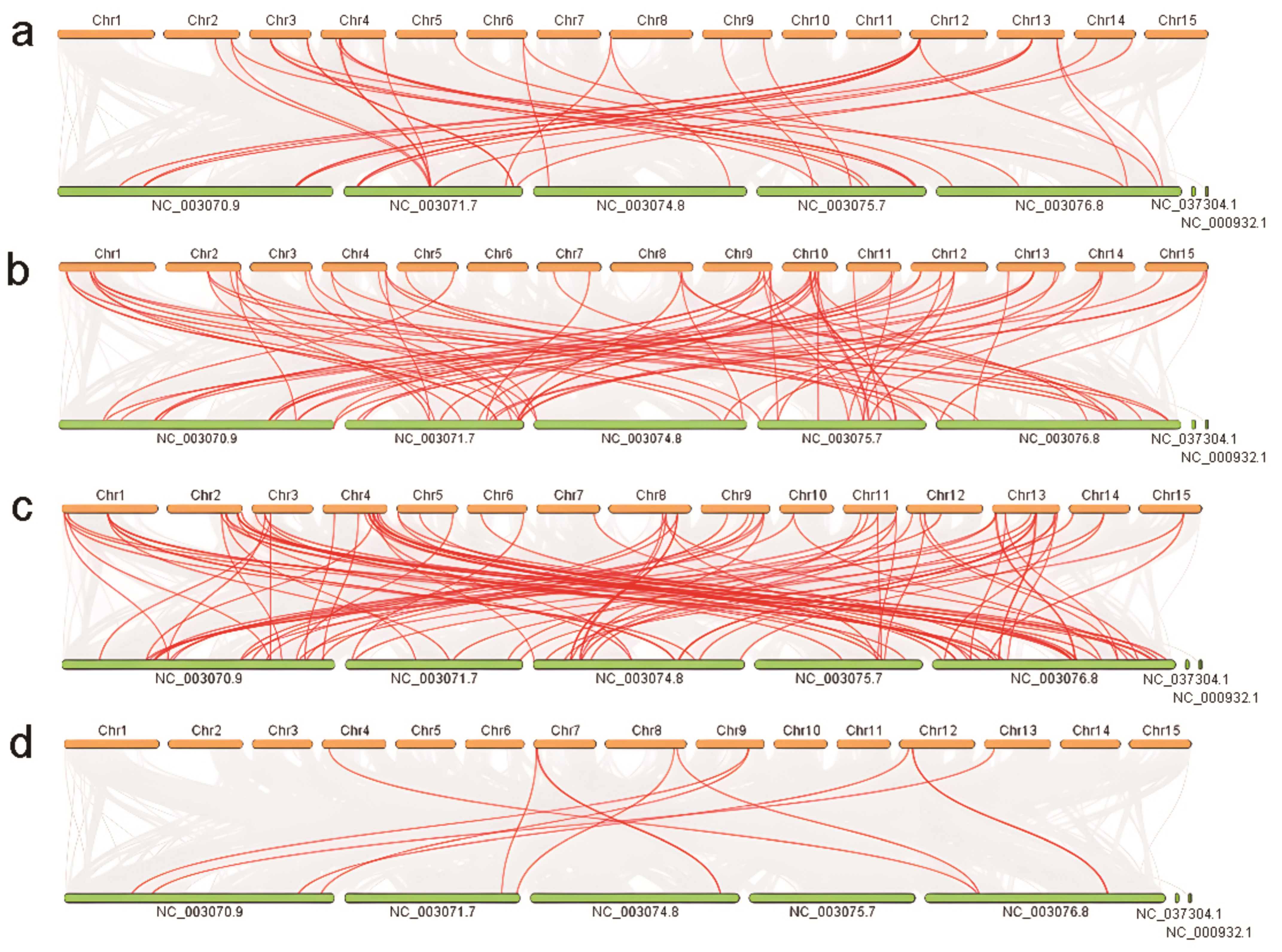
2.2. Phylogenetic Analysis and Gene Duplication Analysis
2.3. Protein Structure, Conserved Domains, and Conserved Motifs Analysis of Transcription Factor Family in D. oleifera
2.4. Promoter cis-Regulatory Element Analysis
2.5. Expression Analysis of the Transcription Factor Families
2.6. Go enrichment Analysis and Predicted Interaction Network of Transcription Factor Family
3. Results
3.1. Identification and Analysis of MADS-box Transcription Factor Family Members in D. oleifera
3.1.1. Identification of MADS-box Members of D. oleifera
3.1.2. Phylogenetic and Gene Duplication Analysis of the MADS-box Members
3.1.3. Conserved Motif, Gene Structure, and Conserved Domain Analyses for the DolMADSs
3.1.4. Analysis of Promoter cis-Acting Elements of the DolMADSs
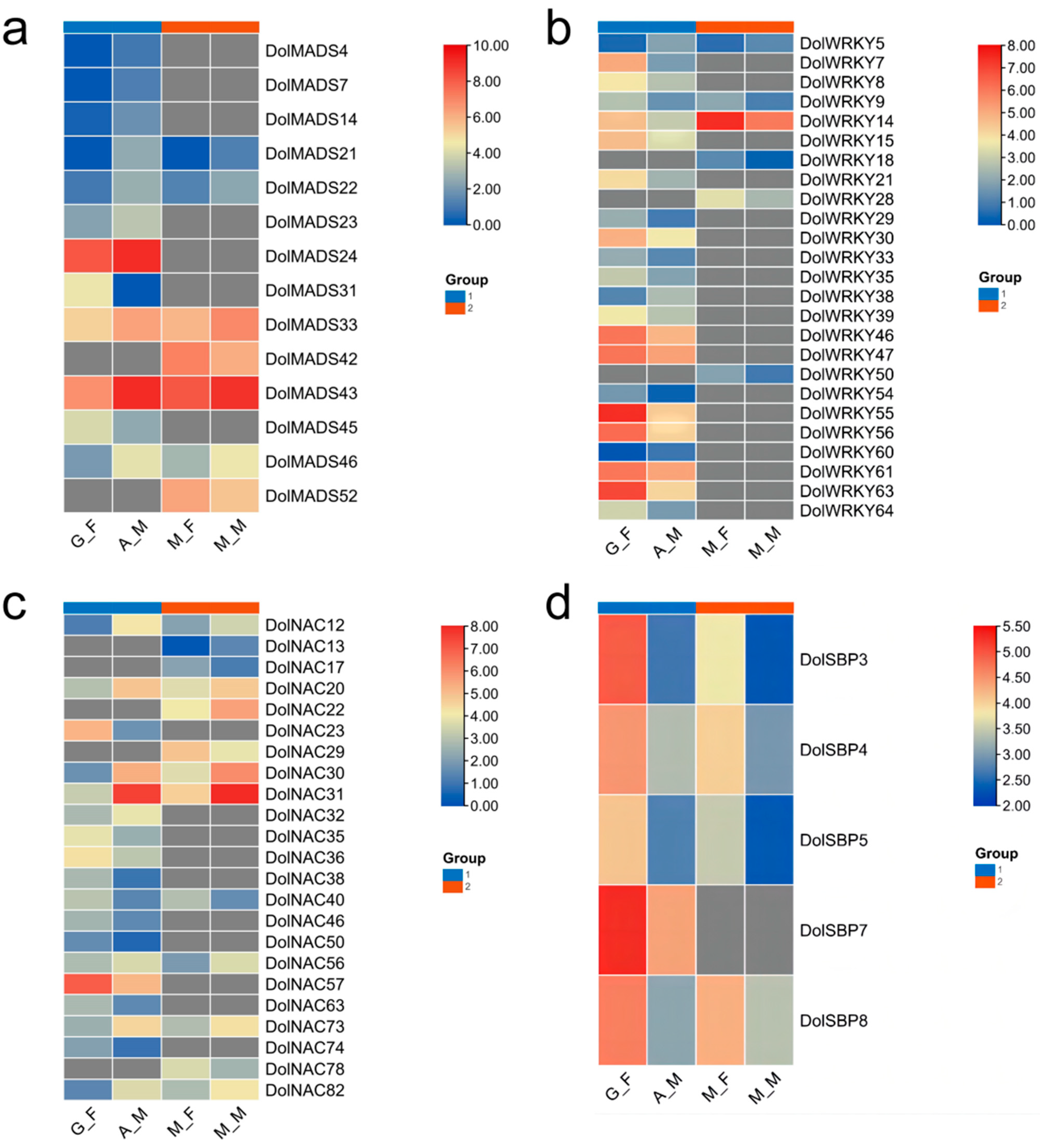
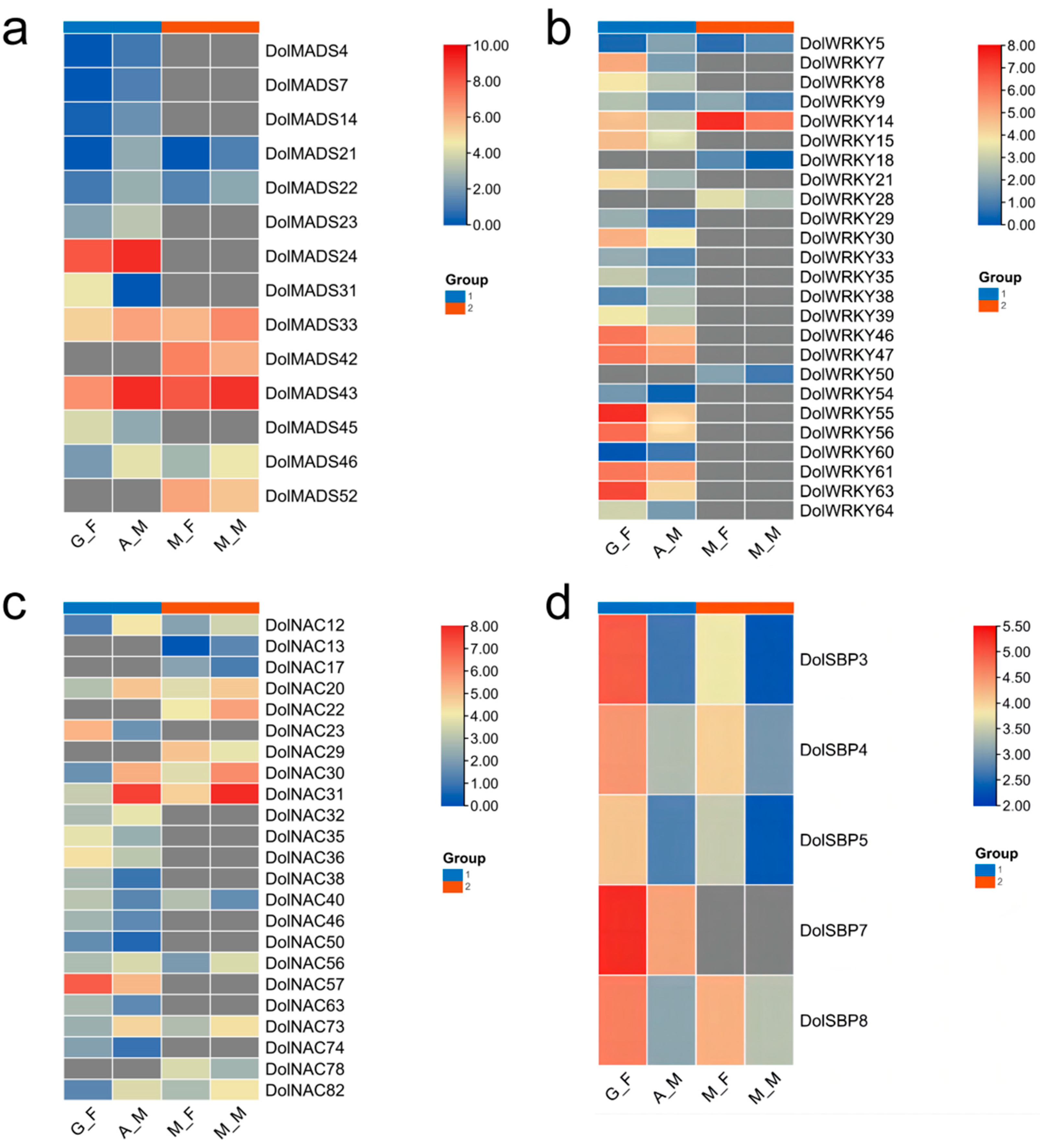
3.1.5. Expression Analysis of the DolMADSs
3.2. Identification and Analysis of WRKY Transcription Factor Family Members
3.2.1. Identification of WRKY Members in D. oleifera
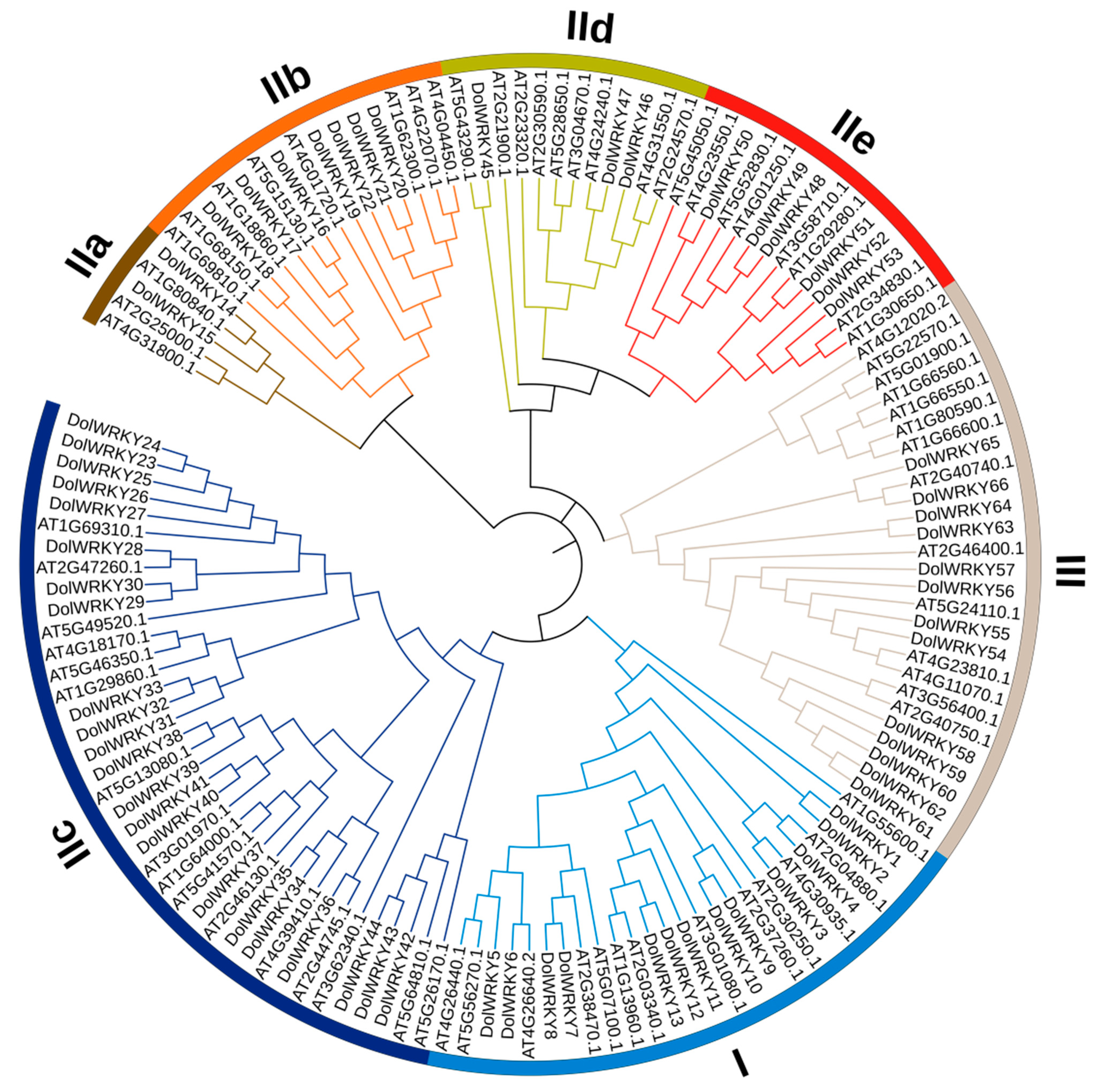
3.2.2. Phylogenetic and Gene Duplication Analysis of the WRKY Members
3.2.3. Conserved Motif, Gene Structure, and Conserved Domain Analyses for the DolWRKYs
3.2.4. Analysis of Promoter cis-Acting Elements of the DolWRKYs
3.2.5. Expression Analysis of the DolWRKYs
3.3. Identification and Analysis of NAC Transcription Factor Family Members in D. oleifera
3.3.1. Identification of NAC Members in D. oleifera
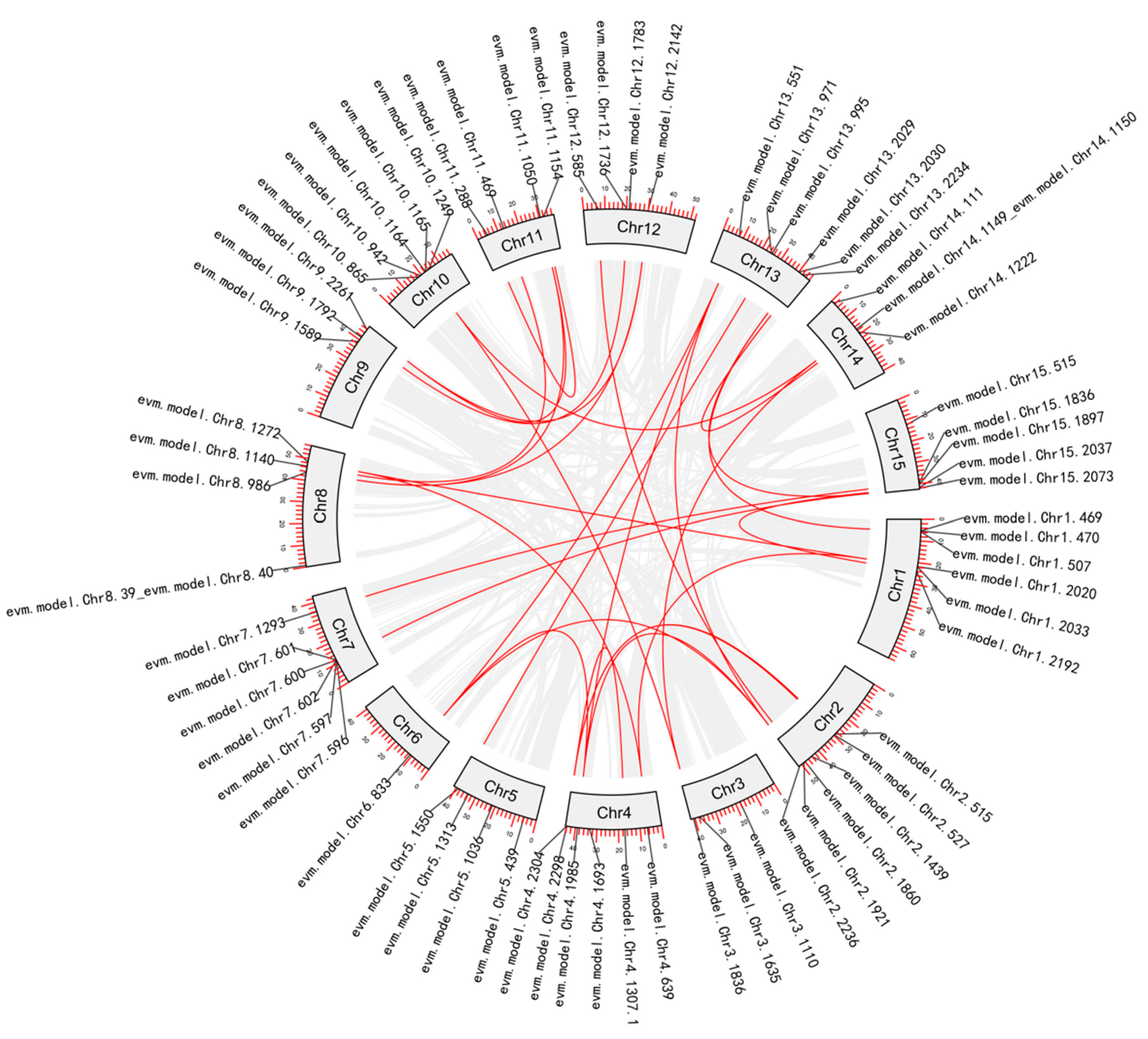
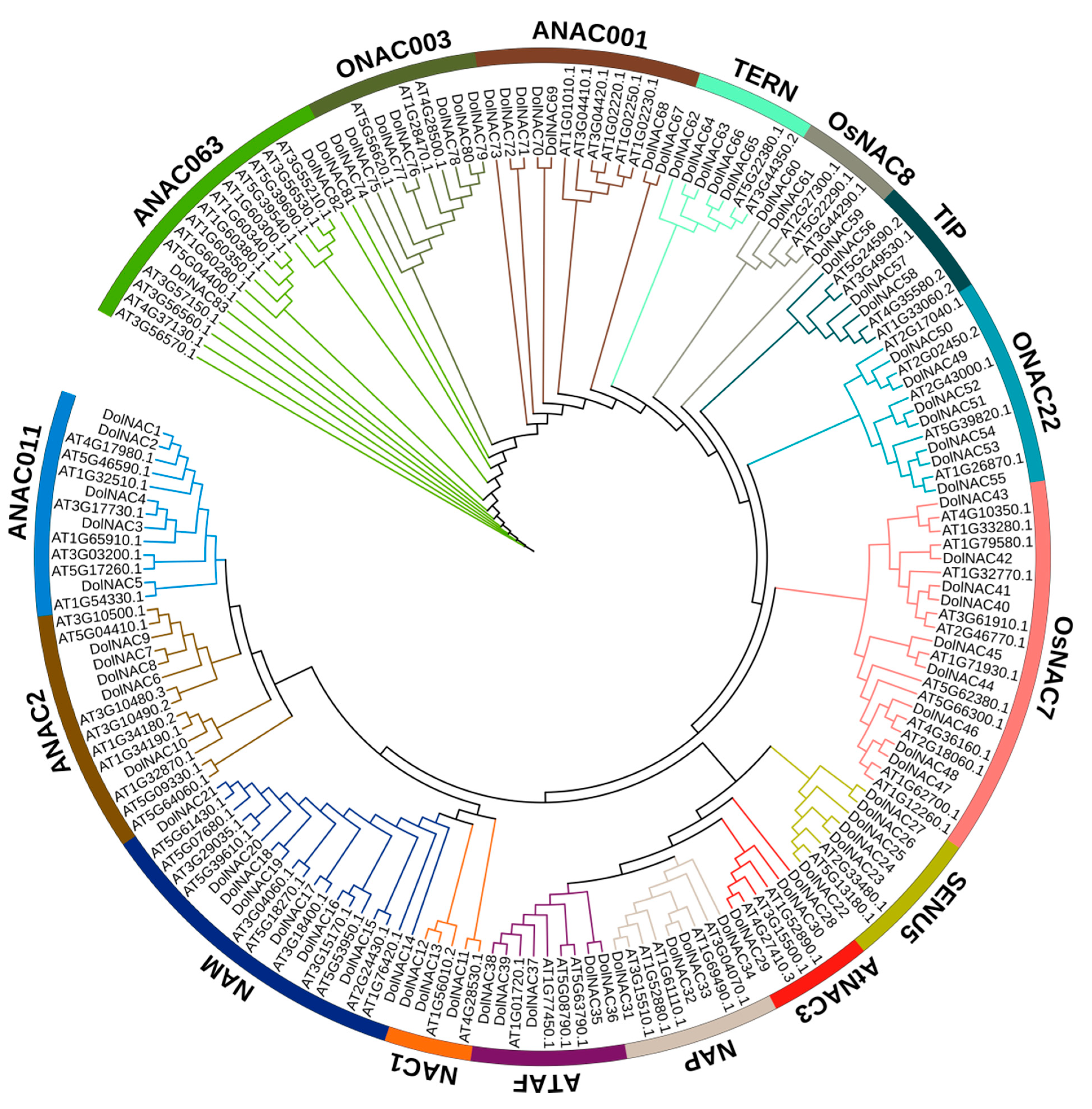
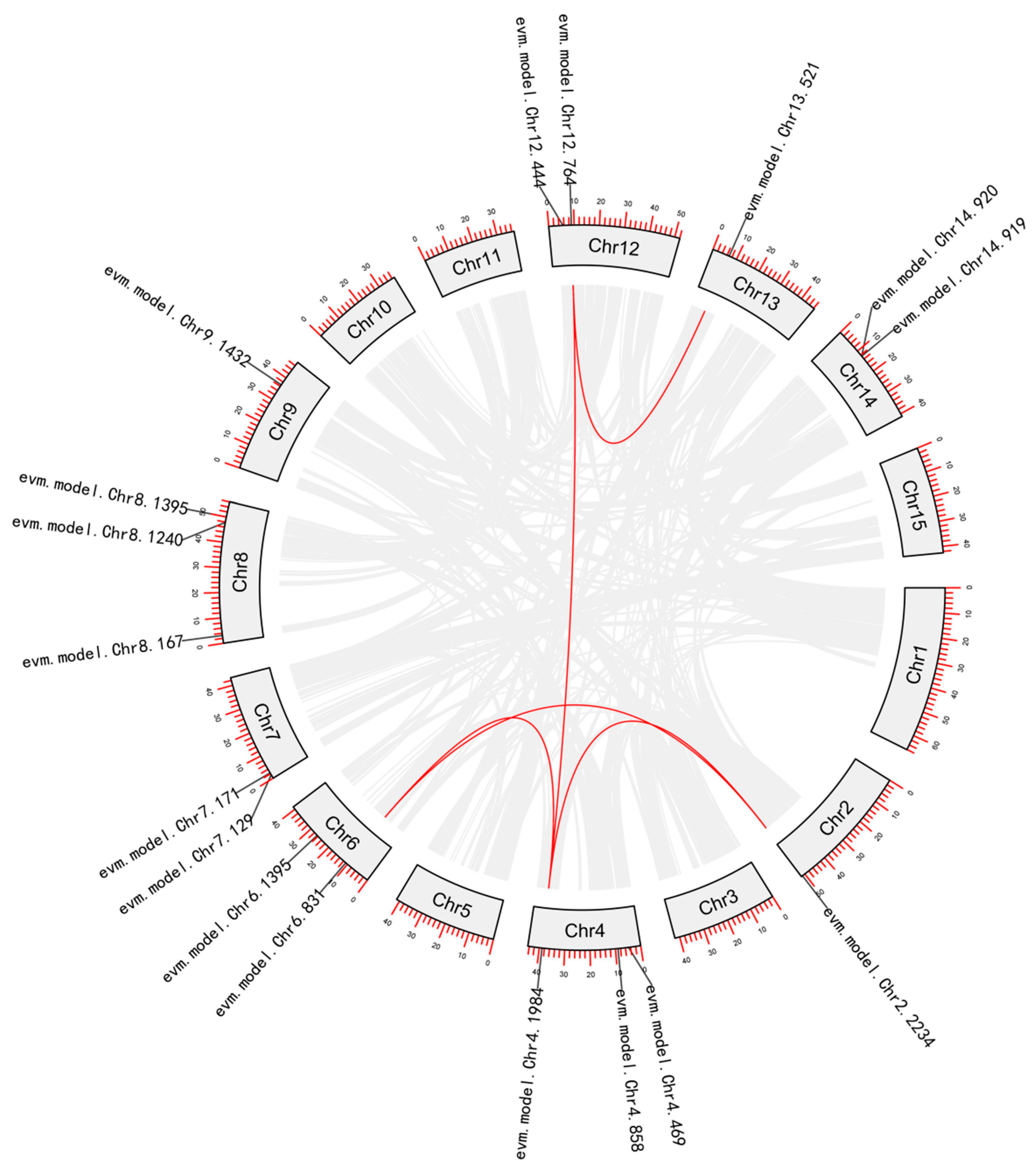
3.3.2. Phylogenetic and Gene Duplication Analysis of the NAC Members
3.3.3. Protein Structure, Conserved Domains, and Conserved Motifs Analysis for the DolNACs
3.3.4. Analysis of Promoter cis-Acting Elements of the DolNACs
3.3.5. Expression Analysis of DolNACs
3.4. Identification and Analysis of SBP-box Transcription Factor Family Members in D. oleifera
3.4.1. Identification of the SBP-box Members in D. oleifera
3.4.2. Phylogenetic and Gene Duplication Analysis of the SBP-box Members
3.4.3. Conserved Motifs, Gene Structure, and Conserved Domains Analysis for DolSBPs
3.4.4. Analysis of Promoter cis-Acting Elements of the DolSBPs
3.4.5. Expression Analysis of DolSBPs
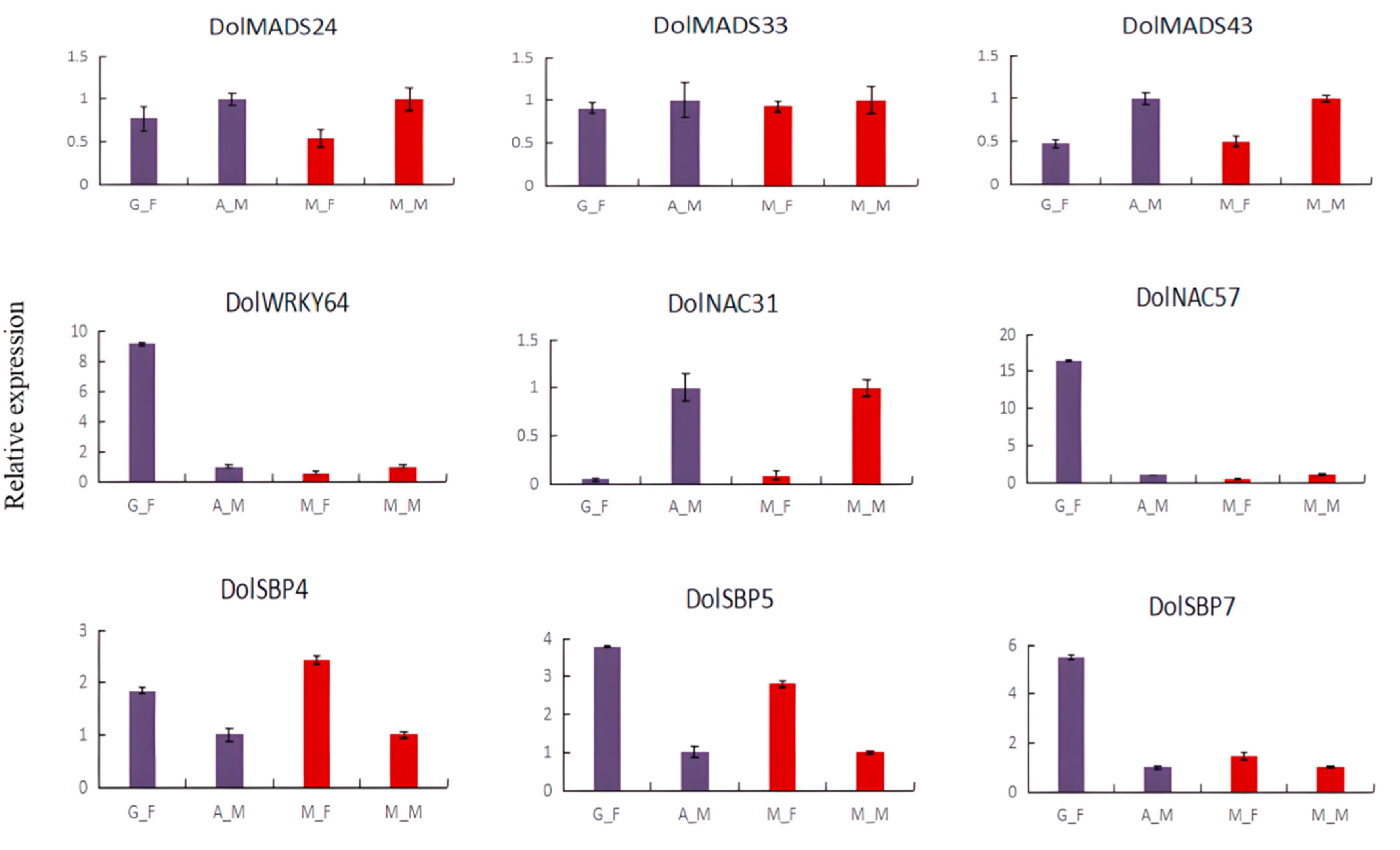
