
Genome-Wide Comparative Analysis of Transposable Elements by Matrix-TE Method Revealed Indica and Japonica Rice Evolution



Abstract
:1. Introduction
2. Materials and Methods
2.1. Indica and Japonica Genomes Used for TE Matrix Generation and GPDF Analysis
2.2. LTR/TE ORF Matrix Generation
2.3. Phylogenetic Analysis of TE ORFs
2.4. Whole Genome and Centromere Scanning with TE ORF Reference Sequences and GPDF Analysis of Individual TE Peaks
2.5. TE Insertion Events, Whole Genome, and Centromere Evolution Analysis
3. Results
3.1. Development of Matrix-TE Approach Pipeline
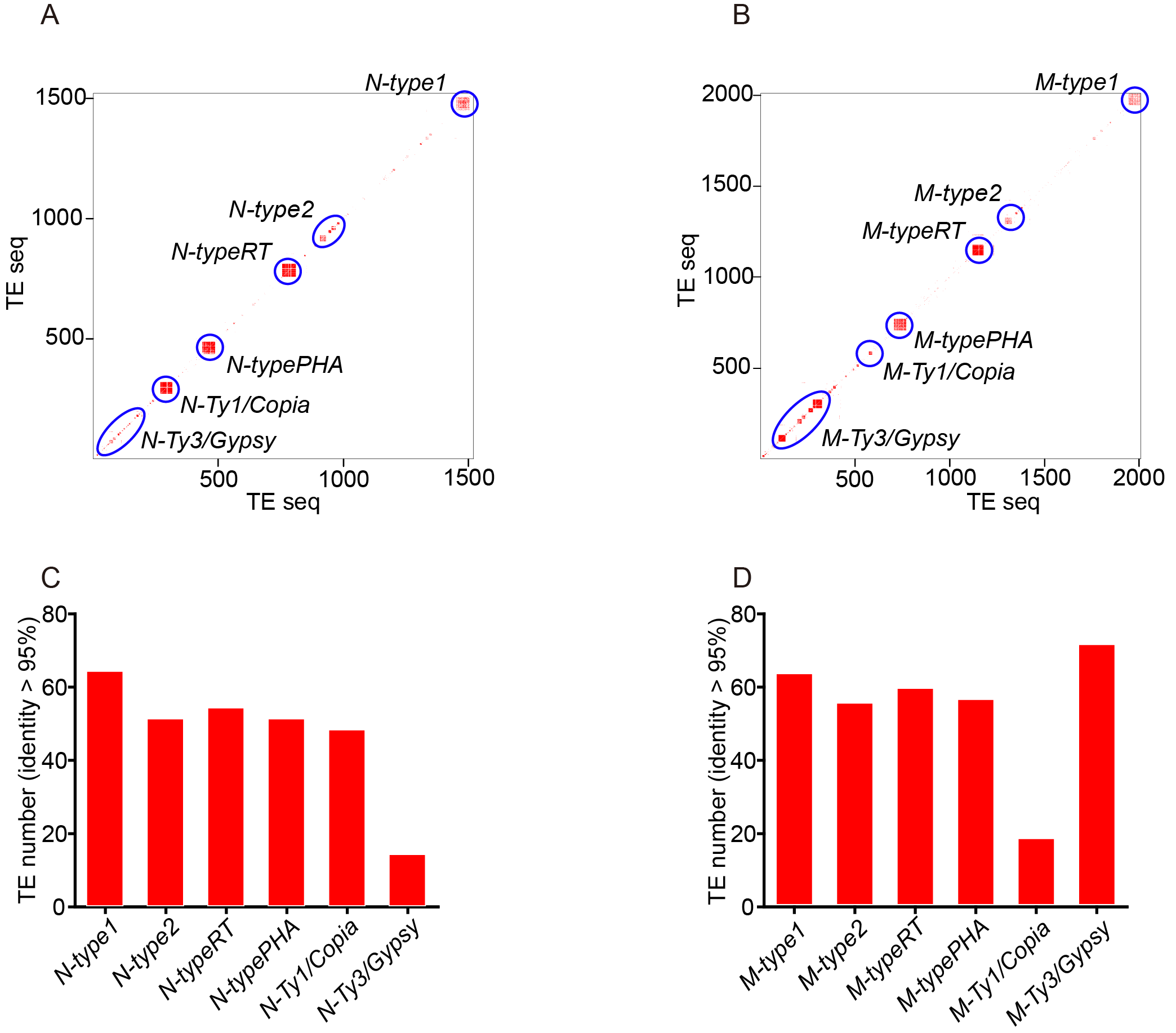
3.2. TE Matrix and Cluster Generation for Indica and Japonica Rice Whole Genomes
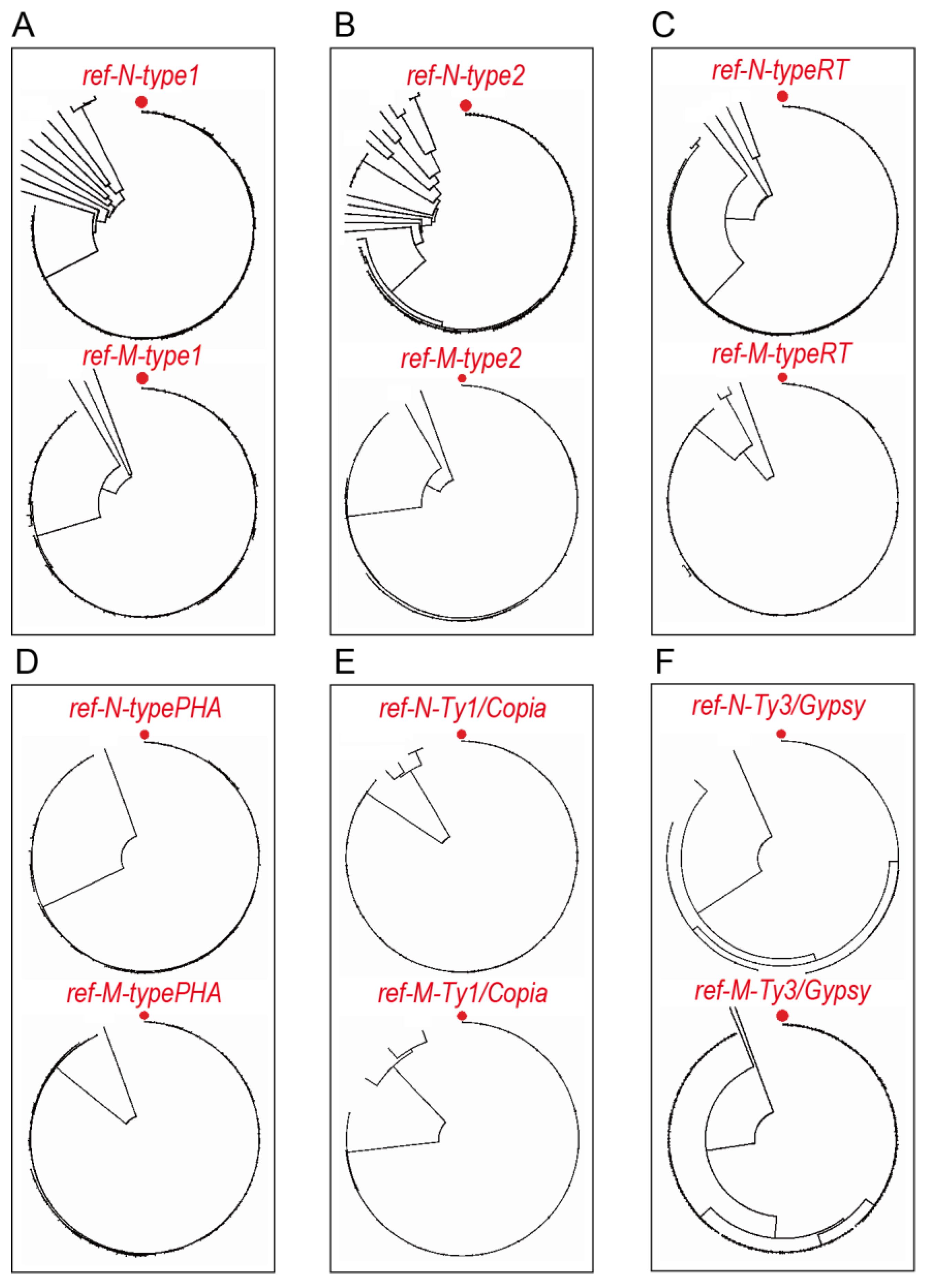
3.3. Phylogenetic Trees and ORF Reference Sequences of TE Clusters
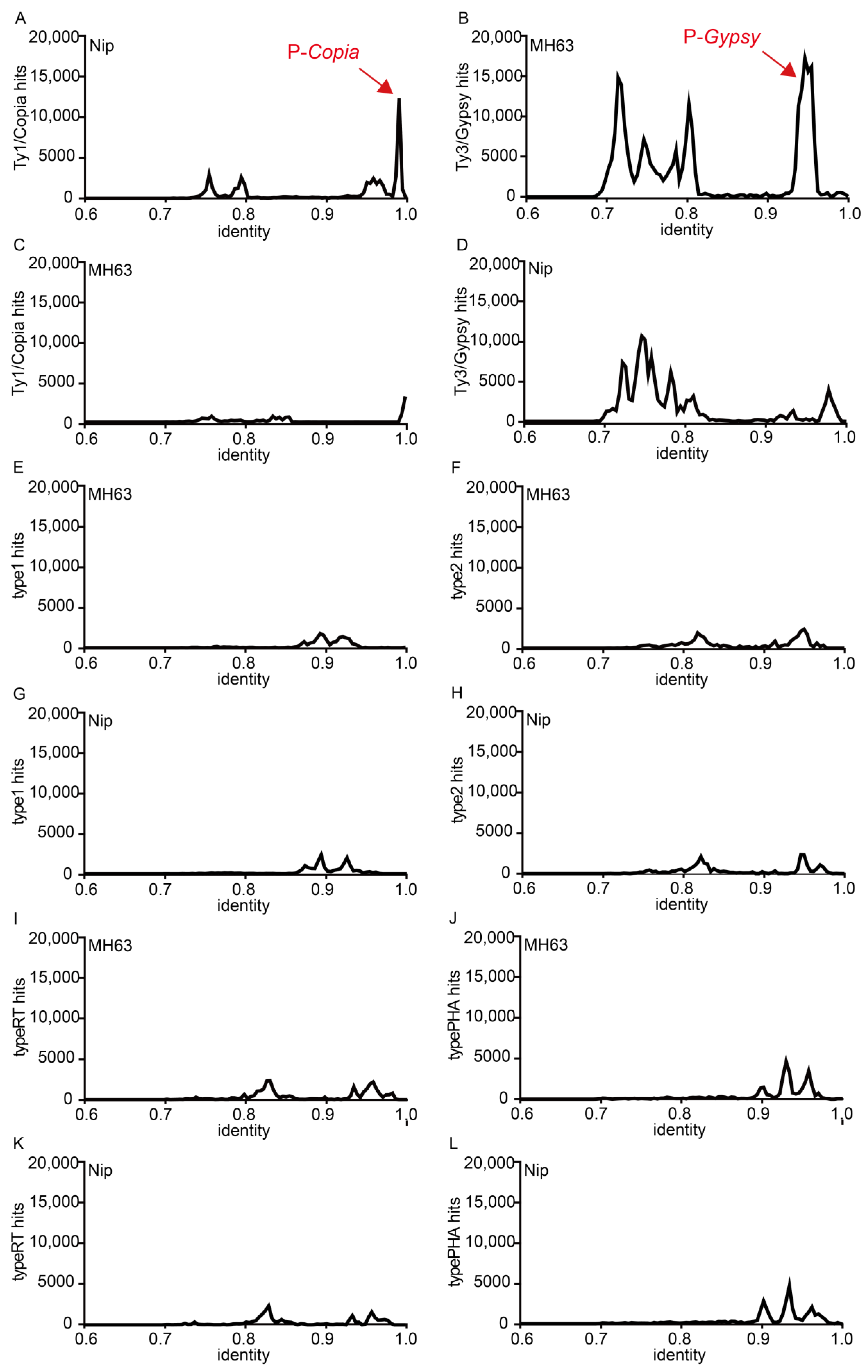
3.4. Whole Genome and Centromere Scanning by TE ORF Reference Sequences
3.5. Stochastic SNP Distribution in TE ORFs, and GPDF Analysis of P-Copia and P-Gypsy
3.6. LTR/TE Analysis in Nip and MH63 Centromeric Regions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cai, Y.; Huang, G.; Yang, Y.; Li, Y.; Wang, K.; Wu, Z. Analysis of the chromatin binding affinity of retrotransposases reveals novel roles in diploid and tetraploid cotton. J. Integr. Plant Biol. 2019, 61, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Wu, Z.; Percy, R.G.; Bai, M.; Li, Y.; Frelichowski, J.E.; Hu, J.; Wang, K.; Yu, J.; Zhu, Y. