
Searching of Novel Herbicides for Paddy Field Weed Management—A Case Study with Acetyl-CoA Carboxylase


Abstract
1. Introduction
2. Materials and Methods
2.1. Homology Modeling and Protein Preparation
2.2. Ligand Collection and Preparation
2.3. Tanimoto Coefficient Similarity
2.4. Binding Site Analysis and Molecular Docking
2.5. MM/GBSA Screening
2.6. Herbicide-Likeness
2.7. MD Simulation
2.8. MM/PBSA
3. Results and Discussion
3.1. Homology Validation
3.2. Tanimoto-Similarity-Based Screening
3.3. Binding Site Prediction and Molecular Docking
3.4. MM/GBSA Screening Analysis
3.5. Herbicide-Likeness
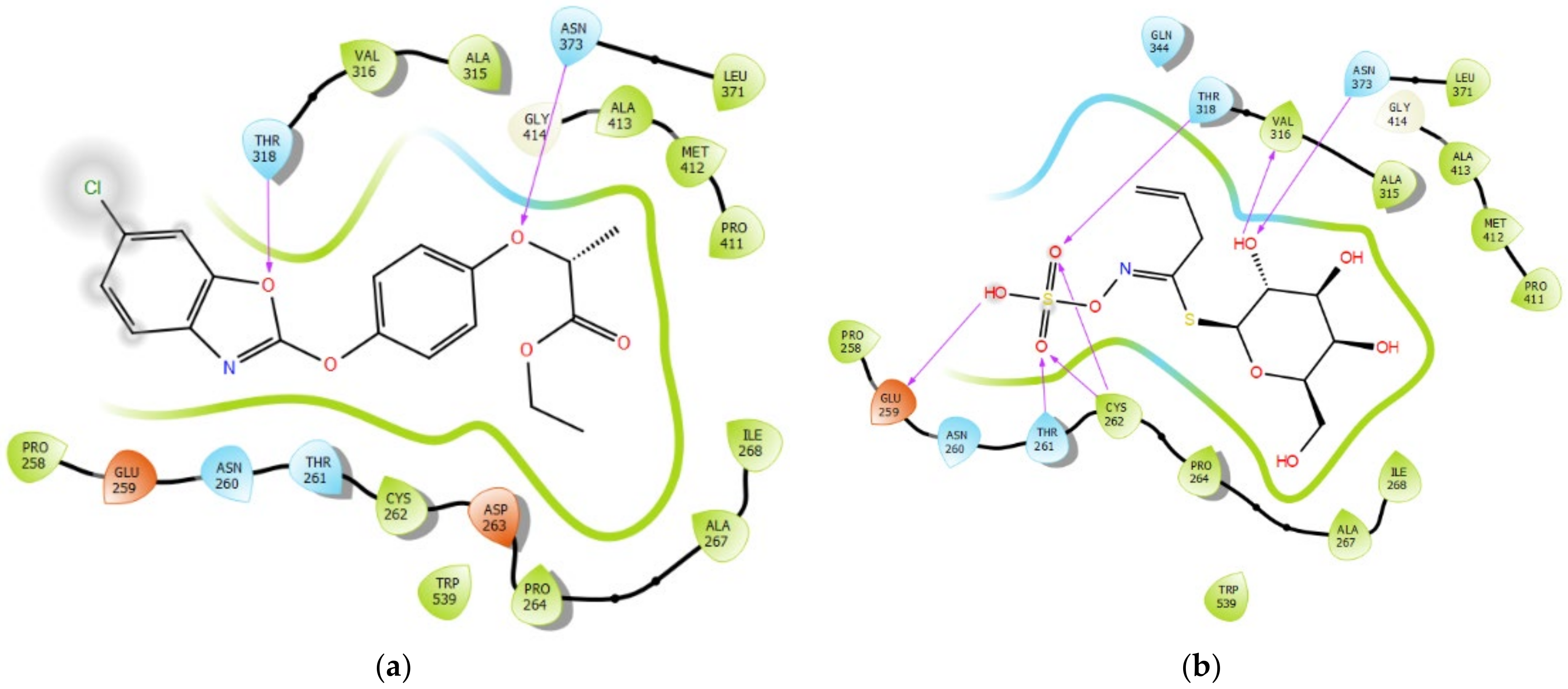
3.6. Interaction Profile of FPPE and Sinigrin with ACCase
3.7. MD Simulation Analysis
3.7.1. Structural Stability of Protein–Ligand Complexes
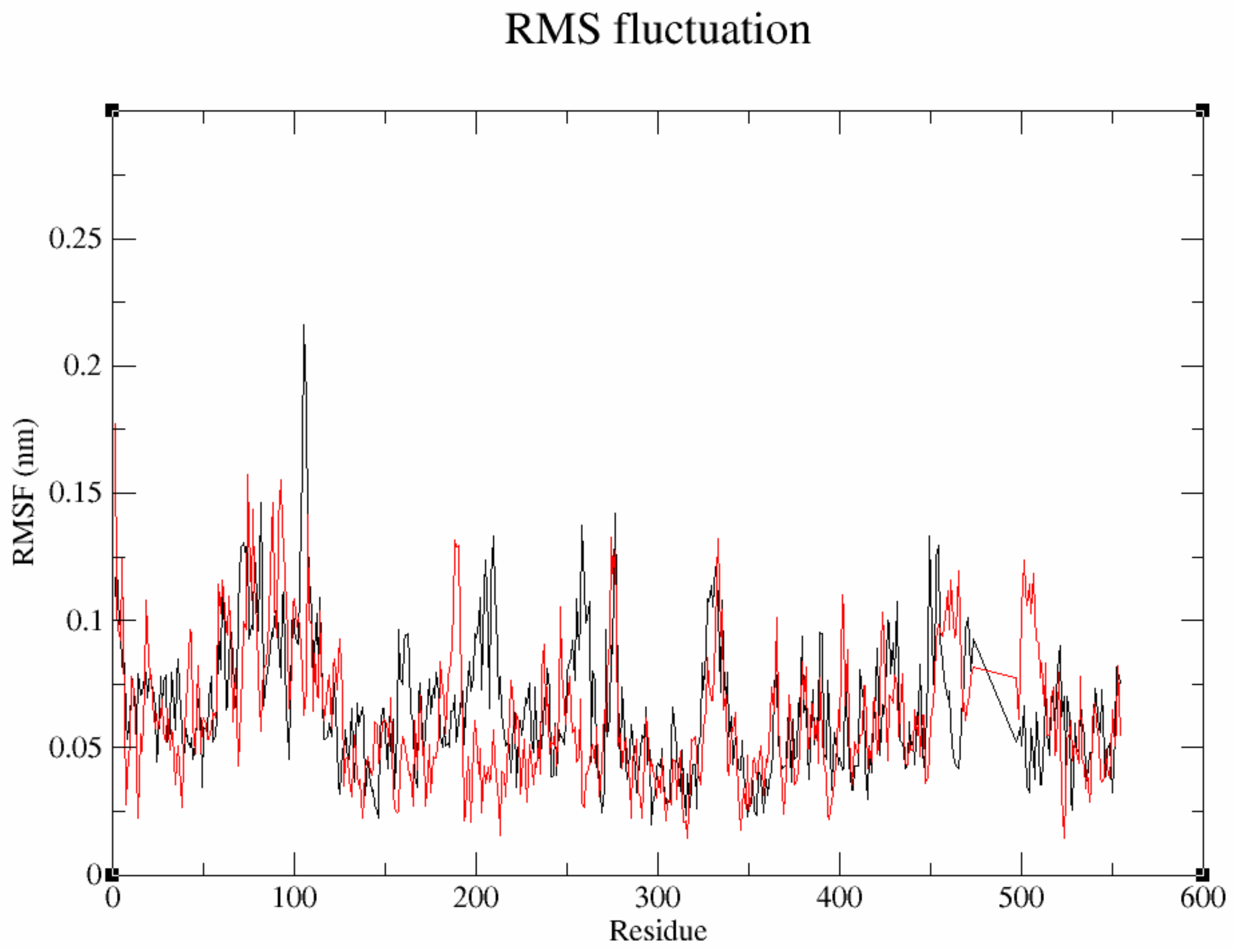
3.7.2. Flexibility Analysis of Protein–Ligand Complexes
3.7.3. Hydrogen Bond Occupancy
3.7.4. Essential Dynamics
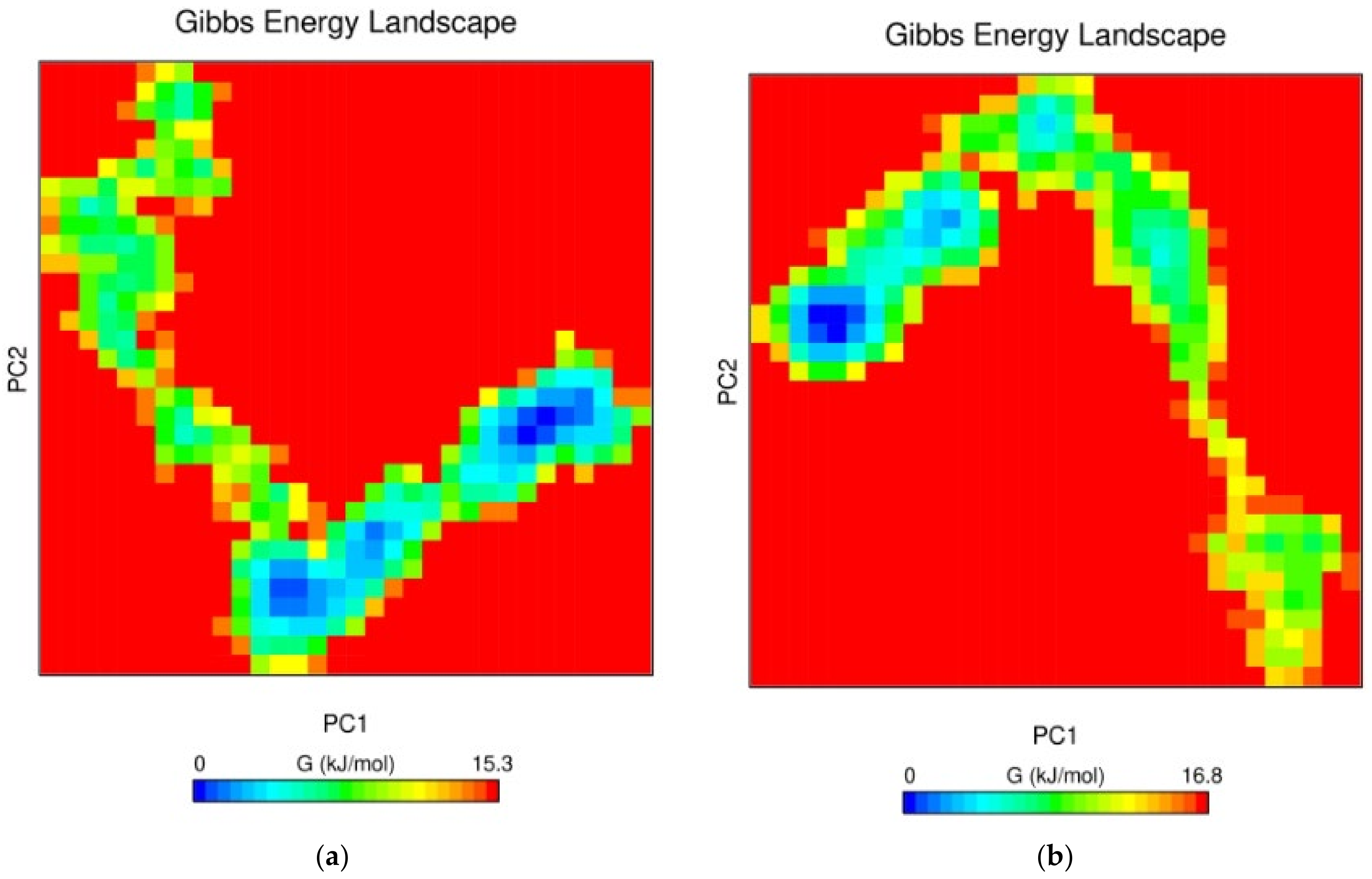
3.7.5. Free Energy Landscape (FEL) Analysis
3.8. MM/PBSA Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anami, B.S.; Malvade, N.N.; Palaiah, S. Classification of yield affecting biotic and abiotic paddy crop stresses using field images. Inf. Process. Agric. 2020, 7, 272–285. [Google Scholar] [CrossRef]
- Fahad, S.; Adnan, M.; Noor, M.; Arif, M.; Alam, M.; Khan, I.A.; Ullah, H.; Wahid, F.; Mian, I.A.; Jamal, Y.; et al. Major constraints for global rice production. In Advances in Rice Research for Abiotic Stress Tolerance; Woodhead Publishing: Sawston, UK, 2019; pp. 1–22. [Google Scholar] [CrossRef]
- Biswas, B.; Timsina, J.; Garai, S.; Mondal, M.; Banerjee, H.; Adhikary, S.; Kanthal, S. Weed control in trans-planted rice with post-emergence herbicides and their effects on subsequent rapeseed in Eastern India. Int. J. Pest Manag. 2020, 64, 1–13. [Google Scholar] [CrossRef]
- Gharde, Y.; Singh, P.K.; Dubey, R.P.; Gupta, P.K. Assessment of yield and economic losses in agriculture due to weeds in India. Crop Prot. 2018, 107, 12–18. [Google Scholar] [CrossRef]
- Golmohammadi, M.J.; Chamanabad, H.R.M.; Yaghoubi, B.; Oveisi, M. Study of postharvest weed population in paddy fields. Sarhad J. Agric. 2017, 34, 395–399. [Google Scholar] [CrossRef]
- Sardana, V.; Mahajan, G.; Jabran, K.; Chauhan, B.S. Role of competition in managing weeds: An introduction to the special issue. Crop Prot. 2017, 95, 1–7. [Google Scholar] [CrossRef]
- Weerarathne, L.V.Y.; Suriyagoda, L.D.; Marambe, B. Barnyard grass (Echinochloa crus-galli (L.) P. Beauv) is less competitive on rice (Oryza sativa L.) when phosphorus (P) is applied to deeper layers in P-deficient and moisture-limited soils. Plant Soil 2015, 391, 1–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Cen, H.; Lu, Y.; Yu, X.; He, Y.; Pieters, J.G. Automated spectral feature extraction from hyperspectral images to differentiate weedy rice and barnyard grass from a rice crop. Comput. Electron. Agric. 2019, 159, 42–49. [Google Scholar] [CrossRef]