3.5. RT-qPCR Analysis of the DolMADSs, DolWRKYs, DolNACs, and DolSBPs
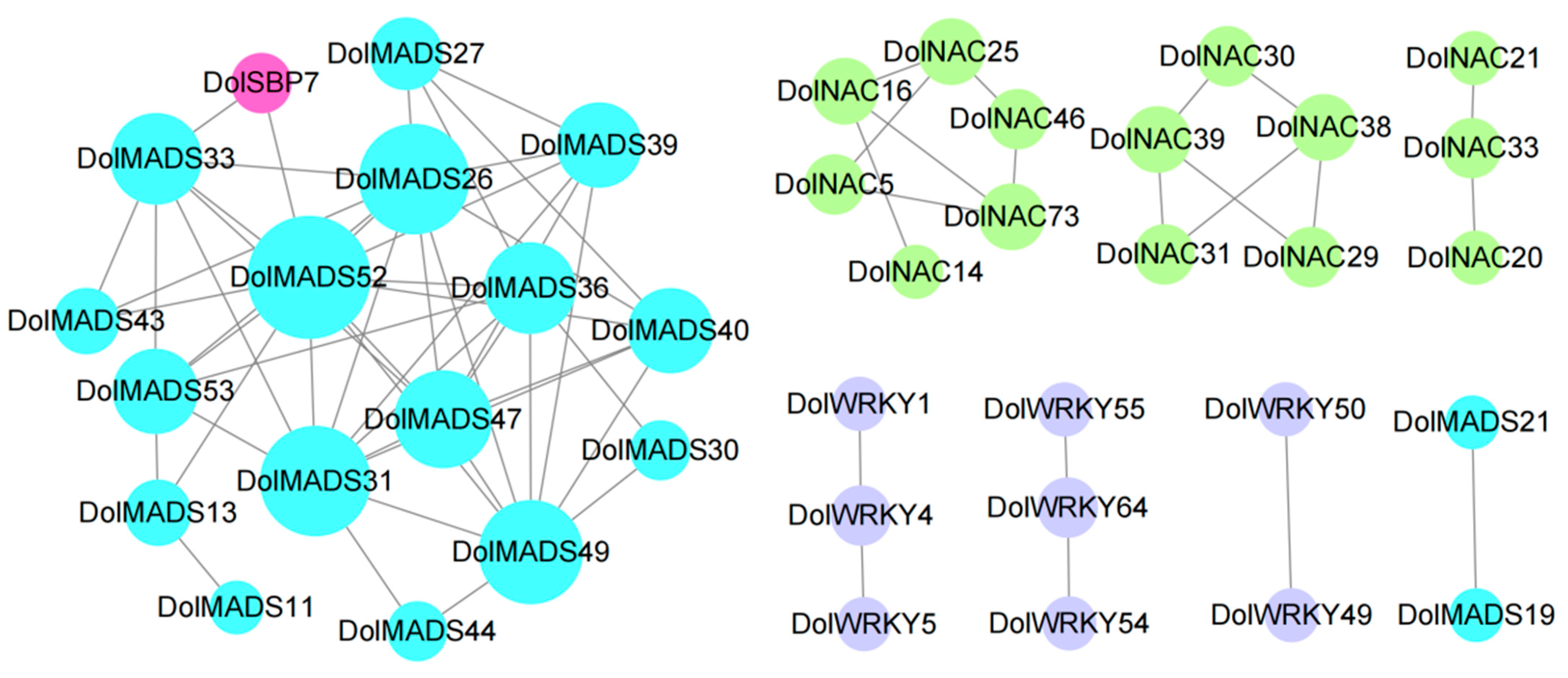
3.6. Go Enrichment Analysis and Protein Interaction Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mai, Y.N.; Li, H.W.; Suo, Y.J.; Fu, J.M.; Sun, P.; Han, W.J.; Diao, S.F.; Li, F.D. High temperature treatment generates unreduced pollen in persimmon (Diospyros kaki Thunb.). Sci. Hortic. 2019, 258, 108774. [Google Scholar] [CrossRef]
- Li, H.W.; Wang, L.Y.; Mai, Y.N.; Han, W.J.; Suo, Y.J.; Diao, S.F.; Sun, P.; Fu, J.M. Phytohormone and integrated mRNA and miRNA transcriptome analyses and differentiation of male between hermaphroditic floral buds of andromonoecious Diospyros kaki Thunb. BMC Genom. 2021, 22, 203. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Li, H.W.; Suo, Y.J.; Han, W.J.; Diao, S.F.; Mai, Y.N.; Sun, P.; Li, F.D.; Fu, J.M. Programmed cell death facilitates the formation of unisexual male and female flowers in persimmon (Diospyros kaki Thunb). Agronomy 2020, 10, 234. [Google Scholar] [CrossRef]
- Fu, J.M.; Liu, H.M.; Hu, J.J.; Liang, Y.Q.; Liang, J.J.; Wuyun, T.N.; Tan, X.F. Five complete chloroplast genome sequences from Diospyros: Genome organization and comparative analysis. PLoS ONE 2017, 11, e0159566. [Google Scholar] [CrossRef]
- Suo, Y.J.; Sun, P.; Cheng, H.H.; Han, W.J.; Diao, S.F.; Li, H.W.; Mai, Y.N.; Zhao, X.; Li, F.D.; Fu, J.M. A high-quality chromosomal genome assembly of Diospyros oleifera Cheng. GigaScience 2020, 9, 1–10. [Google Scholar] [CrossRef]
- Hu, R.; Qi, G.; Kong, Y.; Kong, D.; Gao, Q.; Zhou, G. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol. 2010, 10, 145. [Google Scholar] [CrossRef]
- Yuan, H.M.; Guo, W.D.; Zhao, L.J.; Yu, Y.; Chen, S.; Tao, L.; Cheng, L.L.; Kang, Q.H.; Song, X.X.; Wu, J.Z.; et al. Genome-wide identification and expression analysis of the WRKY transcription factor family in flax (Linum usitatissimum L.). BMC Genom. 2021, 22, 375. [Google Scholar] [CrossRef]
- Theißen, G.; Gramzow, L. Structure and Evolution of Plant MADS Domain Transcription Factors. Plant Transcription Factors: Evolutionary, Structural and Functional Aspects; Elsevier: Philadelphia, PA, USA, 2016; Volume 8, pp. 127–138. [Google Scholar]
- Parenicová, L.; De Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef]