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 2020, 52, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.M.; Xie, W.Z.; Wang, S.; Guo, Y.X.; Koo, D.H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.W.; Zhang, W.; et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant. 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Li, K.; Jiang, W.; Hui, Y.; Kong, M.; Feng, L.; Gao, L.; Li, P.; Lu, S. Gapless Indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol. Plant. 2021, 14, 1745–1756. [Google Scholar] [CrossRef]
- Möller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L.; et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ma, D.; Tang, L.; Zhao, M.; Zhang, G.; Wang, W.; Song, J.; Li, X.; Liu, Z.; Zhang, W.; et al. Population Genomic Analysis and De Novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game. Mol. Plant. 2019, 12, 632–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Springer Nathan, M.; Anderson Sarah, N.; Andorf Carson, M.; Ahern Kevin, R.; Bai, F.; Barad, O.; Barbazuk, W.B.; Bass Hank, W.; Baruch, K.; Ben Zvi, G.; et al. The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat. Genet. 2018, 50, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Haberer, G.; Kamal, N.; Bauer, E.; Gundlach, H.; Fischer, I.; Seidel Michael, A.; Spannagl, M.; Marcon, C.; Ruban, A.; Urbany, C.; et al. European maize genomes highlight intraspecies variation in repeat and gene content. Nat. Genet. 2020, 52, 950–957. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.; Zhao, H.; Zhao, H.; Song, W.; Zhang, M.; Cui, Y.; Dong, X.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Goerner-Potvin, P.; Bourque, G. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018, 19, 688–704. [Google Scholar] [CrossRef]