- Amaro-Blanco, I.; Romano, Y.; Palmerin, J.A.; Gordo, R.; Palma-Bautista, C.; De Prado, R.; Osuna, M.D. Different Mutations Providing Target Site Resistance to ALS and ACCase-Inhibiting Herbicides in Echinochloa spp. from Rice Fields. Agriculture 2021, 11, 382. [Google Scholar] [CrossRef]
- Wang, X.L.; Zhang, Z.Y.; Xu, X.M.; Li, G. The density of barnyard grass affects photosynthesis and physio-logical characteristics of rice. Photosynthetica 2019, 57, 705–711. [Google Scholar] [CrossRef]
- Shekhawat, K.; Rathore, S.S.; Chauhan, B.S. Weed management in dry direct-seeded rice: A review on challenges and opportunities for sustainable rice production. Agronomy 2020, 10, 1264. [Google Scholar] [CrossRef]
- Hu, L.; Huang, Y.; Ding, B.; Cai, R.; Bai, L. Selective Action Mechanism of Fenclorim on Rice and Echinochloa crus-galli Is Associated with the Inducibility of Detoxifying Enzyme Activities and Antioxidative Defense. J. Agric. Food Chem. 2021, 69, 5830–5839. [Google Scholar] [CrossRef]
- Wenger, J.; Niderman, T.; Mathews, C.; Wailes, S. Acetyl-CoA carboxylase inhibitors. In Modern Crop Protection Compounds; Wiley-VCH: Hoboken, NJ, USA, 2019; Volume 3, pp. 501–528. [Google Scholar] [CrossRef]
- Fang, J.; He, Z.; Liu, T.; Li, J.; Dong, L. A novel mutation Asp-2078-Glu in ACCase confers resistance to ACCase herbicides in barnyardgrass (Echinochloa crus-galli). Pestic. Biochem. Physiol. 2020, 168, 104634. [Google Scholar] [CrossRef]
- Xia, X.; Tang, W.; He, S.; Kang, J.; Ma, H.; Li, J. Mechanism of metamifop inhibition of the carboxyltransferase domain of acetyl-coenzyme A carboxylase in Echinochloa crus-galli. Sci. Rep. 2016, 6, 34066. [Google Scholar] [CrossRef]
- Takano, H.K.; Ovejero, R.F.L.; Belchior, G.G.; Maymone, G.P.L.; Dayan, F.E. ACCase-inhibiting herbicides: Mechanism of action, resistance evolution and stewardship. Sci. Agric. 2020, 78, e20190102. [Google Scholar] [CrossRef]
- Sabet Zangeneh, H.; Mohammaddust Chamanabad, H.R.; Zand, E.; Asghari, A.; Alamisaeid, K.; Travlos, I.S.; Ale-brahim, M.T. Study of fitness cost in three rigid ryegrass populations susceptible and resistant to acetyl-CoA carboxylase inhibiting herbicides. Front. Ecol. Evol. 2016, 4, 142. [Google Scholar] [CrossRef]
- Chen, Z.J.; Qiao, Y.X.; Zhang, N.; Liu, J.; Yang, H. Insight into metabolism pathways of pesticide fomesafen in rice: Reducing cropping and environmental risks. Environ. Pollut. 2021, 283, 117128. [Google Scholar] [CrossRef]
