
Differing Roles of Bacterial and Fungal Communities in Cotton Fields by Growth Stage



Abstract
:1. Introduction
2. Materials and Methods
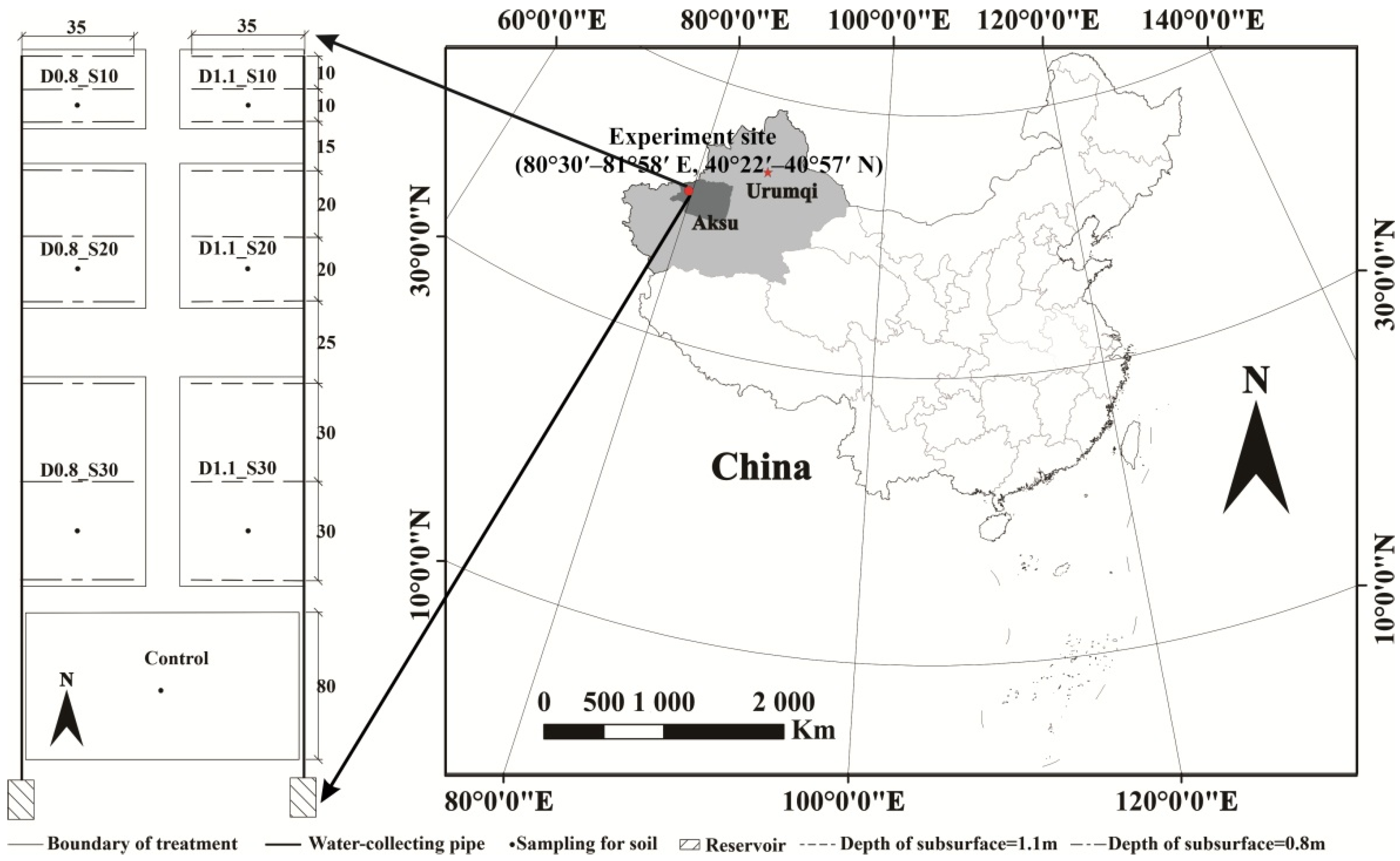
2.1. Experiment Description and Sample Collection
2.2. Soil Properties and Cotton Growth Indices
2.3. Sequencing of Soil Microbial Communities
2.4. Statistical Analyses
3. Results
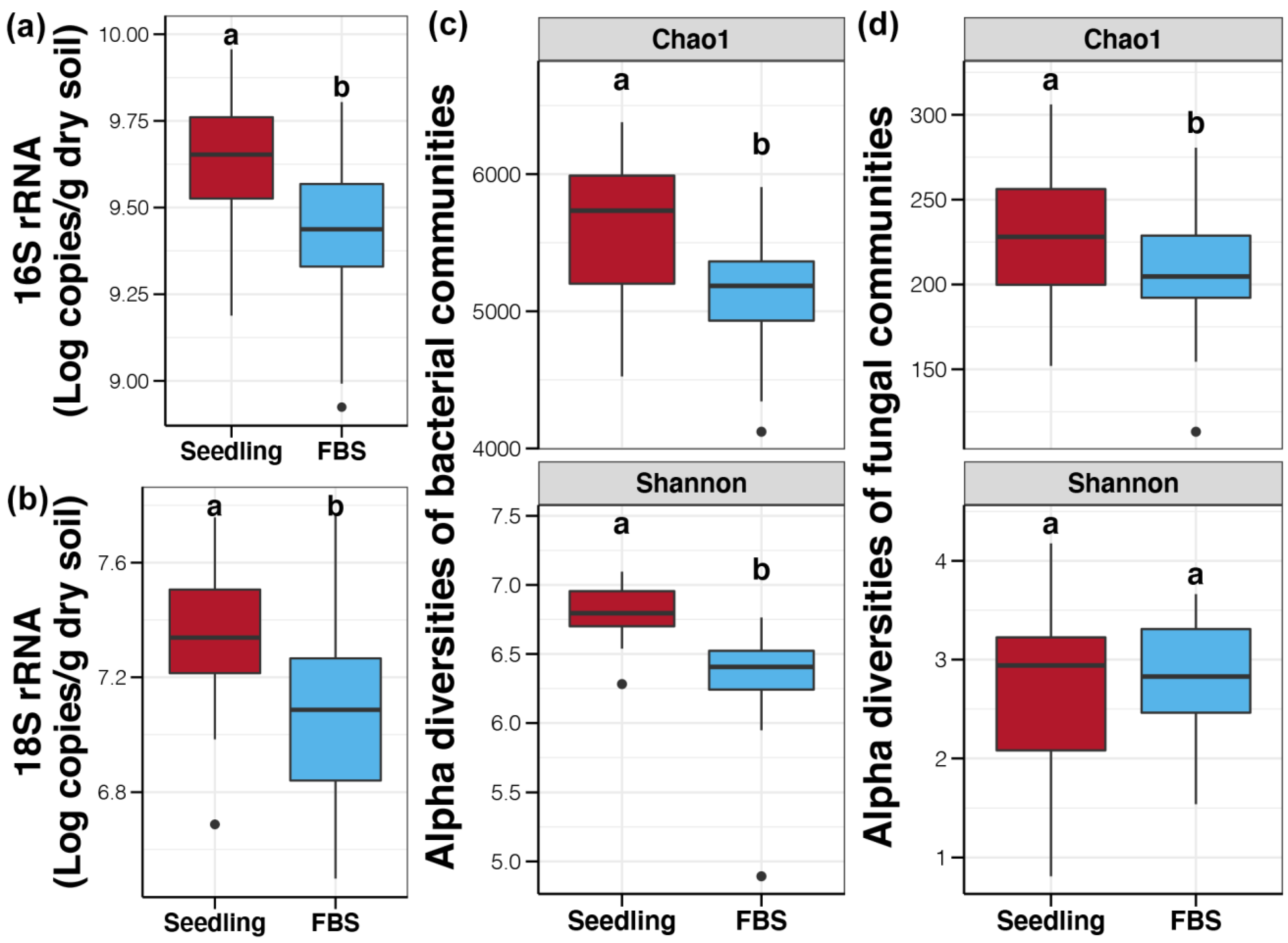
3.1. Biomass and Alpha Diversity of Soil Bacterial and Fungal Communities
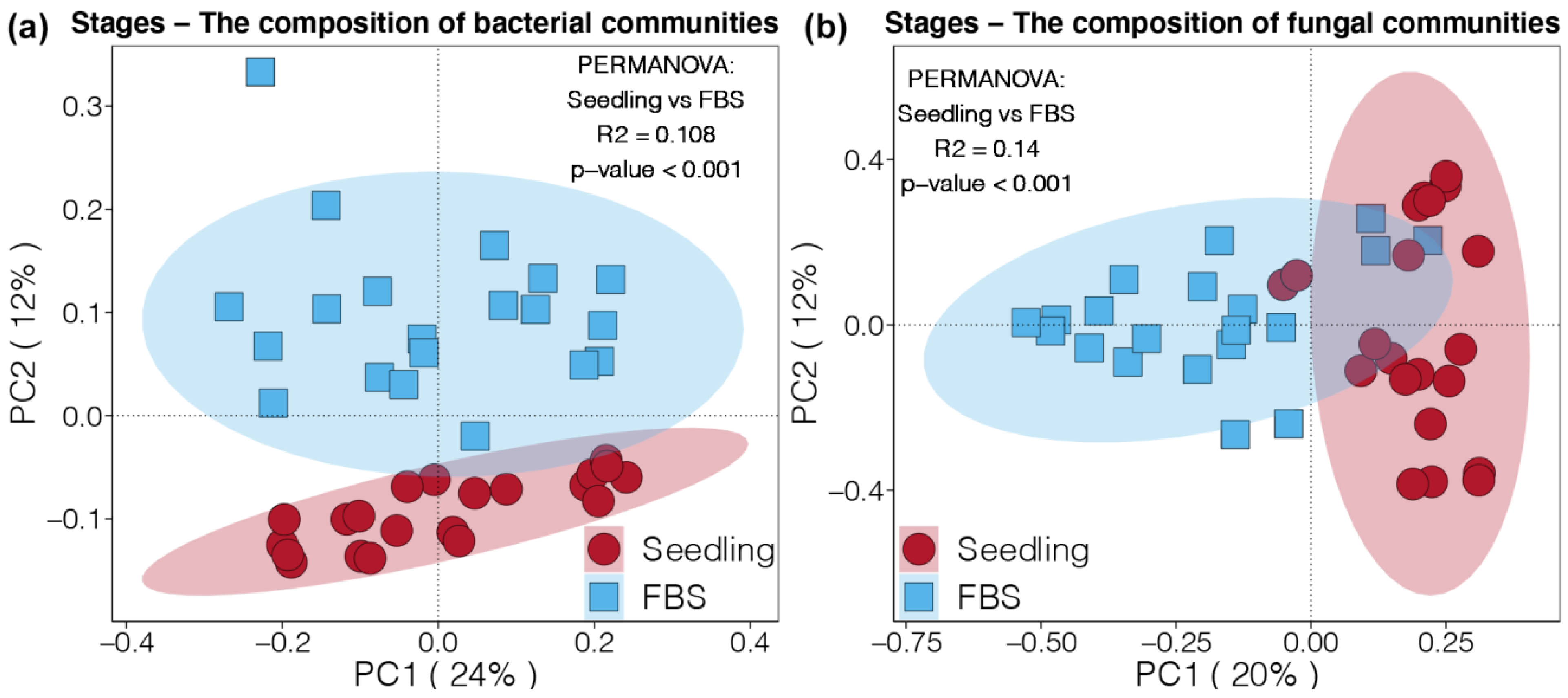
3.2. Beta Diversity of Soil Bacterial and Fungal Communities
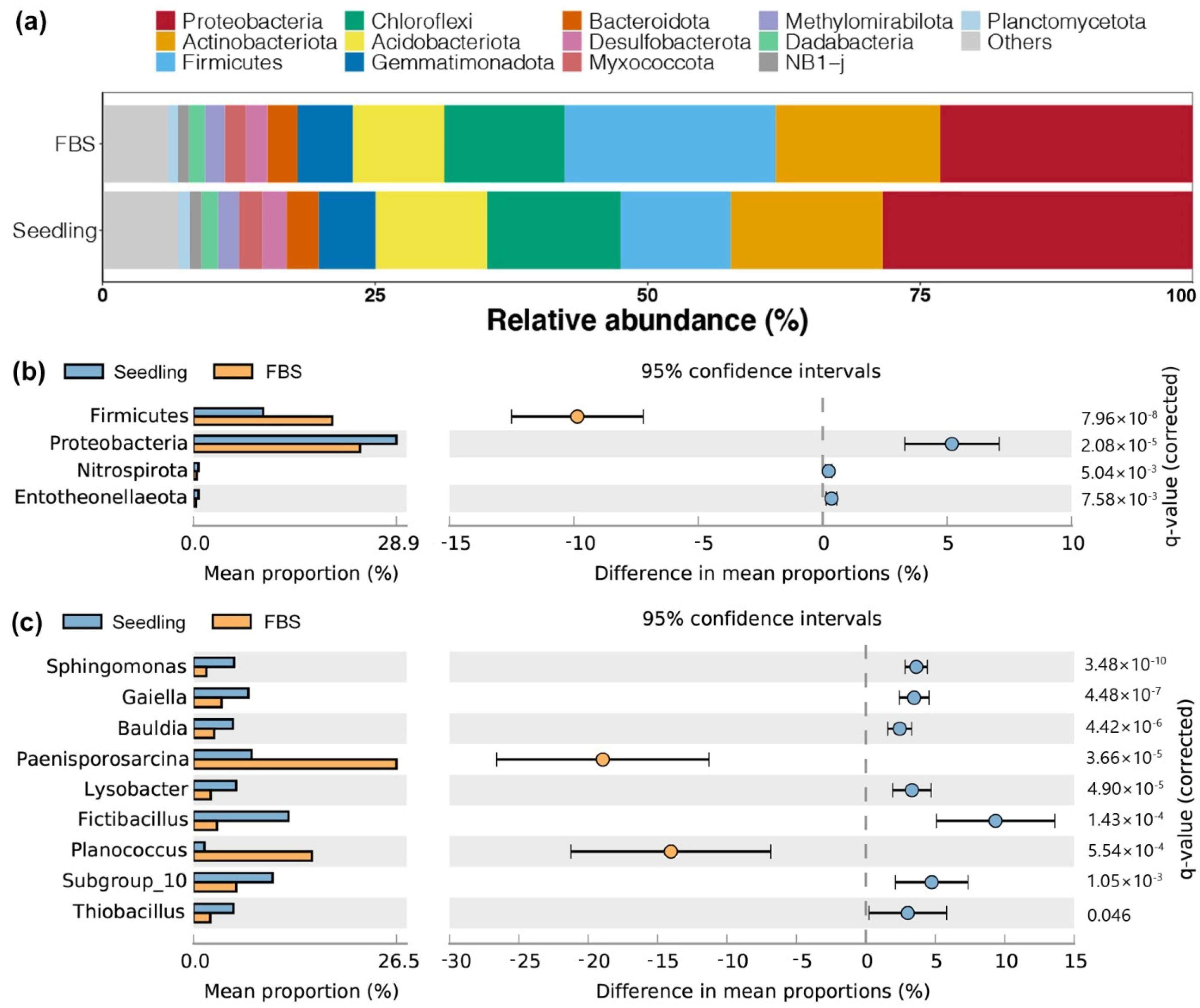
3.3. Difference in Soil Bacterial Abundance between the Seedling and FBS Stages
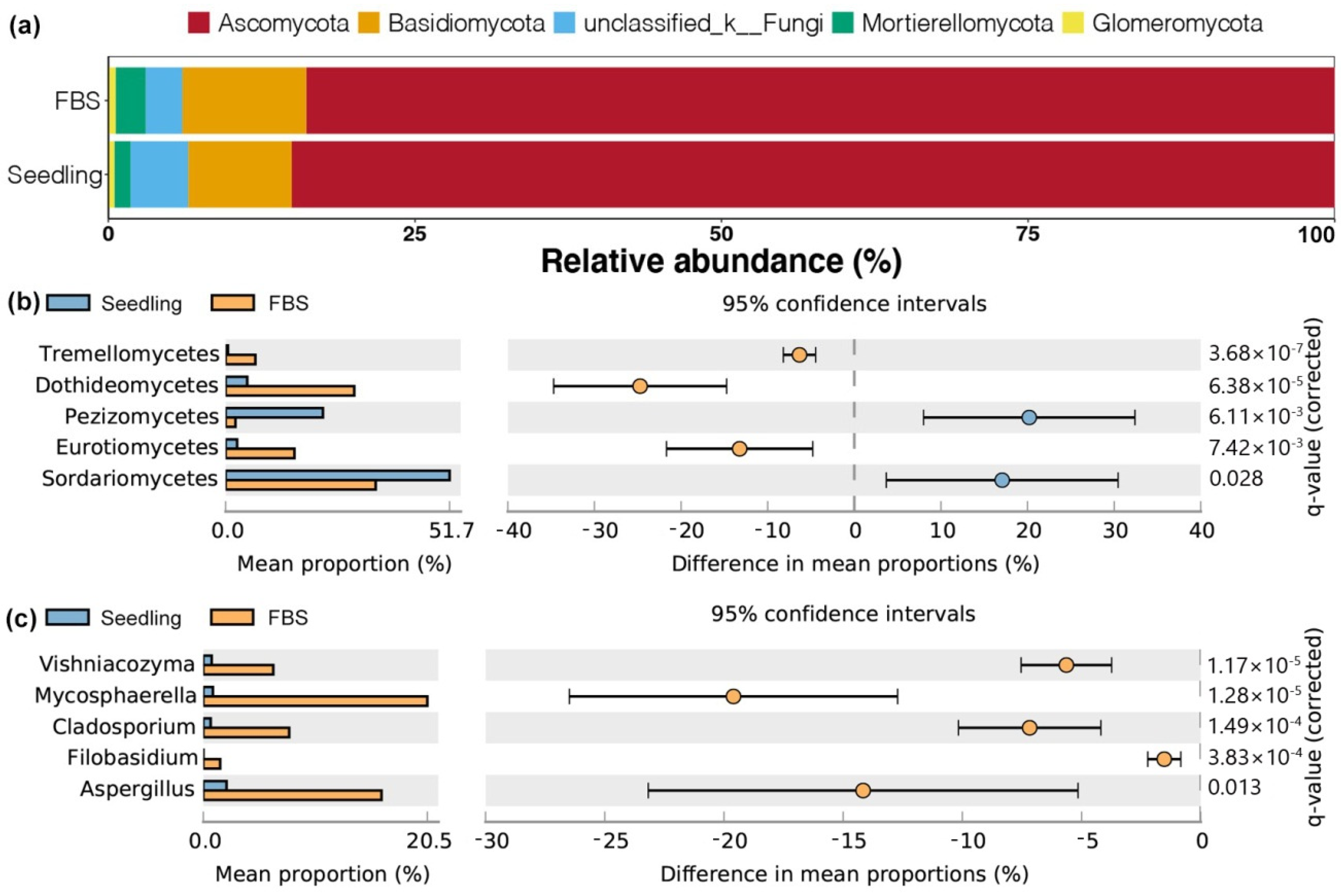
3.4. Differences in Soil Fungal Abundance between the Seedling and FBS Stages
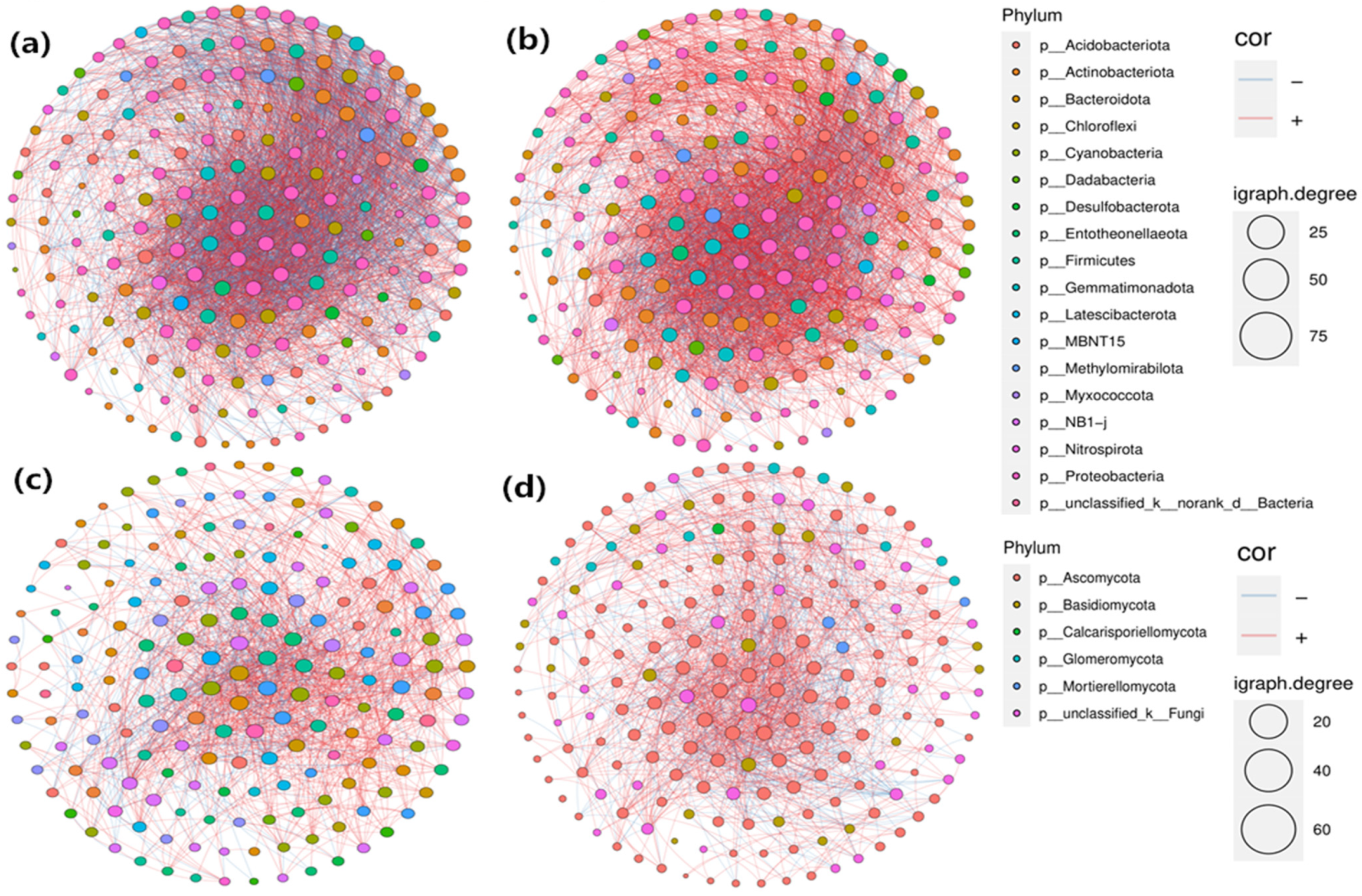
3.5. Co-Occurrence Network Analysis of Soil Bacterial and Fungal Communities
3.6. Microbial Communities in Relation to Soil Properties and Plant Growth
4. Discussion
4.1. Variations in Soil Microbial Communities during Crop Development
4.2. Different Responses of Bacterial and Fungal Communities during Cotton Growth
4.3. Stronger Bacterial Association with Cotton Growth
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Akhtar, F.; Tischbein, B.; Awan, U.K. Optimizing deficit irrigation scheduling under shallow groundwater conditions in lower reaches of Amu Darya River Basin. Water Res. Manag. 2013, 27, 3165–3178. [Google Scholar] [CrossRef]
- Ning, S.R.; Shi, J.C.; Zuo, Q.; Wang, S.; Ben-Gal, A. Generalization of the root length density distribution of cotton under film mulched drip irrigation. Field Crops Res. 2015, 177, 125–136. [Google Scholar] [CrossRef]
- Wang, G.Y.; Zhou, X.M.; Zhang, J.W.; Shen, J. Salt tolerance of cucumber cultivar satger mination stage. J. Plant Genet. Resour. 2004, 5, 299–303. [Google Scholar]