- Tang, Y.; Ma, X.; Zhao, S.; Xue, W.; Zheng, X.; Sun, H.; Gu, P.; Zhu, Z.; Sun, C.; Liu, F.; et al. Identification of an active miniature inverted-repeat transposable element mJing in rice. Plant J. 2019, 98, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef] [PubMed]
- El Baidouri, M.; Kim, K.D.; Abernathy, B.; Arikit, S.; Maumus, F.; Panaud, O.; Meyers, B.C.; Jackson, S.A. A new approach for annotation of transposable elements using small RNA mapping. Nucleic Acids Res. 2015, 43, e84. [Google Scholar] [CrossRef]
- Jiang, N.; Bao, Z.; Zhang, X.; Hirochika, H.; Eddy, S.R.; McCouch, S.R.; Wessler, S.R. An active DNA transposon family in rice. Nature 2003, 421, 163–167. [Google Scholar] [CrossRef]
- Chen, J.; Lu, L.; Benjamin, J.; Diaz, S.; Hancock, C.N.; Stajich, J.E.; Wessler, S.R. Tracking the origin of two genetic components associated with transposable element bursts in domesticated rice. Nat. Commun. 2019, 10, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, M.C.; Manfroi, E.; Wei, F.J.; Wu, H.P.; Lasserre, E.; Llauro, C.; Debladis, E.; Akakpo, R.; Hsing, Y.I.; Panaud, O. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nat. Commun. 2019, 10, 24. [Google Scholar] [CrossRef]
- Liao, Y.; Zhang, X.; Li, B.; Liu, T.; Chen, J.; Bai, Z.; Wang, M.; Shi, J.; Walling, J.G.; Wing, R.A.; et al. Comparison of Oryza sativa and Oryza brachyantha genomes reveals selection-driven gene escape from the centromeric regions. Plant Cell 2018, 30, 1729–1744. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Nati. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Nijhawan, A.; Tyagi, A.K.; Khurana, J.P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2006, 345, 646–651. [Google Scholar] [CrossRef]
- Meyers, B.C.; Tingey, S.V.; Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11, 1660–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, T.; Zamborlini, A.; Cristofari, G.; Lesage, P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017, 18, 292–308. [Google Scholar] [CrossRef] [PubMed]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Biémont, C.; Vieira, C. Genetics: Junk DNA as an evolutionary force. Nature 2006, 443, 521–524. [Google Scholar] [CrossRef]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef]
- Huang, G.; Huang, J.Q.; Chen, X.; Zhu, Y.X. Recent advances and future perspectives in cotton research. Annu. Rev. Plant. Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef]
- Wang, K.; Huang, G.; Zhu, Y. Transposable elements play an important role during cotton genome evolution and fiber cell development. Sci. China Life Sci. 2016, 59, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Qi, Y.; Pan, H.; Tang, H.; Wang, G.; Hua, X.; Wang, Y.; Lin, L.; Li, Z.; Li, Y.; et al. Genomic insights into the recent chromosome reduction of autopolyploid sugarcane Saccharum spontaneum. Nat. Genet. 2022, 54, 885–896. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Japonica (Nip) | Indica (MH63) |
|---|---|---|
| Genome size (Mb) | 380 | 395 |
| Total TE content (%) | 44.1 | 45.9 |
