
Genome-Wide Association Mapping for Seed Weight in Soybean with Black Seed Coats and Green Cotyledons




Abstract
:1. Introduction
2. Materials and Methods
2.1. Growth Conditions of BLG Germplasm and Phenotype Collection
2.2. DNA Extraction and Determination of Genotypes for BLG Accessions
2.3. Genome-Wide Association Studies
2.4. Linkage Disequilibrium Estimation
2.5. Online Tool and Phenotypic Data Set from GRIN
2.6. Statistical Data Analysis
3. Results
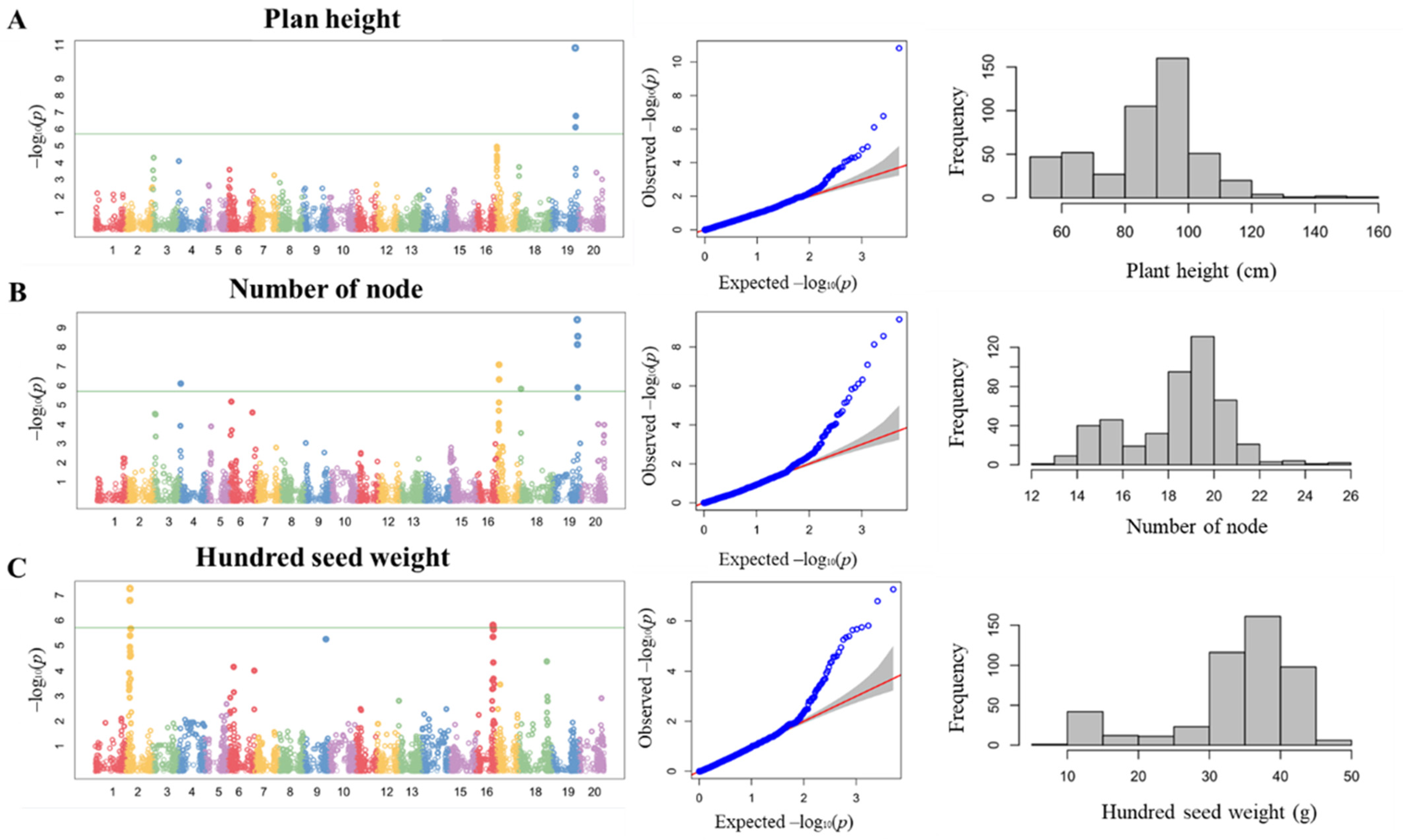
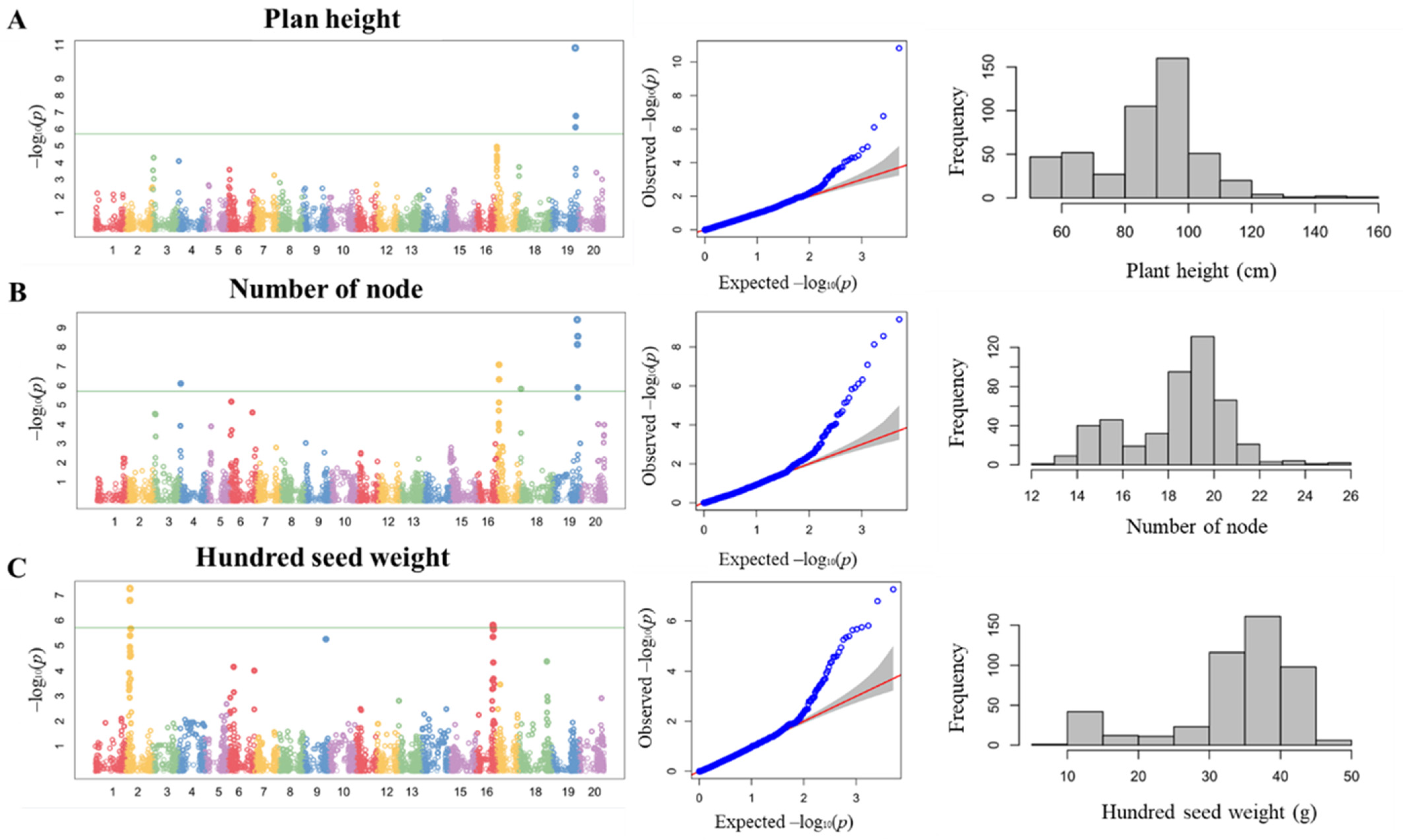
3.1. Phenotypic Distribution of Agronomic Traits in BLG Germplasm
3.2. GWAS with Plant Height and Number of Nodes