- Bagavathiannan, M.V.; Norsworthy, J.K.; Smith, K.L.; Neve, P. Modeling the simultaneous evolution of resistance to ALS-and ACCase-inhibiting herbicides in barnyardgrass (Echinochloa crus-galli) in Clearfield® rice. Weed Technol. 2014, 28, 89–103. [Google Scholar] [CrossRef]
- Ye, F.; Ma, P.; Zhang, Y.Y.; Li, P.; Yang, F.; Fu, Y. Herbicidal activity and molecular docking study of novel ACCase inhibitors. Front. Plant Sci. 2018, 9, 1850. [Google Scholar] [CrossRef]
- Tandon, S. Dissipation dynamics of fenoxaprop-p-ethyl and fenoxaprop acid under Indian rice field conditions. Int. J. Environ. Anal. Chem. 2017, 97, 1352–1361. [Google Scholar] [CrossRef]
- Dash, S.; Duary, B.; Sar, K. Efficacy of fenoxaprop-p-ethyl and penoxsulam for weed management with special emphasis on Echinochloa spp. in transplanted summer rice. Indian J. Weed Sci. 2021, 53, 78–80. [Google Scholar] [CrossRef]
- Dubovik, V.; Dalinova, A.; Berestetskiy, A. Effect of adjuvants on herbicidal activity and selectivity of three phytotoxins produced by the fungus, Stagonospora Cirsii. Plants 2020, 9, 1621. [Google Scholar] [CrossRef]
- Shen, C.; Tang, W.; Zeng, D.; Xu, H.; Su, W.; Wu, R. Isoxadifen-ethyl derivatives protect rice from fenoxaprop-p-ethyl–associated injury during the control of weedy rice. Weed Sci. 2017, 65, 579–587. [Google Scholar] [CrossRef]
- Wu, J.X.; Zhang, Y.; Wang, K.; Zhang, H.Y. Residue analysis and dissipation of fenoxaprop-P-ethyl and its metabolite fenoxaprop-P in rice ecosystem. J. Anal. Chem. 2015, 70, 897–902. [Google Scholar] [CrossRef]
- Wu, X.; Huang, T. Recent development in acetyl-CoA carboxylase inhibitors and their potential as novel drugs. Future Med. Chem. 2019, 12, 533–561. [Google Scholar] [CrossRef]
- Dayan, F.E.; Duke, S.O. Natural compounds as next-generation herbicides. Plant Physiol. 2014, 166, 1090–1105. [Google Scholar] [CrossRef]
- Wang, L.; Wang, M.; Fu, Y.; Huang, P.; Kong, D.; Niu, G. Engineered biosynthesis of thaxtomin phytotoxins. Crit. Rev. Biotechnol. 2020, 40, 1163–1171. [Google Scholar] [CrossRef]
- Bordin, E.R.; Frumi Camargo, A.; Stefanski, F.S.; Scapini, T.; Bonatto, C.; Zanivan, J.; Preczeski, K.; Modkovski, T.A.; Reichert Junior, F.; Mossi, A.J.; et al. Current production of bioherbicides: Mechanisms of action and technical and scientific challenges to improve food and environmental security. Biocatal. Biotransform. 2021, 39, 346–359. [Google Scholar] [CrossRef]
- Chen, H.; Singh, H.; Bhardwaj, N.; Bhardwaj, S.K.; Khatri, M.; Kim, K.H.; Peng, W. An exploration on the toxicity mechanisms of phytotoxins and their potential utilities. Crit. Rev. Biotechnol. 2022, 52, 395–435. [Google Scholar] [CrossRef]
- Dass, A.; Shekhawat, K.; Choudhary, A.K.; Sepat, S.; Rathore, S.S.; Mahajan, G.; Chauhan, B.S. Weed management in rice using crop competition-a review. Crop Prot. 2017, 95, 45–52. [Google Scholar] [CrossRef]