- Yang, L.; Yan, L.L.; Li, H.E.; Du, Z.H.; Zheng, Q.Y. Identification and Analysis of MADS Gene Family in Rhododendron simsii. Mol. Plant Breed. 2021, 19, 6290–6301. [Google Scholar]
- Lin, Z.Y.; Cao, D.D.; Damaris, R.N.; Yang, P.F. Genome-wide identification of MADS-box gene family in sacred lotus (Nelumbo nucifera) identifies a SEPALLATA homolog gene involved in floral development. BMC Plant Biol. 2020, 20, 497. [Google Scholar] [CrossRef]
- Huo, S.J.; Li, Y.F.; Li, R.P.; Chen, R.H.; Xing, H.T.; Wang, J.; Zhao, Y.; Song, X.Q. Genome-wide analysis of the MADS-box gene family in Rhododendron hainanense Merr. and expression analysis under heat and waterlogging stresses. Ind. Crops Prod. 2021, 172, 114007. [Google Scholar] [CrossRef]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol. Gen. Genet. 1994, 244, 563–571. [Google Scholar] [CrossRef]
- Dong, J.; Chen, C.; Chen, Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant. Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef]
- Xu, R.R.; Zhang, S.Z.; Cao, H.; Shu, H.R. Bioinformatics analysis of WRKY transcription factor genes family in apple. Acta Hortic. Sin. 2012, 39, 2049–2060. [Google Scholar]
- Ren, Y.; Zhao, Y.J.; Zhang, X.H.; Zhao, X.Q.; Yuan, Z.H. Genome-wide identification and expression analysis of WRKY gene family in Punica granatum. Acta Bot. Boreal.-Occident. Sin. 2020, 40, 2018–2031. [Google Scholar]
- He, H.S.; Dong, Q.; Shao, Y.H.; Jiang, H.Y.; Zhu, S.W.; Cheng, B.J.; Xiang, Y. Genome -wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012, 31, 1199–1217. [Google Scholar] [CrossRef]
- Wei, W.; Hu, Y.; Han, Y.T.; Zhang, K.; Zhao, F.L.; Feng, J.Y. The WRKY transcription factors in the diploid woodland strawberry Fragaria vesca: Identification and expression analysis under biotic and abiotic stresses. Plant Physiol. Biochem. 2016, 105, 129–144. [Google Scholar] [CrossRef]
- Jing, Z.B.; Liu, Z.D. Genome-wide identification of WRKY transcription factors in kiwifruit (Actinidia spp.) and analysis of WRKY expression in responses to biotic and abiotic stresses. Genes Genom. 2018, 40, 429–446. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Souer, E.; Vanhouwelingen, A.; Kloos, D.; Mol, J.; Koes, R. The no apical meristem gene of petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell 1996, 85, 159–170. [Google Scholar] [CrossRef]
