
SNP Genotyping for Purity Assessment of a Forage Oat (Avena sativa L.) Variety from Colombia

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials Used
2.2. Agronomic Locations Used
2.3. Leaf Tissue and DNA Extraction
2.4. SNP Genotyping
2.5. Data Analysis
3. Results
3.1. Oat Selections Based on Harvest Date
3.2. Oat SNP Chip Success Rate
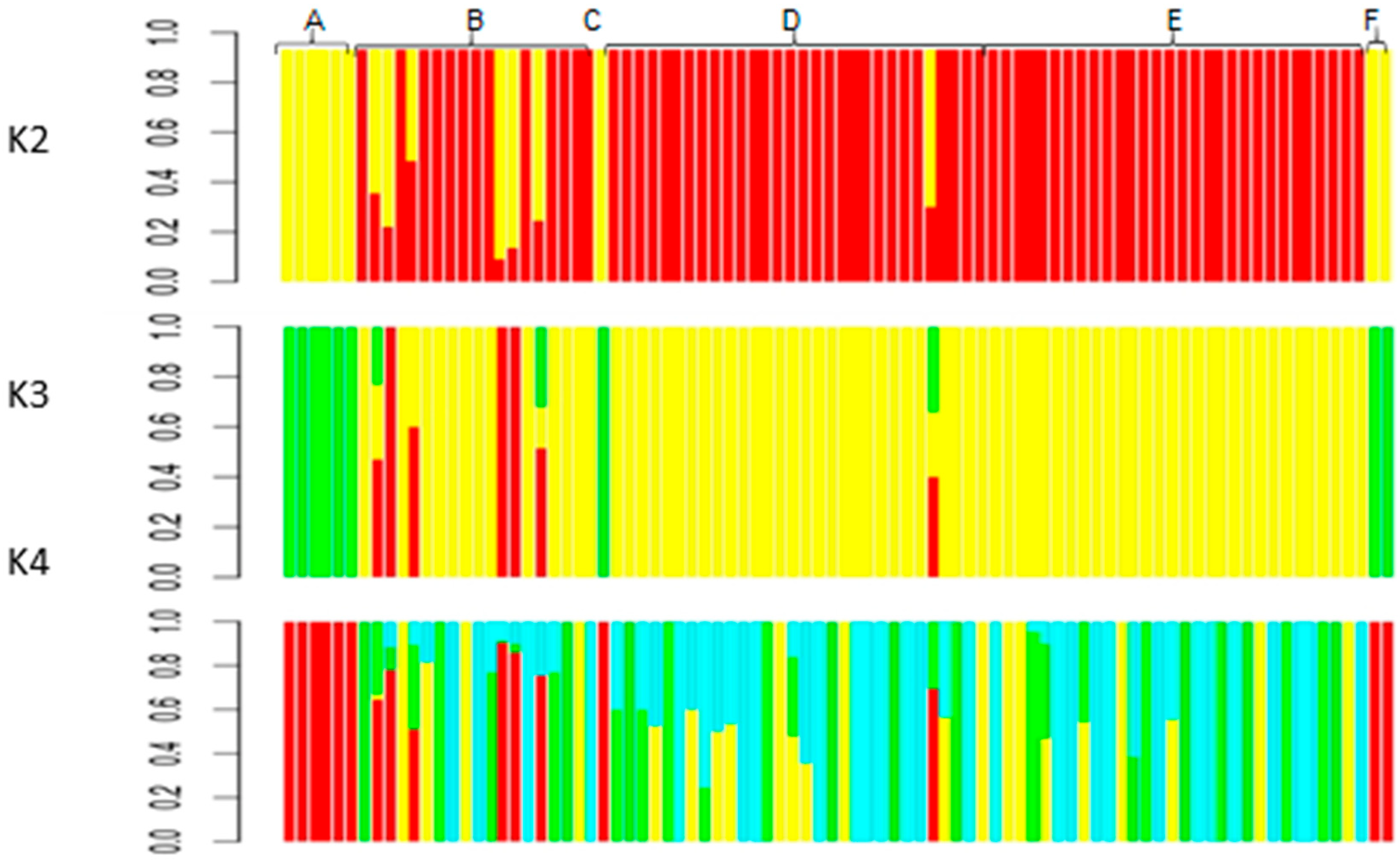
3.3. Clustering of Individual Plants
3.4. Principal Component Analysis
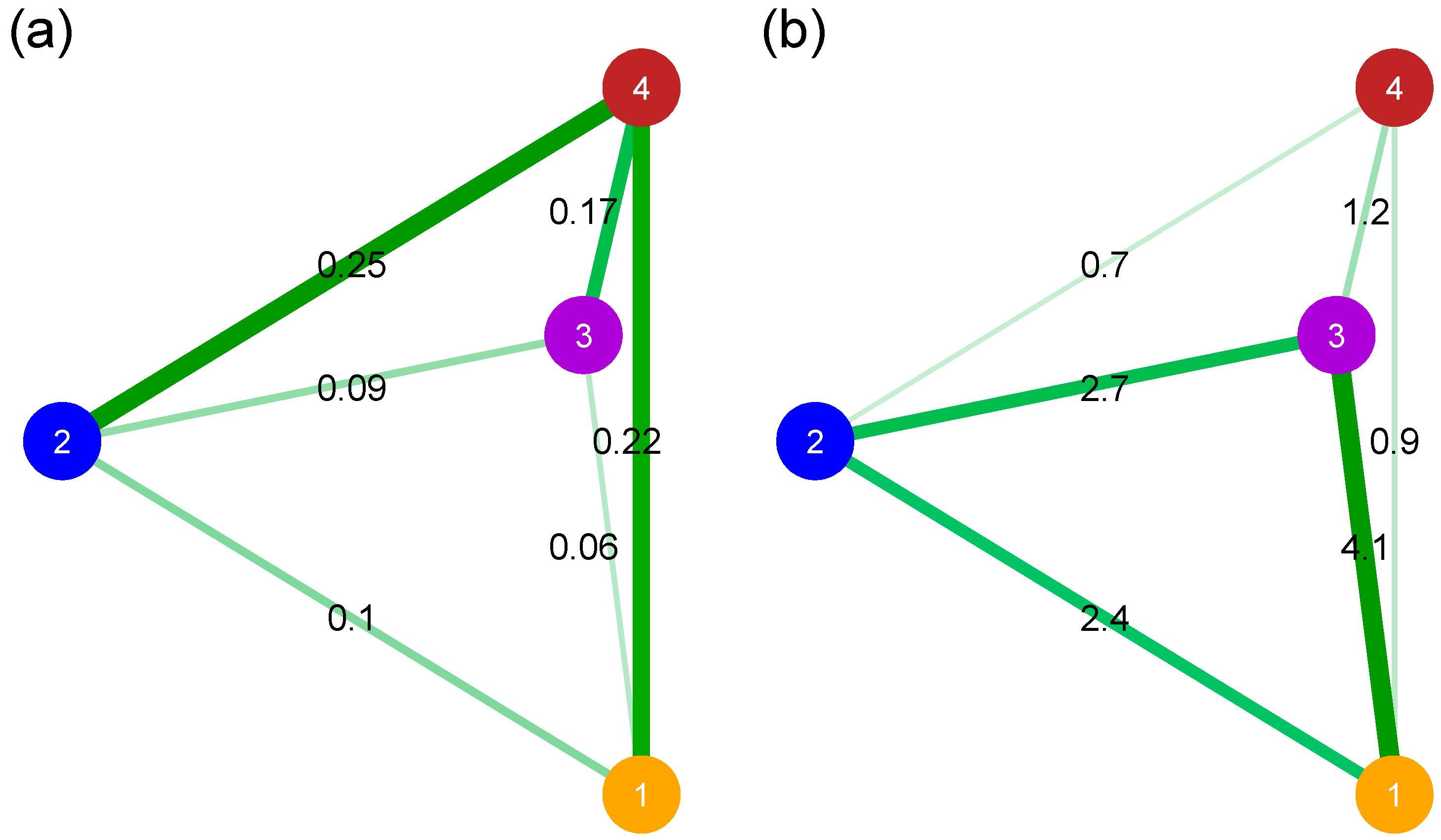
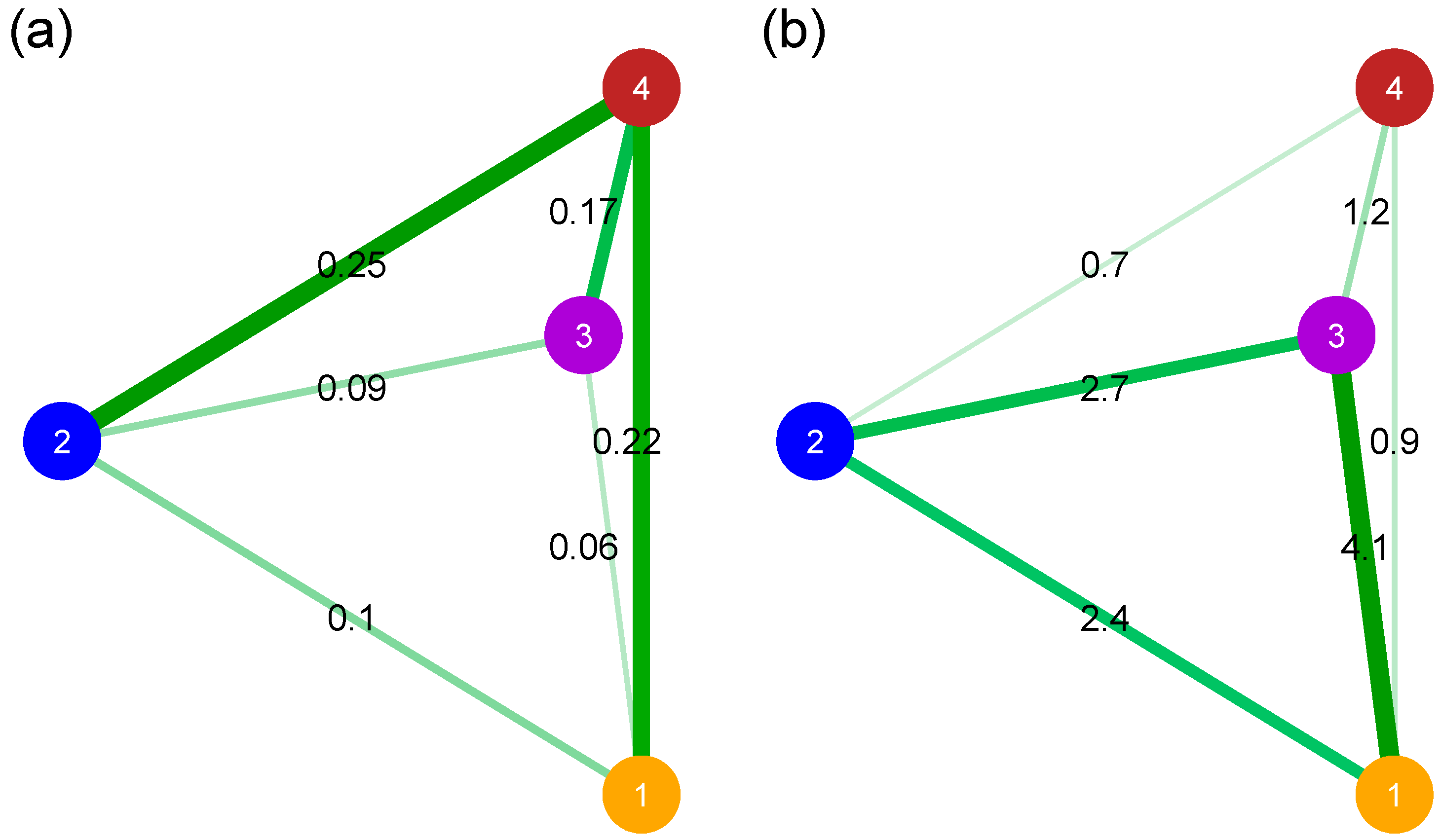
3.5. Relative Divergence and Genetic Patterns of Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, H.; Martin, S.L.; Bekele, W.A.; Latta, R.G.; Diederichsen, A.; Peng, Y.; Tinker, N.A. Genome size variation in the genus Avena. Genome 2016, 59, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango-Quispe, S.J.S.; Viera-Valencia, M.; Gómez Bravo, C.A. Agronomic Evaluation and Nutritive Value of Oat (Avena Sativa) under Rainfall Restriction in the Andes. Cienc. Tecnol. Agropecu. 2022, 23. [Google Scholar] [CrossRef]
- Campuzano-Duque, L.F.; Castro-Rincón, E.; Torres-Cuesta, D.; Castillo-Sierra, J.; Nieto-Sierra, D.; Portillo-Lopez, P.A. Altoandina: Nueva variedad forrajera para la zona andina en Colombia. Agron. Mesoam. 2020, 31, 581–591. [Google Scholar] [CrossRef]
- McCouch, S.R.; Wright, M.H.; Tung, C.W.; Maron, L.G.; McNally, K.L.; Fitzgerald, M.; Singh, N.; DeClerck, G.; Agosto-Perez, F.; Korniliev, P.; et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 2016, 7, 10532. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sánchez-Villeda, H.; Sorrells, M.; et al. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome J. 2012, 5, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Tinker, N.A.; Chao, S.; Lazo, G.R.; Oliver, R.E.; Huang, Y.F.; Poland, J.A.; Jellen, E.; Maughan, P.J.; Kilian, A.; Jackson, E.W. A SNP genotyping array for hexaploid oat. Plant Genome 2014, 7, 1–8. [Google Scholar] [CrossRef]