- Jain, C.K.; Gupta, M.; Prasad, Y.; Wadhwa, G.; Sharma, S.K. Homology modelling and molecular dynamics simulations of a protein serine/threonine phosphatase stp1 in Staphylococcus aureus N315: A potential drug target. Mol. Simul. 2015, 41, 592–599. [Google Scholar] [CrossRef]
- Sobolev, O.V.; Afonine, P.V.; Moriarty, N.W.; Hekkelman, M.L.; Joosten, R.P.; Perrakis, A.; Adams, P.D. A global Ramachandran score identifies protein structures with unlikely stereochemistry. Structure 2020, 28, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Saxena, S.; Guruprasad, L. Homology modeling, docking and structure-based virtual screening for new inhibitor identification of Klebsiella pneumoniae heptosyltransferase-III. J. Biomol. Struct. 2019, 38, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Idris, M.O.; Yekeen, A.A.; Alakanse, O.S.; Durojaye, O.A. Computer-aided screening for potential TMPRSS2 inhibitors: A combination of pharmacophore modeling, molecular docking and molecular dynamics simulation approaches. J. Biomol. Struct. 2021, 39, 5638–5656. [Google Scholar] [CrossRef] [PubMed]
- Bathula, R.; Lanka, G.; Muddagoni, N.; Dasari, M.; Nakkala, S.; Bhargavi, M.; Somadi, G.; Sivan, S.K.; Potlapally, S.R. Identification of potential Aurora kinase-C protein inhibitors: An amalgamation of energy minimization, virtual screening, prime MMGBSA and AutoDock. J. Biomol. Struct. 2019, 38, 2314–2325. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Muddagoni, N.; Bathula, R.; Dasari, M.; Potlapally, S.R. Homology Modeling, Virtual Screening, Prime-MMGBSA, AutoDock-Identification of Inhibitors of FGR Protein. Biointerface Res. Appl. Chem. 2020, 11, 11088–11103. [Google Scholar] [CrossRef]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Abhirami, R.; Boura, E.; Singh, S.K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral in-hibitors against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 4582–4593. [Google Scholar] [CrossRef]
- Hemmati, S.A.; Karam Kiani, N.; Serrão, J.E.; Jitonnom, J. Inhibitory potential of a designed peptide inhibitor based on zymogen structure of trypsin from Spodoptera frugiperda: In silico insights. Int. J. Pept. Res. Ther. 2021, 27, 1677–1687. [Google Scholar] [CrossRef]
- Gunthardt, B.F.; Hollender, J.; Hungerbuhler, K.; Scheringer, M.; Bucheli, T.D. Comprehensive toxic plants–phytotoxins database and its application in assessing aquatic micropollution potential. J. Agric. Food Chem. 2018, 66, 7577–7588. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of open natural products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 36. [Google Scholar] [CrossRef]