- Bischoff, A.; Vonlanthen, B.; Steinger, T.; Müller-Schärer, H. Seed provenance matters—Effects on germination of four plant species used for ecological restoration. Basic Appl. Ecol. 2006, 7, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.J.; Ma, X.L.; Wan, P.; Liu, L.Y. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- Parkes, R.J.; Sass, H. Deep Sub-Surface in Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 64–79. [Google Scholar] [CrossRef]
- Zhou, Y.; Bastida, F.; Zhou, B.; Sun, Y.; Li, Y. Soil fertility and crop production are fostered by micronano bubble irrigation with associated changes in soil bacterial community. Soil Biol. Biochem. 2019, 141, 107663. [Google Scholar] [CrossRef]
- Niu, J.; Chao, J.; Xiao, Y.; Chen, W.; Dai, L. Insight into the effects of different cropping systems on soil bacterial community and tobacco bacterial wilt rate. J. Basic Microbiol. 2017, 57, 3–11. [Google Scholar] [CrossRef]
- She, S.; Niu, J.; Zhang, C.; Xiao, Y.; Chen, W.; Dai, L.; Liu, X.D.; Yin, H.Q. Significant relationship between soil bacterial community structure and incidence of bacterial wilt disease under con-tinuous cropping system. Arch. Microbiol. 2017, 199, 267–275. [Google Scholar] [CrossRef]
- Shen, D.; Langenheder, S.; Jürgens, K. Dispersal modifies the diversity and composition of active bacterial communities in response to a salinity disturbance. Front. Microbiol. 2018, 9, 2188. [Google Scholar] [CrossRef] [Green Version]
- Huber, P.; Metz, S.; Unrein, F.; Mayora, G.; Sarmento, H.; Devercelli, M. Environmental heterogeneity determines the ecological processes that govern bacterial metacommunity assembly in a floodplain river system. ISME J. 2020, 14, 2951–2966. [Google Scholar] [CrossRef] [PubMed]