- You, Q.; Yang, X.; Peng, Z.; Xu, L.; Wang, J. Development and applications of a high throughput genotyping tool for polyploid crops: Single nucleotide polymorphism (SNP) Array. Front. Plant Sci. 2018, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass Genomic Diversity, Ploidy, and Evolution: Novel Insights from a Network-Based SNP Discovery Protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinker, N.A.; Kilian, A.; Wight, C.P.; Heller-Uszynska, K.; Wenzl, P.; Rines, H.W.; Bjørnstad, A.; Howarth, C.J.; Jannink, J.-L.; Anderson, J.M.; et al. New DArT markers for oat provide enhanced map coverage and global germplasm characterization. BMC Genom. 2009, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Korir, P.C.; Zhang, J.; Wu, K.; Zhao, T.; Gai, J. Association mapping combined with linkage analysis for aluminum tolerance among soybean cultivars released in Yellow and Changjiang River Valleys in China. Theor. Appl. Genet. 2013, 126, 1659–1675. [Google Scholar] [CrossRef]
- Bayer, M.M.; Rapazote-Flores, P.; Ganal, M.; Hedley, P.; Macaulay, M.; Plieske, J.; Ramsay, L.; Russell, J.; Shaw, P.D.; Thomas, W.; et al. Development and evaluation of a barley 50k iSelect SNP array. Front. Plant Sci. 2017, 8, 1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekele, W.A.; Wight, C.P.; Chao, S.; Howarth, C.; Tinker, N.A. Haplotype based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 2018, 16, 1452–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, D.; Bundock, P.; Henry, R. Genotyping by allele-specific PCR. In Plant Genotyping II: SNP Technology; Henry, R., Ed.; CABI: Oxfordshire, UK, 2008; pp. 88–97. [Google Scholar]