- Ooka, H.; Satoh, K.; Doi, K.; Nagata, T.; Kikuchi, S. Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2003, 10, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Zhao, L.Y.; Song, X.F.; Lin, Z.K.; Gu, B.J.; Yan, J.X.; Zhang, S.L.; Tao, S.T.; Huang, X.S. Genome-wide analyses and expression patterns under abiotic stress of NAC transcription factors in white pear (Pyrus bretschneideri). BMC Plant Biol. 2019, 19, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranwal, V.K.; Khurana, P. Genome-wide analysis, expression dynamics and varietal comparison of NAC gene family at various developmental stages in Morus notabilis. Mol. Genet. Genom. 2016, 291, 1305–1317. [Google Scholar] [CrossRef]
- Duval, M.; Hsieh, T.F.; Kim, S.Y.; Thomas, T.L. Molecular characterization of AtNAM: A member of the Arabidopsis NAC domain superfamily. Plant Mol. Biol. 2002, 50, 237–248. [Google Scholar] [CrossRef]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Gang, L.; Tian, A.G.; Zhang, W.K. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 2011, 68, 302–313. [Google Scholar] [CrossRef]
- Mao, C.; Ding, W.; Wu, Y.; Yu, J.; He, X.; Shou, H.; Wu, P. Over-expression of a NAC-domain protein promotes shoot branching in rice. New Phytol. 2007, 176, 288–298. [Google Scholar] [CrossRef]
- Birkenbihl, R.P.; Jach, G.; Saedler, H.; Huijser, P. Functional dissection of the plant-specific SBP-domain: Overlap of the DNA-binding and nuclear localization domains. J. Mol. Biol. 2005, 352, 585–596. [Google Scholar] [CrossRef]
- Song, S.; Zhou, H.Y.; Sheng, S.B.; Cao, M.; Li, Y.Y.; Pang, X.M. Genome-Wide Organization and Expression Profiling of the SBP-box Gene Family in Chinese Jujube (Ziziphus jujuba Mill.). Int. J. Mol. Sci. 2017, 18, 1734. [Google Scholar] [CrossRef]
- Yang, Z.F.; Wang, X.F.; Gu, S.L.; Hu, Z.Q.; Xu, H.; Xu, C.W. Comparative study of SBP-box gene family in Arabidopsis and rice. Gene 2007, 407, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Z.L.; Yang, Y.X.; Chen, X.Q.; Chen, G.P. Bioinformatics analysis of SBP-box gene family in rice. China J. Bioinform. 2011, 9, 82–87. [Google Scholar]
- Hou, H.M.; Li, J.; Gao, M.; Singer, S.D.; Wang, H.; Mao, L.; Fei, Z.; Wang, X.P. Genomic organization, phylogenetic comparison and differential expression of the SBP-box family genes in grape. PLoS ONE 2012, 235, 1171–1184. [Google Scholar] [CrossRef]