| Intact LTR/TE | 4744 | 5146 |
| ORFs in matrix | 1520 | 2010 |
| Genome and Centromere Scanning by TE ORFs | |||||||
|---|---|---|---|---|---|---|---|
| Type1 | Type2 | TypeRT | TypePHA | Ty1/Copia | Ty3/Gypsy | ||
| Number of TE ORFs in genomes | Indica (MH63) | 728 | 1086 | 998 | 2379 | 316 | 6277 |
| Japonica (Nip) | 820 | 1039 | 859 | 2219 | 1479 | 3328 | |
| Number of TE ORFs in centromeres | Indica (MH63) | / | / | / | / | 23 | 758 |
| Japonica (Nip) | / | / | / | / | 57 | 345 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Xi, W.; Han, Z.; Wu, Y.; Guan, Y.; Zhu, Y. Genome-Wide Comparative Analysis of Transposable Elements by Matrix-TE Method Revealed Indica and Japonica Rice Evolution. Agronomy 2022, 12, 1490. https://doi.org/10.3390/agronomy12071490
Wu Z, Xi W, Han Z, Wu Y, Guan Y, Zhu Y. Genome-Wide Comparative Analysis of Transposable Elements by Matrix-TE Method Revealed Indica and Japonica Rice Evolution. Agronomy. 2022; 12(7):1490. https://doi.org/10.3390/agronomy12071490
Chicago/Turabian StyleWu, Zhiguo, Wei Xi, Zixuan Han, Yanhua Wu, Yongzhuo Guan, and Yuxian Zhu. 2022. "Genome-Wide Comparative Analysis of Transposable Elements by Matrix-TE Method Revealed Indica and Japonica Rice Evolution" Agronomy 12, no. 7: 1490. https://doi.org/10.3390/agronomy12071490
APA StyleWu, Z., Xi, W., Han, Z., Wu, Y., Guan, Y., & Zhu, Y. (2022). Genome-Wide Comparative Analysis of Transposable Elements by Matrix-TE Method Revealed Indica and Japonica Rice Evolution. Agronomy, 12(7), 1490. https://doi.org/10.3390/agronomy12071490