- Chen, H.; Xie, W.; He, H.; Yu, H.; Chen, W.; Li, J.; Yu, R.; Yao, Y.; Zhang, W.; He, Y.; et al. A high-density SNP genotyping array for rice biology and molecular breeding. Mol. Plant 2014, 7, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Felcher, K.J.; Coombs, J.J.; Massa, A.N.; Hansey, C.N.; Hamilton, J.P.; Veilleux, R.E.; Buell, C.R.; Douches, D.S. Integration of two diploid potato linkage maps with the potato genome sequence. PLoS ONE 2012, 7, e36347. [Google Scholar] [CrossRef] [Green Version]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A.; Clarke, J.D.; Graner, E.-M.; Hansen, M.; Joets, J.; et al. A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 2011, 6, e28334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, W.E.; Higgins, E.E.; Plieske, J.; Wieseke, R.; Sidebottom, C.; Khedikar, Y.; Batley, J.; Edwards, D.; Meng, J.; Li, R.; et al. A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor. Appl. Genet. 2016, 129, 1887–1899. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Xie, W.; Li, J.; Zhou, F.; Zhang, Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol. J. 2013, 12, 28–37. [Google Scholar] [CrossRef]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP Array for Cotton and High-Density Mapping of Intraspecific and Interspecific Populations of Gossypium spp. Genes|Genomes|Genet. 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [Green Version]
- Unterseer, S.; Bauer, E.; Haberer, G.; Seidel, M.; Knaak, C.; Ouzunova, M.; Meitinger, T.; Strom, T.M.; Fries, R.; Pausch, H.; et al. A powerful tool for genome analysis in maize: Development and evaluation of the high density 600 k SNP genotyping array. BMC Genom. 2014, 15, 823. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Cortés, A.J.; Blair, M.W. Genotyping by Sequencing and Genome—Environment Associations in Wild Common Bean Predict Widespread Divergent Adaptation to Drought. Front. Plant Sci. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, M.W.; Cortes, A.J.; Farmer, A.D.; Huang, W.; Ambachew, D.; Penmetsa, R.V.; Carrasquilla-Garcia, N.; Assefa, T.; Cannon, S.B. Uneven recombination rate and linkage disequilibrium across a reference SNP map for common bean (Phaseolus vulgaris L.). PLoS ONE 2018, 13, e0189597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, L.; Cestaro, A.; Linsmith, G.; Muranty, H.; Denance, C.; Théron, A.; Troggio, M. Development and validation of the Axiom Apple 480k SNP genotyping array. Plant J. 2016, 86, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, K.; Farmer, A.; Berkman, P.; Muller, C.; Wei, X.; Demano, E.; Jackson, P.; Magwire, M.; Dietrich, B.; Kota, R. Generation of a 345K sugarcane SNP chip. Proc. Int. Soc. Sugar Cane Technol. 2016, 29, 1165–1172. Available online: http://www.issct.org/pdf/proceedings/2016/Molecular-Biology-papers/document-4.pdf (accessed on 12 July 2022).
- Bassil, N.V.; Davis, T.M.; Zhang, H.; Ficklin, S.; Mittmann, M.; Webster, T.; Mahoney, L.L.; Wood, D.; Alperin, E.S.; Rosyara, U.R.; et al. Development and preliminary evaluation of a 90K Axiom SNP array for the allo-octoploid cultivated strawberry Fragaria x ananassa. BCM Genom. 2015, 16, 155. Available online: https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-1310-1 (accessed on 12 July 2022). [CrossRef] [Green Version]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Agarwal, G.; Bertioli, D.J.; Leal-Bertioli, S.C.M.; Pandey, M.K.; Vaughn, J.; Abernathy, B.; Barkley, N.A.; et al. Genome-wide SNP Genotyping Resolves Signatures of Selection and Tetrasomic Recombination in Peanut. Mol. Plant 2017, 10, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Agarwal, G.; Kale, S.M.; Clevenger, J.; Nayak, S.N.; Sriswathi, M.; Chitikineni, A.; Chavarro, C.; Chen, X.; Upadhyaya, H.D.; et al. Development and evaluation of a high-density genotyping “axiom_arachis” array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci. Rep. 2017, 7, 40577. [Google Scholar] [CrossRef]