- Salcher, M.M.; Posch, P.; Pernthaler, J. In situ substrate preferences of abundant bacterioplankton populations in a prealpine freshwater lake. ISME J. 2013, 7, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Frey-Klett, P.; Burlinson, P.; Deveau, A.; Barret, M.; Tarkka, M.; Sarniguet, A. Bacterial-Fungal Interactions: Hyphens between Agricultural, Clinical, Environmental, and Food Microbiologists. Am. Soc. Microbiol. 2011, 75, 583–609. [Google Scholar] [CrossRef] [Green Version]
- Logares, R.; Tesson, S.V.M.; Canbäck, B.; Pontarp, M.; Hedlund, K.; Rengefors, K. Contrasting prevalence of selection and drift in the community structuring of bacteria and microbial eukaryo-tes. Environ. Microbiol. 2018, 20, 2231–2240. [Google Scholar] [CrossRef]
- Wu, W.; Lu, H.P.; Sastri, A.; Yeh, Y.C.; Gong, G.C.; Chou, W.C.; Hsieh, C.H. Contrasting the relative importance of species sorting and dispersal limitation in shaping marine bacterial versus protist communities. ISME J. 2018, 12, 285–494. [Google Scholar] [CrossRef] [Green Version]
- Romaní, A.M.; Fischer, H.; Mille-Lindblom, C.; Tranvik, L.J. Interactions of bacteria and fungi on decomposing litter: Differential extracellular enzyme activities. Ecology 2006, 87, 2559–2569. [Google Scholar] [CrossRef] [Green Version]
- Clausen, C.A. Bacterial associations with decaying wood: A review. Int. Biodeterior. Biodegrad. 1996, 37, 101–107. [Google Scholar] [CrossRef]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.; Fitter, A.H.; Hodge, A. Growth and symbiotic effectiveness of an arbuscular mycorrhizal fungus in organic matter in competition with soil bacteria. FEMS Microbiol. Ecol. 2011, 76, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, M.; Liu, Y.; Zhang, F.; Hodge, A.; Feng, G. Carbon and phosphorus exchange may enable cooperation between an arbuscular mycorrhizal fungus and a phosphate-solubilizing bacterium. New Phytol. 2016, 210, 1022–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Legrand, F.; Picot, A.; Cobo-Díaz, J.F.; Carof, M.; Chen, W.; Le Floch, G. Effect of tillage and static abiotic soil properties on microbial diversity. Appl. Soil Ecol. 2018, 132, 135–145. [Google Scholar] [CrossRef]