- Sun, P.; Fu, J.M. A BioNano optical mapping-assisted chromosomal genome assembly of Diospyros oleifera. figshare. Dataset 2022. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Li, J.R.; Sun, P.; Han, W.J.; Li, F.D.; Fu, J.M.; Diao, S.F. Morphological key period study on floral sex differentiation in pollination-constant and non-astringent persimmon‘Zenjimaru’. Acta Hortic. Sin. 2016, 43, 451–461. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.P.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, 607–613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Shipra, K.; Kumar, K.B.; Juyoung, A.; Hwa, K.J.; GeungJoo, L. Genome-wide transcriptomic identification and functional insight of lily WRKY genes responding to botrytis fungal disease. Plants 2021, 10, 776. [Google Scholar]
- Li, B.; Fan, R.Y.; Yang, Q.S.; Hu, C.H.; Sheng, O.; Deng, G.M.; Dong, T.; Li, C.Y.; Peng, X.X.; Bi, F.C.; et al. Genome-Wide identification and characterization of the NAC transcription factor family in Musa Acuminata and expression analysis during fruit ripening. Int. J. Mol. Sci. 2020, 21, 634. [Google Scholar] [CrossRef]
- Riese, M.; Höhmann, S.; Saedler, H.; Münster, T.; Huijser, P. Comparative analysis of the SBP-box gene families in P. patens and seed plants. Gene 2007, 401, 28–37. [Google Scholar] [CrossRef]
- Zeng, H.; Du, L.; Zou, H.M.; Lu, C.Z.; Luo, L.F.; Zhang, H.Z. Changes of endogenous hormones in macadamia during flower bud differentiation. J. Anhui Agri. Sci. 2008, 36, 14949–14953. [Google Scholar]
- Ming, R.; Bendahmane, A.; Renner, S.S. Sex chromosomes in land plants. Annu. Rev. Plant Biol. 2011, 62, 485–514. [Google Scholar] [CrossRef]
- Huang, F.Y.; Zhang, Y.H.; Hou, X.L. BcAP3, a MADS box gene, controls stamen development and male sterility in Pak-choi (Brassica rapa ssp. chinensis). Gene 2020, 747, 144698. [Google Scholar] [CrossRef]
- Yamada, K.; Saraike, T.; Shitsukawa, N.; Hirabayashi, C.; Takumi, S.; Murai, K. Class D and B (sister) MADS-box genes are associated with ectopic ovule formation in the pistil-like stamens of alloplasmic wheat (Triticum aestivum L.). Plant Mol. Biol. 2009, 71, 114. [Google Scholar] [CrossRef]
- Shulga, O.A.; Mitiouchkina, T.Y.; Shchennikova, A.V.; Skryabin, G.K.; Dolgov, V.S. Chrysanthemum modification via ectopic expression of sunflower MADS-box gene HAM59. Acta Hortic. 2015, 8, 1069–1089. [Google Scholar] [CrossRef]