- Oliver, R.E.; Tinker, N.A.; Lazo, G.R.; Chao, S.; Jellen, E.N.; Carson, M.L.; Rines, H.W.; Obert, D.E.; Lutz, J.D.; Shackelford, I.; et al. SNP discovery and chromosome anchoring provide the first physically anchored hexaploid oat map and reveal synteny with model species. PLoS ONE 2013, 8, e58068. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-base estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 1999, 152, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Watterson, G.A. Number of segregating sites in genetic models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, R.M.; Ramdoss, Y.; Buckler, E.S. TASSELL Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1461589 (accessed on 12 July 2022). [CrossRef]
- Tumino, G.; Voorrips, R.E.; Rizza, F.; Badeck, F.W.; Morcia, C.; Ghizzoni, R.; Germeier, C.U.; Paulo, M.-J.; Terzi, V.; Smulders, M.J.M. Population structure and genome-wide association analysis for frost tolerance in oat using continuous SNP array signal intensity ratios. Theor. Appl. Genet. 2016, 129, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Maughan, P.J.; Smith, S.M.; Rojas-Beltran Elzinga, D.; Raney, J.A.; Jellen, E.N.; Fairbanks, J.D. Single nucleotide polymorphisms identification, characterization, and linkage mapping in Chenopodium quinua. Plant Genome 2012, 5, 1–7. [Google Scholar] [CrossRef]
- Winfield, M.O.; Allen, A.M.; Burridge, A.J.; Barker, G.L.A.; Benbow, H.R.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; Scopes, G.; et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 2015, 14, 1195–1206. [Google Scholar] [CrossRef]