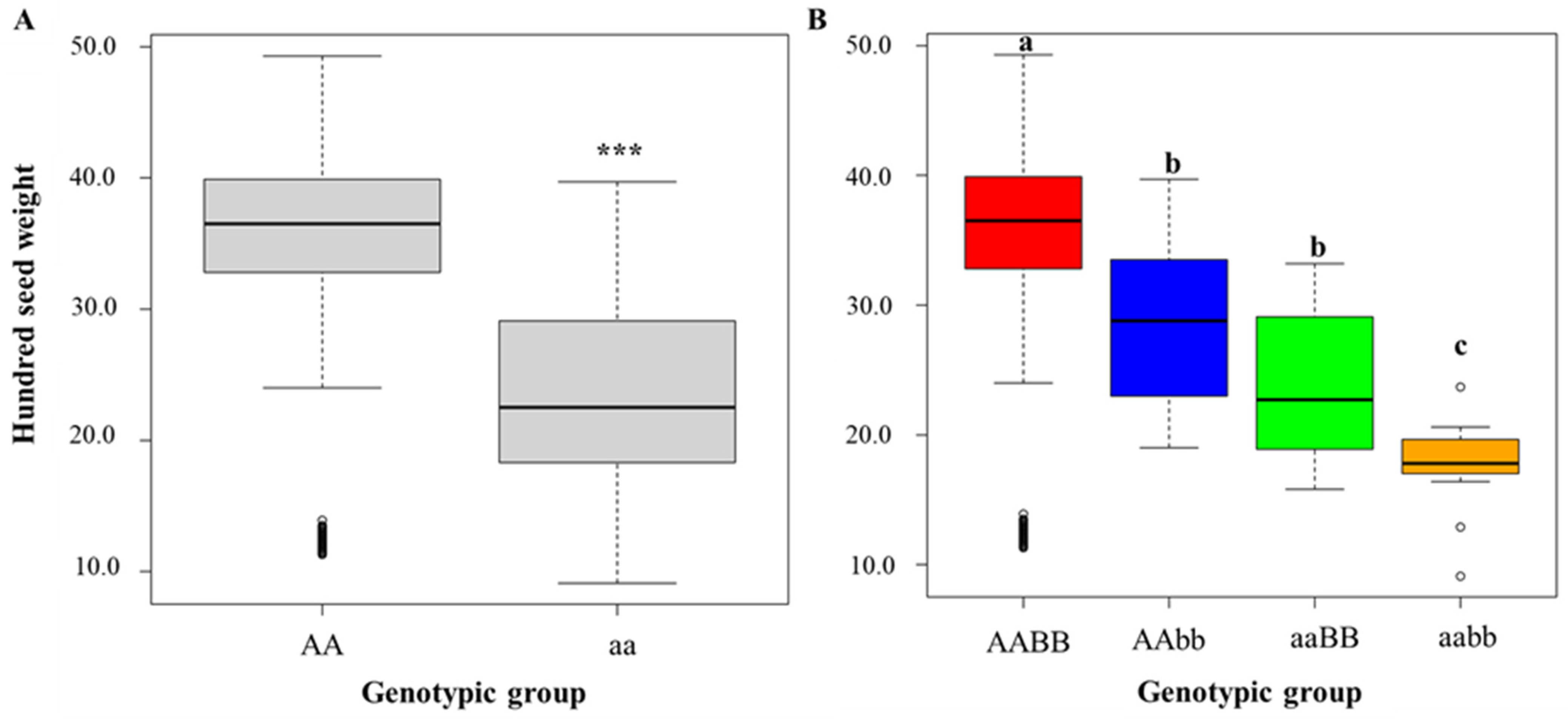
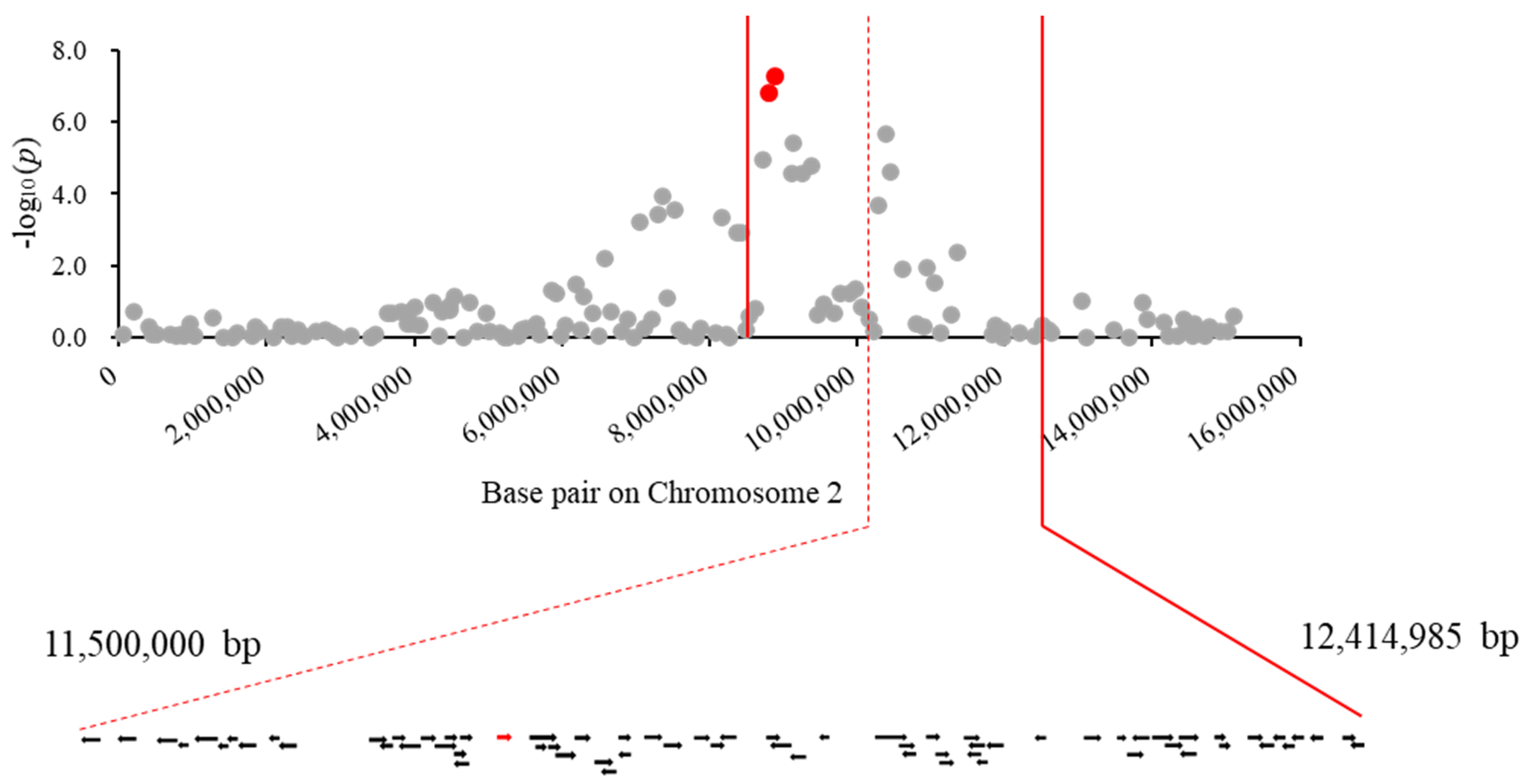
3.3. GWAS of HSW in BLG Accessions and Candidate Gene Prediction
3.4. Allelic Variation of Candidate Gene Analyzed with Publicly Available Genome Sequencing Data
3.5. Phenotypic Data Set of HSW from GRIN
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, S.; Zhang, M.; Feng, F.; Tian, Z. Toward a “green revolution” for soybean. Mol. Plant 2020, 13, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Liu, X.B.; Wang, C.; Li, Y.S.; Jin, J.; Herbert, S.J. Soybean yield and yield component distribution across the main axis in response to light enrichment and shading under different densities. Plant Soil Environ. 