- Chen, R.; Shen, L.P.; Wang, D.H.; Wang, F.G.; Zeng, H.Y.; Chen, Z.S.; Peng, Y.B.; Lin, Y.N.; Tang, X.; Deng, M.H.; et al. A gene expression profiling of early rice stamen development that reveals inhibition of photosynthetic genes by OsMADS58. Mol. Plant. 2015, 8, 1069–1089. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.S.; Liu, X.F.; Wang, D.H.; Chen, R.; Zhang, X.L.; Xu, Z.H.; Bai, S.N. Transcription factor OsTGA10 is a target of the MADS protein OsMADS8 and is required for tapetum development. Plant Physiol. Preview 2017, 176, 01419. [Google Scholar] [CrossRef]
- Hama, E.; Takumi, S.; Ogihara, Y.; Murai, K. Pistillody is caused by alterations to the class-B MADS-box gene expression pattern in alloplasmic wheats. Planta 2004, 218, 712–720. [Google Scholar]
- Tanaka, Y.; Oshima, Y.; Yamamura, T.; Sugiyama, M.; Terakawa, T. Multi-petal cyclamen flowers produced by AGAMOUS chimeric repressor expression. Sci. Rep. 2013, 3, 2641. [Google Scholar] [CrossRef]
- Noor, S.H.; Ushijima, K.; Murata, A.; Yoshida, K.; Nakano, R. Double flower formation induced by silencing of C-class MADS-box genes and its variation among petunia cultivars. Sci. Hortic. 2014, 178, 1–7. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; He, S.P.; Gao, Y.; Wang, N.N.; Lu, R.; Li, X.B. A cotton (Gossypium hirsutum) WRKY transcription factor (GhWRKY22) participates in regulating anther/pollen development. Plant Physiol. Biochem. 2019, 141, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Jiang, W.; Yu, D. Male gametophyte-specific WRKY34 transcription factor mediates cold sensitivity of mature pollen in Arabidopsis. J. Exp. Bot. 2010, 61, 3901–3914. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.S.; Liu, X.; Somssich, I.E. Elucidating the role of WRKY27 in male sterility in Arabidopsis. Plant Signal. Behav. 2017, 12, e1363945. [Google Scholar] [CrossRef]
- Zhang, J. Functional Studies of WRKY Transcription Factor OsWRKY31 and OsWRKY51 Genes in Rice; China Agricultural University: Beijing, China, 2007. [Google Scholar]
- Zheng, Y.P.; Deng, X.X.; Qu, A.L.; Zhang, M.M.; Tao, Y.; Yang, L.Y.; Liu, Y.D.; Xu, J.; Zhang, S.Q. Regulation of pollen lipid body biogenesis by MAP kinases and downstream WRKY transcription factors in Arabidopsis. PLoS Genet. 2018, 14, e1007880. [Google Scholar] [CrossRef]