- Bangarwa, S.K.; Norsworthy, J.K. Glucosinolate and Isothiocyanate Production for Weed Control in Plasticulture Production System. In Glucosinolates; Mérillon, J.M., Ramawat, K., Eds.; Reference Series in Phytochemistry; Springer: Cham, Switzerland, 2016; pp. 1–35. [Google Scholar] [CrossRef]
- Mellor, C.L.; Robinson, R.M.; Benigni, R.; Ebbrell, D.; Enoch, S.J.; Firman, J.W.; Madden, J.C.; Pawar, G.; Yang, C.; Cronin, M.T.D. Molecular fingerprint-derived similarity measures for toxicological read-across: Recommendations for optimal use. Regul. Toxicol. Pharmacol. 2019, 101, 121–134. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef]
- Bragina, M.E.; Daina, A.; Perez, M.A.; Michielin, O.; Zoete, V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics, GitHub. 2019. Available online: https://github.com/rdkit/rdkit (accessed on 28 January 2022).
- Gudipati, S.; Muttineni, R.; Mankad, A.U.; Pandya, H.A.; Jasrai, Y.T. Molecular docking-based screening of Noggin inhibitors. Bioinformation 2018, 14, 15. [Google Scholar] [CrossRef]
- Srinivasan, P.; Vijayakumar, S.; Kothandaraman, S.; Palani, M. Anti-diabetic activity of quercetin extracted from Phyllanthus emblica L. fruit: In silico and in vivo approaches. J. Pharm. Anal. 2018, 8, 109–118. [Google Scholar] [CrossRef]
- Lanka, G.; Bathula, R.; Dasari, M.; Nakkala, S.; Bhargavi, M.; Somadi, G.; Potlapally, S.R. Structure-based identification of potential novel inhibitors targeting FAM3B (PANDER) causing type 2 diabetes mellitus through virtual screening. J. Recept. Signal Transduct. 2019, 39, 253–263. [Google Scholar] [CrossRef]
- Yadav, M.; Dhagat, S.; Eswari, J.S. Structure Based Drug Design and Molecular Docking Studies of Anticancer Molecules Paclitaxel, Etoposide and Topotecan using Novel Ligands. Curr. Drug Discov. Technol. 2020, 17, 183–190. [Google Scholar] [CrossRef]
- Poopandi, S.; Sundaraj, R.; Rajmichael, R.; Thangaraj, S.; Dhamodharan, P.; Biswal, J.; Malaisamy, V.; Pandian, C.J.; Jeyaraman, J. Computational screening of potential inhibitors targeting MurF of Brugia malayi Wolbachia through multi-scale molecular docking, molecular dynamics and MM-GBSA analysis. Mol. Biochem. Parasitol. 2021, 246, 111427. [Google Scholar] [CrossRef]
- Shahroz, M.M.; Sharma, H.K.; Altamimi, A.S.; Alamri, M.A.; Ali, A.; Ali, A.; Alqahtani, S.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; et al. Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study. Molecules 2022, 27, 1159. [Google Scholar] [CrossRef]
- Kellici, T.F.; Ntountaniotis, D.; Liapakis, G.; Tzakos, A.G.; Mavromoustakos, T. The dynamic properties of angiotensin II type 1 receptor inverse agonists in solution and in the receptor site. Arab. J. Chem. 2019, 12, 5062–5078. [Google Scholar] [CrossRef]