2010, 56, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Burris, J.S.; Edje, O.T.; Wahab, A.H. Effect of seed size on seedling performance in soybeans. II. Seedling growth and photosynthesis and field performance. Crop Sci. 1973, 13, 207–210. [Google Scholar] [CrossRef]
- Smith, T.J.; Camper, H.M. Effect of seed size on soybean performance. Agron. J. 1975, 67, 681–684. [Google Scholar] [CrossRef]
- Maughan, P.J.; Maroof, M.S.; Buss, G.R. Molecular marker analysis of seed-weight: Genomic locations, gene action, and evidence for orthologous evolution among three legume species. Theor. Appl. Genet. 1996, 93, 574–579. [Google Scholar] [CrossRef]
- Mian, M.A.; Bailey, M.A.; Tamulonis, J.P.; Shipe, E.R.; Carter, T.E.; Parrott, W.A.; Ashley, D.A.; Hussey, R.S.; Boerma, H.R. Molecular markers associated with seed weight in two soybean populations. Theor. Appl. Genet. 1996, 93, 1011–1016. [Google Scholar] [CrossRef]
- Wilson, D. Storage of orthodox seeds. In Seed Quality: Basic Mechanisms, Agricultural Implications, 1st ed.; Basra, A.S., Ed.; CRC Press: New York, NY, USA, 1995; pp. 173–208. [Google Scholar]
- Brim, C.A.; Cockerham, C.C. Inheritance of quantitative characters in soybean. Crop Sci. 1961, 1, 187–190. [Google Scholar] [CrossRef] [Green Version]
- SoyBase. Available online: www.soybase.org (accessed on 11 November 2021).