- Lei, R.H.; Li, X.L.; Ma, Z.B.; Lv, Y.; Hu, Y.; Yu, D. Arabidopsis WRKY2 and WRKY34 transcription factors interact with VQ20 protein to modulate pollen development and function. Plant J. 2017, 91, 962. [Google Scholar] [CrossRef]
- Xun, Z.L.; Li, X.P.; Zhang, D.D.; Lu, H.; Liu, D. Functional analysis of transcription factor WRKY18 in pollen development of Arabidopsis thaliana (L.) Heynh. J. Chin. Electron Microsc. Soc. 2014, 33, 156–162. [Google Scholar]
- Yang, Q.; Zhang, H.P.; Liu, C.; Huang, L.P.; Zhao, L.L.; Zhang, A.Y. A NAC transcription factor ZmNAC84 affects pollen development through the repression of ZmRbohH expression in maize. J. Plant Biol. 2018, 61, 366–373. [Google Scholar] [CrossRef]
- Yan, C.H.; Li, G.R.; Mu, J.Y.; Lou, H.T.; Zhu, Z.G. Subcellular localization and functional analysis of a NAC gene VvDRL1 from Vitis vinifera ‘Yatomi Rosa’. Acta Hortic. Sin. 2016, 43, 643–652. [Google Scholar]
- Sun, Q.W. A Cotton NAC Domain Transcription Factor, GhFSN5, Negatively Regulates Secondary Cell Wall Biosynthesis and Anther Development in Transgenic Arabidopsis; Central China Normal University: Wuhan, China, 2018. [Google Scholar]
- Zheng, G.H.; Wei, W.; Li, Y.P.; Kan, L.J.; Wang, F.X.; Zhang, X.; Li, F.; Liu, Z.C.; Kang, C.Y. Conserved and novel roles of miR164-CUC2 regulatory module in specifying leaf and floral organ morphology in strawberry. New Phytol. 2019, 224, 480–492. [Google Scholar] [CrossRef]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC transcription factors NST1 and NST2 of Arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef]
- Zhao, X.; Gallego-Giraldo, L.; Wang, H.Z.; Zeng, Y.N.; Ding, S.Y.; Chen, F.; Dixon, R.A. An NAC transcription factor orchestrates multiple features of cell wall development in Medicago truncatula. Plant J. 2010, 63, 100–114. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Plant NAC-type transcription factor proteins contain a NARD domain for repression of transcriptional activation. Planta 2010, 232, 1033–1043. [Google Scholar]
- Shih, C.F.; Hsu, W.H.; Peng, Y.J.; Yang, C.H. The NAC-like gene anther indehiscence factor acts as a repressor that controls anther dehiscence by regulating genes in the jasmonate biosynthesis pathway in Arabidopsis. J. Exp. Bot. 2014, 65, 621–639. [Google Scholar] [CrossRef]