- Zhang, Y.; Lorsbach, B.A.; Castetter, S.; Lambert, W.T.; Kister, J.; Wang, N.X.; Klittich, C.J.; Roth, J.; Sparks, T.C.; Loso, M.R. Physicochemical property guidelines for modern agrochemicals. Pest Manag. Sci. 2018, 74, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Funar-Timofei, S.; Borota, A.; Chennamaneni, S.R.; Manchala, A.K.; Muresan, S. Quantitative estimation of pesticide-likeness for agrochemical discovery. J. Cheminform. 2014, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Isa, M.A.; Majumdar, R.S.; Haider, S. In silico identification of potential inhibitors against shikimate dehydrogenase through virtual screening and toxicity studies for the treatment of tuberculosis. Int. J. Microbiol. 2019, 22, 7–17. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open-Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.K.; Shin, W.H.; Karuppasamy, R. Exploring safe and potent bioactives for the treatment of non-small cell lung cancer. 3 Biotech 2021, 11, 241. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Beuming, T.; van Veen, C.; Massink, A.; Sherman, W.; van Vlijmen, H.W.; IJzerman, A.P. In search of novel ligands using a structure-based approach: A case study on the adenosine A2A receptor. J. Comput. Aided Mol. Des. 2016, 30, 863–874. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.Q. TargetHunter: An in-silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013, 15, 395–406. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef]
- Motmainna, M.; Juraimi, A.S.; Uddin, M.K.; Asib, N.B.; Islam, A.K.M.M.; Hasan, M. Assessment of allelopathic compounds to develop new natural herbicides: A review. Allelopath. J. 2021, 52, 21–40. [Google Scholar] [CrossRef]
- Mazumder, A.; Dwivedi, A.; Du Plessis, J. Sinigrin and its therapeutic benefits. Molecules 2016, 21, 416. [Google Scholar] [CrossRef]
- Manallack, D.T. The acid/base profile of agrochemicals. SAR QSAR Environ. Res. 2017, 28, 621–628. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Sneha, P.; George Priya Doss, C.; Siva, R.; Zayed, H. A profound computational study to pri-oritize the disease-causing mutations in PRPS1 gene. Metab. Brain Dis. 2018, 33, 589–600. [Google Scholar] [CrossRef]
- Ononamadu, C.J.; Abdalla, M.; Ihegboro, G.O.; Li, J.; Owolarafe, T.A.; John, T.D.; Tian, Q. In silico identification and study of potential anti-mosquito juvenile hormone binding protein (MJHBP) compounds as candidates for dengue virus-Vector insecticides. Biochem. Biophys. Rep. 2021, 28, 101178. [Google Scholar] [CrossRef]
- Prabhu, N.; Sharp, K. Protein−solvent interactions. Chem. Rev. 2006, 106, 1616–1623. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Nicolotti, O.; Miteva, M.A.; Villoutreix, B.O. Analysis of solvent-exposed and buried co-crystallized ligands: A case study to support the design of novel protein–protein interaction inhibitors. Drug Discov. Today 2019, 24, 551–559. [Google Scholar] [CrossRef]