- Ren, C.; Zhang, W.; Zhong, Z.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Differential responses of soil microbial biomass, diversity, and compositions to altitudinal gradients depend on plant and soil characteristics. Sci. Total Environ. 2018, 610, 750–758. [Google Scholar] [CrossRef]
- Berg, J.; Brandt, K.K.; Al-Soud, W.A.; Holm, P.E.; Hansen, L.H.; Sorensen, S.J.; Nybroe, O. Selection for Cu-Tolerant Bacterial Communities with Altered Composition, but Unaltered Richness, via Long-Term Cu Exposure. Appl. Environ. Microbiol. 2012, 78, 7438–7446. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.H.; Yin, S.X.; Liu, X.; Zhang, W.M.; Gu, T.Y.; Shen, Q.R.; Qiu, H.Z. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTOP)”. Taxonom-ic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, P.F.; Aller, J.Y. Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 2004, 47, 161–177. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Goslee, S.C.; Urban, D.L. The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 2007, 22, 1–19. [Google Scholar] [CrossRef]
- Paul, E.; Clark, F. Occurrences and Distribution of Soil Organics. Soil Microbiol. Biochem. 1989, 81–84. [Google Scholar] [CrossRef]
- Chen, Z.M.; Wang, H.Y.; Liu, X.W.; Zhao, X.L.; Lu, D.J.; Zhou, J.M.; Li, C.Z. Changes in soil microbial community and organic carbon fractions under short-term straw return in a rice–wheat cropping system. Soil Tillage Res. 2017, 165, 121–127. [Google Scholar] [CrossRef]
- Li, H.; Yang, X.; Weng, B.; Su, J.; Nie, S.A.; Gilbert, J.A.; Zhu, Y.G. The phenological stage of rice growth determines anaerobic ammonium oxidation activity in rhizosphere soil. Soil Biol. Biochem. 2016, 100, 59–65. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Wieland, G.; Neumann, R.; Backhaus, H. Variation of microbial communities in soil, rhizosphere, and rhizoplane in response to crop species, soil type, and crop development. Appl. Environ. Microbiol. 