- Yang, L. Analysis of SPL8-Like Genes in Arabidopsis and Beyond; Shanxi University: Taiyuan, China, 2018. [Google Scholar]
- Unte, U.S.; Sorensen, A.M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef]
- Lännenpää, M.; Jänönen, I.; Hölttä-Vuori, M.; Gardemeister, M.; Porali, I.; Sopanen, T. A new SBP-box gene BpSPL1 in silver birch (Betula pendula). Physiol. Plant 2004, 120, 491–500. [Google Scholar] [CrossRef]
- Schwarz, S.; Grande, A.V.; Bujdoso, N.; Saedler, H.; Huijser, P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol. Biol. 2008, 67, 183–195. [Google Scholar] [CrossRef]
- Cao, D.; Li, Y.; Wang, J.L.; Nan, H.Y.; Wang, Y.N.; Lu, S.J.; Jiang, Q.; Li, X.M.; Shi, D.N.; Fang, C.; et al. GmmiR156b overexpression delays flowering time in soybean. Plant Mol. Biol. 2015, 89, 353–363. [Google Scholar] [CrossRef]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell. 2005, 8, 517–527. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, Y.; Diao, S.; Yuan, J.; Wang, L.; Suo, Y.; Li, H.; Han, W.; Wang, Y.; Ye, L.; Liu, Y.; et al. Identification and Analysis of MADS-box, WRKY, NAC, and SBP-box Transcription Factor Families in Diospyros oleifera Cheng and Their Associations with Sex Differentiation. Agronomy 2022, 12, 2100. https://doi.org/10.3390/agronomy12092100
Mai Y, Diao S, Yuan J, Wang L, Suo Y, Li H, Han W, Wang Y, Ye L, Liu Y, et al. Identification and Analysis of MADS-box, WRKY, NAC, and SBP-box Transcription Factor Families in Diospyros oleifera Cheng and Their Associations with Sex Differentiation. Agronomy. 2022; 12(9):2100. https://doi.org/10.3390/agronomy12092100
Chicago/Turabian StyleMai, Yini, Songfeng Diao, Jiaying Yuan, Liyuan Wang, Yujing Suo, Huawei Li, Weijuan Han, Yiru Wang, Lingshuai Ye, Yang Liu, and et al. 2022. "Identification and Analysis of MADS-box, WRKY, NAC, and SBP-box Transcription Factor Families in Diospyros oleifera Cheng and Their Associations with Sex Differentiation" Agronomy 12, no. 9: 2100. https://doi.org/10.3390/agronomy12092100
APA StyleMai, Y., Diao, S., Yuan, J., Wang, L., Suo, Y., Li, H., Han, W., Wang, Y., Ye, L., Liu, Y., Pu, T., Zhang, Q., Sun, P., & Fu, J. (2022). Identification and Analysis of MADS-box, WRKY, NAC, and SBP-box Transcription Factor Families in Diospyros oleifera Cheng and Their Associations with Sex Differentiation. Agronomy, 12(9), 2100. https://doi.org/10.3390/agronomy12092100