- Shukla, R.; Shukla, H.; Kalita, P.; Sonkar, A.; Pandey, T.; Singh, D.B.; Tripathi, T. Identification of potential inhibitors of Fasciola gigantica thioredoxin1: Computational screening, molecular dynamics simulation, and binding free energy studies. J. Biomol. Struct. Dyn. 2018, 36, 2147–2162. [Google Scholar] [CrossRef]
- Rampogu, S.; Lemuel, M.R.; Lee, K.W. Virtual screening, molecular docking, molecular dynamics simulations and free energy calculations to discover potential DDX3 inhibitors. Adv. Cancer Biol. Metastasis 2022, 4, 100022. [Google Scholar] [CrossRef]
- Li, T.; Pang, W.; Wang, J.; Zhao, Z.; Cheng, L. Docking-based 3D-QSAR, molecular dynamics simulation studies and virtual screening of novel ONC201 analogues targeting Mitochondrial ClpP. J. Mol. Struct. 2021, 1245, 131025. [Google Scholar] [CrossRef]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef]
- Kumar, A.; Purohit, R. Computational screening and molecular dynamics simulation of disease associated nsSNPs in CENP-E. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2012, 738, 28–37. [Google Scholar] [CrossRef]
- Joshi, T.; Joshi, T.; Pundir, H.; Sharma, P.; Mathpal, S.; Chandra, S. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J. Biomol. Struct. 2021, 39, 6728–6746. [Google Scholar] [CrossRef]
- Pitaloka, D.A.E.; Ramadhan, D.S.F.; Chaidir, L.; Fakih, T.M. Docking-based virtual screening and molecular dynamics simulations of quercetin analogs as enoyl-acyl carrier protein reductase (InhA) inhibitors of Mycobacterium tuberculosis. Sci. Pharm. 2021, 89, 20. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Doss, G.P.C.; Siva, R.; Magesh, R.; Zayed, H. Molecular insights of the G2019S substitution in LRRK2 kinase domain associated with Parkinson’s disease: A molecular dynamics simulation approach. J. Theor. Biol. 2019, 469, 163–171. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, G.; Srivastava, S.; Verma, S.; Negi, A.S.; Sharma, A. Investigation of naphthofuran moiety as potential dual inhibitor against BACE-1 and GSK-3β: Molecular dynamics simulations, binding energy, and network analysis to identify first-in-class dual inhibitors against Alzheimer’s disease. J. Mol. Model. 2017, 23, 239. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound ID | Name | Tanimoto Coefficient | XP GScore (kcal/mol) | ∆Gbind (kcal/mol) | ∆Gbind Coulomb (kcal/mol) | ∆Gbind Covalent (kcal/mol) | ∆Gbind H-bond (kcal/mol) | ∆Gbind Lipophilicity (kcal/mol) | ∆Gbind van der Waals (vdW) (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 91707 | Fenoxaprop-P-ethyl | 1 | −5.977 | −77.03 | −5.13 | 6.07 | −0.64 | −42.32 | −44.46 |