2001, 67, 5849–5854. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Chaparro, J.M.; Manter, D.K.; Zhang, R.; Vivanco, J.M.; Shen, Q. Roots from disti-nct plant developmental stages are capable of rapidly selecting their own microbiome without the influence of environmental and soil edaphic factors. Soil Biol. Biochem. 2015, 89, 206–209. [Google Scholar] [CrossRef]
- Wang, J.; Xue, C.; Song, Y.; Wang, L.; Huang, Q.; Shen, Q. Wheat and rice growth stages and fertilization regimes alter soil bacterial community structure, but not diversity. Front. Microbiol. 2016, 7, 1207. [Google Scholar] [CrossRef] [PubMed]
- Pershina, E.; Valkonen, J.; Kurki, P.; Ivanova, E.; Chirak, E.; Korvigo, I.; Provorov, N.; Andronov, E. Comparative analysis of prokaryotic communities associated with organic and conventional farming systems. PLoS ONE 2015, 10, e0145072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Luo, X.; Chen, Y.; Ye, X.; Wang, H.; Cao, Z.; Ran, W.; Cui, Z.L. Succession of composition and function of soil bacterial communities during key rice growth stages. Front. Microbiol. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef]
- Zak, D.R.; Tilman, D.; Parmenter, R.R.; Rice, C.W.; Fisher, F.M.; Vose, J.; Milchunas, D.; Martin, C.W. Plant-production and soilmicroorganisms in late-successional ecosystems—A continentalscale study. Ecology 1994, 75, 2333–2347. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. mBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Bukar, M.; Sodipo, O.; Dawkins, K.; Ramirez, R.; Esiobu, N. Microbiomes of top and sub-layers of semi-arid soils in north-eastern nigeria are rich in firmicutes and proteobacteria with surprisingly high diversity of rare species. Adv. Microbiol. 2019, 9, 102–118. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Moulin, L.; Munive, A.; Dreyfus, B.; Boivin-Masson, C. Nodulation of legumes by members of the β-subclass of Proteobacteria. Nature 2001, 411, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Rasolomampianina, R.; Bailly, X.; Fetiarison, R.; Rabevohitra, R.; Bena, G.; Ramaroson, L.; Raherimand-imby, M.; Moulin, L.; De Lajudie, P.; Dreyfus, B.; et al. Nitrogen-fixing nodules from rose wood legume trees (Dalbergia spp.) endemic to Madagascar host seven different genera belonging to α-and β-Proteobacteria. Mol. Ecol. 2005, 14, 4135–4146. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Chen, L.; Wang, J.; Zheng, M.; Liu, S.; Chen, Q.; Ni, J. Comammox Nitrospira within the Yangtze River continuum: Community, biogeography, and ecological drivers. ISME J. 2020, 14, 2488–2504. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.J.; Liu, W.B.; Zhu, C.; Luo, G.W.; Kong, Y.L.; Ling, N.; Wang, M.; Dai, J.Y.; Shen, Q.R.; Guo, S.W. Bacterial rather than fungal community composition is associated with microbial activities and nutrient-use efficiencies in a paddy soil with short-term organic amendments. Plant Soil 2018, 424, 335–349. [Google Scholar] [CrossRef]
- Zhong, W.H.; Bian, B.Y.; Gao, N.; Min, J.; Shi, W.M.; Lin, X.G.; Shen, W.S. Nitrogen fertilization induced changes in ammonia oxidation are attributable mostly to bacteria rather than archaea in greenhouse-based high N input vegetable soil. Soil Biol. Biochem. 2016, 93, 150–159. [Google Scholar] [CrossRef]
- Shurin, J.B.; Cottenie, K.; Hillebrand, H. Spatial autocorrelation and dispersal limitation in freshwater organisms. Oecologia 2009, 159, 151–159. [Google Scholar] [CrossRef]
- Farjalla, V.F.; Srivastava, D.S.; Marino, N.A.C.; Azevedo, F.D.; Dib, V.; Lopes, P.M.; Rosado, A.S.; Bozelli, R.L.; Esteves, F.A. Ecological determinism increases with organism size. Ecology 2012, 93, 1752–1759. [Google Scholar] [CrossRef]