- Yan, L.; Hofmann, N.; Li, S.; Ferreira, M.E.; Song, B.; Jiang, G.; Ren, S.; Quigley, C.; Fickus, E.; Cregan, P.; et al. Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses. BMC Genom. 2017, 18, 529. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Song, Q.; Cregan, P.B.; Jiang, G.L. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theor. Appl. Genet. 2016, 129, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, E.Y.; Song, Q.; Jia, G.; Specht, J.E.; Hyten, D.L.; Costa, J.; Cregan, P.B. A genome-wide association study of seed protein and oil content in soybean. BMC Genom. 2014, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Tan, R.; Yuan, J.; Bales, C.; Du, W.; Zhang, S.; Chilvers, M.I.; Schmidt, C.; Song, Q.; Cregan, P.B. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom. 2014, 15, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Singh, A.; Mueller, D.S.; Singh, A.K. Genome-wide association and epistasis studies unravel the genetic architecture of sudden death syndrome resistance in soybean. Plant J. 2015, 84, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; He, J.; Wang, Y.; Xing, G.; Zhao, J.; Li, Y.; Yang, S.; Palmer, R.; Zhao, T.; Gai, J. Establishment of a 100-seed weight quantitative trait locus–allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. J. Exp. Bot. 2015, 66, 6311–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaler, A.S.; Purcell, L.C. Association mapping identifies and confirms loci for soybean seed weight. Crop Sci. 2021, 61, 1289–1300. [Google Scholar] [CrossRef]
- Karikari, B.; Wang, Z.; Zhou, Y.; Yan, W.; Feng, J.; Zhao, T. Identification of quantitative trait nucleotides and candidate genes for soybean seed weight by multiple models of genome-wide association study. BMC Plant Biol. 2020, 20, 404. [Google Scholar] [CrossRef]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527. [Google Scholar] [CrossRef] [Green Version]
- Teng, W.; Feng, L.; Li, W.; Wu, D.; Zhao, X.; Han, Y.; Li, W. Dissection of the genetic architecture for soybean seed weight across multiple environments. Crop Pasture Sci. 2017, 68, 358–365. [Google Scholar] [CrossRef]
- Han, Y.; Li, D.; Zhu, D.; Li, H.; Li, X.; Teng, W.; Li, W. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor. Appl. Genet. 2012, 125, 671–683. [Google Scholar] [CrossRef]
- Yan, L.; Li, Y.H.; Yang, C.Y.; Ren, S.X.; Chang, R.Z.; Zhang, M.C.; Qiu, L.J. Identification and validation of an over-dominant QTL controlling soybean seed weight using populations derived from Glycine max × Glycine soja. Plant Breed. 2014, 133, 632–637. [Google Scholar] [CrossRef]
- Kulkarni, K.P.; Kim, M.; Shannon, J.G.; Lee, J.D. Identification of quantitative trait loci controlling soybean seed weight in recombinant inbred lines derived from PI 483463 (Glycine soja) × ‘Hutcheson’ (G. max). Plant Breed. 2016, 135, 614–620. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Z.; Jiang, S.; Xu, N.; Chen, Q.; Qi, Z.; Lv, W. QTL mapping and candidate gene mining for soybean seed weight per plant. Biotechnol. Biotechnol. Equip. 2018, 32, 908–914. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xu, R.; Li, Y. Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Baekelandt, A.; Pauwels, L.; Wang, Z.; Li, N.; De Milde, L.; Natran, A.; Vermeersch, M.; Li, Y.; Goossens, A.; Inzé, D.; et al. Arabidopsis leaf flatness is regulated by PPD2 and NINJA through repression of CYCLIN D3 genes. Plant Physiol. 2018, 178, 217–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, N.; Pauwels, L.; Baekelandt, A.; De Milde, L.; Van Leene, J.; Besbrugge, N.; Heyndrickx, K.S.; Pérez, A.C.; Durand, A.N.; De Clercq, R.; et al. A repressor protein complex regulates leaf growth in Arabidopsis. Plant Cell 2015, 27, 2273–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, N.; Jiang, S.; Gonzalez, N.; Huang, X.; Wang, Y.; Inze, D.; Li, Y. SCFSAP controls organ size by targeting PPD proteins for degradation in Arabidopsis thaliana. Nat. Commun. 2016, 7, 11192. [Google Scholar] [CrossRef] [Green Version]