| 2 | CNP0166854 | Ligustaloside A | 0.29 | −10.974 | −83.78 | −37.71 | 1.96 | −4.36 | −35.05 | −50.49 |
| 3 | CNP0293269 | Glucoiberverin | 0.22 | −9.093 | −83.51 | 3.30 | 5.38 | −1.47 | −42.56 | −35.90 |
| 4 | CNP0288601 | Isoacteoside | 0.27 | −12.829 | −81.18 | −53.07 | 9.29 | −3.84 | −37.25 | −53.40 |
| 5 | CNP0233323 | Ophiohayatone B | 0.32 | −7.689 | −81.05 | −34.39 | 6.72 | −2.57 | −41.46 | −41.75 |
| 6 | CNP0232558 | Iridin | 0.33 | −10.393 | −79.61 | −31.46 | 11.66 | −3.52 | −42.17 | −49.75 |
| 7 | CNP0259095 | Sinigrin | 0.22 | −9.068 | −79.21 | 6.98 | 3.65 | −1.56 | −39.38 | −34.54 |
| 8 | CNP0332060 | Glucolepidiin | 0.21 | −10.593 | −77.29 | −9.08 | 8.06 | −1.49 | −37.78 | −27.22 |
| S. No. | Compound ID | MW (150–500) * Da | LogP (≤3.5) * | HBD (≤3) * | HBA (2–12) * | RB (<12) * |
|---|---|---|---|---|---|---|
| 1 | 91707 | 361.8 | 4.9 | 0 | 6 | 7 |
| 2 | CNP0166854 | 556.51 | −0.74 | 2 | 7 | 13 |
| 3 | CNP0293269 | 407.48 | −3.48 | 5 | 12 | 9 |
| 4 | CNP0288601 | 624.58 | −0.23 | 9 | 15 | 11 |
| 5 | CNP0233323 | 564.49 | −0.93 | 8 | 14 | 6 |
| 6 | CNP0232558 | 522.45 | 0.07 | 6 | 13 | 7 |
| 7 | CNP0259095 | 358.36 | −4.52 | 4 | 11 | 6 |
| 8 | CNP0332060 | 347.36 | −3.67 | 5 | 11 | 6 |
| S. No. | Binding Residues | Fenoxaprop-P-Ethyl (Å) | Sinigrin (Å) |
|---|---|---|---|
| 1 | THR 318 | 1.93 | 2.37 |
| 2 | ASN 373 | 2.03 | 2.00 |
| 3 | CYS262 | NI | 2.61 |
| 2.36 | |||
| 4 | VAL 316 | NI | 1.85 |
| 5 | GLU 259 | NI | 1.78 |
| 6 | THR 261 | NI | 2.54 |
| S. No. | Energy | Sinigrin (kJ/mol) | Fenoxaprop−P−Ethyl (kJ/mol) |
|---|---|---|---|
| 1 | Van der Waal energy | −105.790 ± 60.492 | −102.784 ± 57.997 |
| 2 | Electrostatic energy | −4.901 ±5.543 | −5.353 ±8.379 |
| 3 | Polar solvation energy | 46.698 ± 47.823 | 34.996 ± 28.300 |
| 4 | SAV energy | −81.638 ± 64.592 | −56.249 ± 81.862 |
| 5 | Binding energy | −145.631 ± 73.851 | −129.390 ± 111.326 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antony, A.; Karuppasamy, R. Searching of Novel Herbicides for Paddy Field Weed Management—A Case Study with Acetyl-CoA Carboxylase. Agronomy 2022, 12, 1635. https://doi.org/10.3390/agronomy12071635
Antony A, Karuppasamy R. Searching of Novel Herbicides for Paddy Field Weed Management—A Case Study with Acetyl-CoA Carboxylase. Agronomy. 2022; 12(7):1635. https://doi.org/10.3390/agronomy12071635
Chicago/Turabian StyleAntony, Ajitha, and Ramanathan Karuppasamy. 2022. "Searching of Novel Herbicides for Paddy Field Weed Management—A Case Study with Acetyl-CoA Carboxylase" Agronomy 12, no. 7: 1635. https://doi.org/10.3390/agronomy12071635
APA StyleAntony, A., & Karuppasamy, R. (2022). Searching of Novel Herbicides for Paddy Field Weed Management—A Case Study with Acetyl-CoA Carboxylase. Agronomy, 12(7), 1635. https://doi.org/10.3390/agronomy12071635