- Langenheder, S.; Ragnarsson, H. The role of environmental and spatial factors for the composition of aquatic bacterial communities. Ecology 2007, 88, 2154–2161. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, J.N.; Lu, M.; Qin, C.; Chen, Y.H.; Yang, L.; Huang, Q.W.; Wang, J.C.; Shen, Z.; Shen, Q. Microbial communities of an arable soil treated for 8 years with organic and inorganic fertilizers. Biol. Fertil. Soils 2016, 52, 455–467. [Google Scholar] [CrossRef]
- Lucas, J.M.; Mc Bride, S.G.; Strickland, M.S. Trophic level mediates soil microbial community composition and function. Soil Biol. Biochem. 2020, 143, 107756. [Google Scholar] [CrossRef]
- Albassam, B.A. Effect of nitrate nutrition on growth and nitrogen assimilation of pearl millet exposed to sodium chloride stress. J. Plant Nutr. 2001, 24, 1325–1335. [Google Scholar] [CrossRef]
- Esmaili, E.; Kapourchal, S.A.; Malakouti, M.J.; Homaee, M. Interactive effect of salinity and two nitrogen fertilizers on growth and composition of sorghum. Plant Soil Environ. 2008, 54, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.Z.; Li, W.J.; Egrinya, E.A.; Zhang, D.M. Nitrogen rate and plant density effects on yield and late-season leaf senescence of cotton raised on a saline field. Field Crops Res. 2012, 126, 137–144. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria-Seedling | Fungi-Seedling | Bacteria-FBS | Fungi-FBS | |
|---|---|---|---|---|
| Num edges | 4971 | 2106 | 4832 | 2158 |
| Pos edges | 2668 | 1565 | 3610 | 1544 |
| Neg edges | 2303 | 541 | 1222 | 614 |
| Num nodes | 199 | 191 | 199 | 199 |
| Connectance | 0.252 | 0.116 | 0.245 | 0.110 |
| Average degree | 49.960 | 22.052 | 48.563 | 21.688 |
| Average path length | 1.851 | 2.205 | 1.828 | 2.234 |
| Diameter | 1.890 | 2.333 | 1.919 | 2.292 |
| Clustering coefficient | 0.610 | 0.423 | 0.581 | 0.421 |
| Centralization degree | 0.308 | 0.210 | 0.250 | 0.199 |
| Centralization betweenness | 0.010 | 0.025 | 0.013 | 0.018 |