- White, D.W. PEAPOD regulates lamina size and curvature in Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 13238–13243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.X.; Paddock, K.J.; Zhang, Z.; Stacey, M.G. GmKIX8-1 regulates organ size in soybean and is the causative gene for the major seed weight QTL qSw17-1. New Phytol. 2021, 229, 920–934. [Google Scholar] [CrossRef]
- Sun, Z.; Su, C.; Yun, J.; Jiang, Q.; Wang, L.; Wang, Y.; Cao, D.; Zhao, F.; Zhao, Q.; Zhang, M.; et al. Genetic improvement of the shoot architecture and yield in soya bean plants via the manipulation of GmmiR156b. Plant Biotechnol. J. 2019, 17, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.T.; Liu, X.L.; Bi, Y.D.; Li, W.; Zhang, W.K.; Ma, B.; Lai, Y.C.; et al. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, C.A.; Peacock, W.J.; Dennis, E.S. Isolation and functional characterization of cytochrome P450s in gibberellin biosynthesis pathway. Methods Enzymol. 2002, 357, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hull, A.K.; Vij, R.; Celenza, J.L. Arabidopsis cytochrome P450s that catalyze the first step of tryptophan-dependent indole- 3-acetic acid biosynthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, E.; Kenz, S.; Gerstung, M.; MacLean, D.; Timmer, J.; Fleck, C.; Lenhard, M. Control of plant organ size by KLUH/CYP78A5-dependent intercellular signaling. Dev. Cell 2007, 13, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Wang, Z.; Cui, R.; Li, J.; Li, Y. Maternal control of seed size by EOD3/CYP78A6 in Arabidopsis thaliana. Plant J. 2012, 70, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, N.; Hibara, K.I.; Heppard, E.P.; Vander Velden, K.A.; Luck, S.; Beatty, M.; Nagato, Y.; Sakai, H. GIANT EMBRYO encodes CYP78A13, required for proper size balance between embryo and endosperm in rice. Plant J. 2013, 75, 592–605. [Google Scholar] [CrossRef]
- Xu, F.; Fang, J.; Ou, S.; Gao, S.; Zhang, F.; Du, L.; Xiao, Y.; Wang, H.; Sun, X.; Chu, J.; et al. Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ. 2015, 38, 800–811. [Google Scholar] [CrossRef]
- Yang, W.; Gao, M.; Yin, X.; Liu, J.; Xu, Y.; Zeng, L.; Li, Q.; Zhang, S.; Wang, J.; Zhang, X.; et al. Control of rice embryo development, shoot apical meristem maintenance, and grain yield by a novel cytochrome p450. Mol. Plant 2013, 6, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Ahn, B.O.; Kawakatsu, T.; Ito, Y.; Itoh, J.; Nagato, Y.; Kurata, N. PLASTOCHRON1, a timekeeper of leaf initiation in rice, encodes cytochrome P450. Proc. Natl. Acad. Sci. USA 2004, 101, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Dai, A.; Wei, H.; Yang, S.; Wang, B.; Jiang, N.; Feng, X. Arabidopsis KLU homologue GmCYP78A72 regulates seed size in soybean. Plant Mol. Biol. 2016, 90, 33–47. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Zhang, H.; Sun, G.; Zhang, W.; Qiu, L. Evolution and association analysis of GmCYP78A10 gene with seed size/weight and pod number in soybean. Mol. Biol. Rep. 2015, 42, 489–496. [Google Scholar] [CrossRef]