- Saxena, R.K.; Varma Penmetsa, R.; Upadhyaya, H.D.; Kumar, A.; Carrasquilla-Garcia, N.; Schlueter, J.A.; Farmer, A.; Whaley, A.M.; Sarma, B.; May, G.D.; et al. Large-scale development of cost-effective single-nucleotide polymorphism marker assays for genetic mapping in Pigeonpea and comparative mapping in legumes. DNA Res. 2012, 19, 449–461. [Google Scholar] [CrossRef]
- Tinker, N.A.; Bekele, W.A.; Hattori, J. Haplotag: Software for Haplotype-Based Genotyping-by-Sequencing Analysis. Genes|Genomes|Genet. 2016, 6, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.J.; López-Hernández, F. Harnessing Crop Wild Diversity for Climate Change Adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campuzano-Duque, L.F.; Bejarano-Garavito, D.; Castillo-Sierra, J.; Torres-Cuesta, D.R.; Cortés, A.J.; Blair, M.W. SNP Genotyping for Purity Assessment of a Forage Oat (Avena sativa L.) Variety from Colombia. Agronomy 2022, 12, 1710. https://doi.org/10.3390/agronomy12071710
Campuzano-Duque LF, Bejarano-Garavito D, Castillo-Sierra J, Torres-Cuesta DR, Cortés AJ, Blair MW. SNP Genotyping for Purity Assessment of a Forage Oat (Avena sativa L.) Variety from Colombia. Agronomy. 2022; 12(7):1710. https://doi.org/10.3390/agronomy12071710
Chicago/Turabian StyleCampuzano-Duque, Luis Fernando, Diego Bejarano-Garavito, Javier Castillo-Sierra, Daniel Ricardo Torres-Cuesta, Andrés J. Cortés, and Matthew Wohlgemuth Blair. 2022. "SNP Genotyping for Purity Assessment of a Forage Oat (Avena sativa L.) Variety from Colombia" Agronomy 12, no. 7: 1710. https://doi.org/10.3390/agronomy12071710
APA StyleCampuzano-Duque, L. F., Bejarano-Garavito, D., Castillo-Sierra, J., Torres-Cuesta, D. R., Cortés, A. J., & Blair, M. W. (2022). SNP Genotyping for Purity Assessment of a Forage Oat (Avena sativa L.) Variety from Colombia. Agronomy, 12(7), 1710. https://doi.org/10.3390/agronomy12071710