| Centralization closeness | 0.282 | 0.230 | 0.211 | 0.219 |
| Bacteria–Seedling | Bacterial–FBS | Fungi–Seedling | Fungi–FBS | |||||
|---|---|---|---|---|---|---|---|---|
| r | p | r | p | r | p | r | p | |
| Salinity | 0.287 | 0.003 | 0.203 | 0.039 | 0.058 | 0.312 | 0.033 | 0.338 |
| Ammonia | 0.205 | 0.031 | 0.161 | 0.096 | 0.087 | 0.237 | 0.261 | 0.017 |
| Nitrate | 0.579 | 0.001 | 0.234 | 0.008 | 0.255 | 0.008 | 0.029 | 0.309 |
| Moisture | 0.085 | 0.149 | 0.258 | 0.036 | 0.024 | 0.380 | −0.103 | 0.778 |
| Phosphate | 0.158 | 0.032 | 0.194 | 0.032 | 0.185 | 0.026 | −0.028 | 0.587 |
| pH | −0.084 | 0.795 | 0.167 | 0.105 | −0.102 | 0.820 | 0.028 | 0.382 |
| SOM | 0.090 | 0.154 | 0.243 | 0.083 | −0.054 | 0.694 | 0.218 | 0.074 |
| Plant height | 0.238 | 0.013 | 0.424 | 0.002 | −0.037 | 0.614 | 0.065 | 0.256 |
| Leaf area | 0.223 | 0.021 | 0.389 | 0.005 | −0.039 | 0.615 | 0.102 | 0.205 |
| Emergence rate/Yield | 0.398 | 0.004 | 0.419 | 0.002 | 0.183 | 0.048 | 0.089 | 0.181 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Yang, Y.; Zhao, Y.; Tian, G.; Li, M.; Qiu, H.; Zhou, X. Differing Roles of Bacterial and Fungal Communities in Cotton Fields by Growth Stage. Agronomy 2022, 12, 657. https://doi.org/10.3390/agronomy12030657
Li D, Yang Y, Zhao Y, Tian G, Li M, Qiu H, Zhou X. Differing Roles of Bacterial and Fungal Communities in Cotton Fields by Growth Stage. Agronomy. 2022; 12(3):657. https://doi.org/10.3390/agronomy12030657
Chicago/Turabian StyleLi, Dongwei, Yuhui Yang, Yulong Zhao, Guangli Tian, Mingsi Li, Husen Qiu, and Xinguo Zhou. 2022. "Differing Roles of Bacterial and Fungal Communities in Cotton Fields by Growth Stage" Agronomy 12, no. 3: 657. https://doi.org/10.3390/agronomy12030657
APA StyleLi, D., Yang, Y., Zhao, Y., Tian, G., Li, M., Qiu, H., & Zhou, X. (2022). Differing Roles of Bacterial and Fungal Communities in Cotton Fields by Growth Stage. Agronomy, 12(3), 657. https://doi.org/10.3390/agronomy12030657