- Xu, B.; Chang, S.K. Antioxidant capacity of seed coat, dehulled bean, and whole black soybeans in relation to their distributions of total phenolics, phenolic acids, anthocyanins, and isoflavones. J. Agric. Food Chem. 2008, 56, 8365–8373. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, H.J.; Son, C.K.; Bae, J.S.; Jo, H.; Lee, J.D. Genetic diversity of black soybean germplasms with green cotyledon based on agronomic traits and cotyledon pigments. Korean J. Breed. Sci. 2021, 53, 127–139. [Google Scholar] [CrossRef]
- Yan, W.; Yingpeng, H.; Xue, Z.; Yongguang, L.; Weili, T.; Dongmei, L.; Yong, Z.; Wenbin, L. Mapping isoflavone QTL with main, epistatic and QTL × environment effects in recombinant inbred lines of soybean. PLoS ONE. 2015, 10, e0118447. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Lee, S.; Van, K.; Kim, T.H.; Jeong, S.C.; Choi, I.Y.; Kim, D.S.; Lee, Y.S.; Park, D.; Ma, J.; et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc. Natl. Acad. Sci. USA 2010, 107, 22032–22037. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef]
- Kim, M.S.; Lozano, R.; Kim, J.H.; Bae, D.N.; Kim, S.T.; Park, J.H.; Choi, M.S.; Kim, J.; Ok, H.C.; Park, S.K.; et al. The patterns of deleterious mutations during the domestication of soybean. Nat. Commun. 2021, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Valliyodan, B.; Qiu, D.; Patil, G.; Zeng, P.; Huang, J.; Dai, L.; Chen, C.; Li, Y.; Joshi, T.; Song, L.; et al. Landscape of genomic diversity and trait discovery in soybean. Sci. Rep. 2016, 6, 23598. [Google Scholar] [CrossRef] [Green Version]
- Valliyodan, B.; Brown, A.V.; Wang, J.; Patil, G.; Liu, Y.; Otyama, P.I.; Nelson, R.T.; Vuong, T.; Song, Q.; Musket, T.A.; et al. Genetic variation among 481 diverse soybean accessions, inferred from genomic re-sequencing. Sci. Data 2021, 8, 50. [Google Scholar] [CrossRef]
- Phytozome. Available online: https://phytozome-next.jgi.doe.gov/ (accessed on 11 November 2021).
- SoyKB. Available online: https://soykb.org/ (accessed on 11 November 2021).
- Jo, H.; Lee, J.Y.; Cho, H.; Choi, H.J.; Son, C.K.; Bae, J.S.; Bilyeu, K.; Song, J.T.; Lee, J.D. Genetic Diversity of Soybeans (Glycine max (L.) Merr.) with Black Seed Coats and Green Cotyledons in Korean Germplasm. Agronomy 2021, 11, 581. [Google Scholar] [CrossRef]
- Fehr, W.R.; Caviness, C.E.; Burmood, D.T.; Pennington, J.S. Stage of development descriptions for soybeans, Glycine Max (L.) Merrill1. Crop Sci. 1971, 11, 929–931. [Google Scholar] [CrossRef]
- Doyle, J.; Doyle, J.L. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull 1987, 19, 11–15. [Google Scholar]
- Song, Q.; Yan, L.; Quigley, C.; Fickus, E.; Wei, H.; Chen, L.; Dong, F.; Araya, S.; Liu, J.; Hyten, D.; et al. Soybean BARCSoySNP6K: An assay for soybean genetics and breeding research. Plant J. 2020, 104, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 2013, 8, e54985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinform 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.H.; Wei, X.H.; Sang, T.; Zhao, Q.A.; Feng, Q.; Zhao, Y.; Li, C.Y.; Zhu, C.R.; Lu, T.T.; Zhang, Z.W.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–976. [Google Scholar] [CrossRef]
- Soybean Allele Catalog. Available online: https://soykb.org/GenescapeAnalysis/search.php (accessed on 11 November 2021).
- GRIN Data Explorer. Available online: https://soybase.org/grindata/ (accessed on 11 November 2021).
- Do, M.H.; Lee, S.S.; Jung, P.J.; Lee, M.H. Intake of fruits, vegetables, and soy foods in relation to breast cancer risk in Korean women: A case-control study. Nutr. Cancer 2007, 57, 20–27. [Google Scholar] [CrossRef]
- Takahashi, R.; Ohmori, R.; Kiyose, C.; Momiyama, Y.; Ohsuzu, F.; Kondo, K. Antioxidant activities of black and yellow soybeans against low density lipoprotein oxidation. J. Agric. Food Chem. 2005, 53, 4578–4582. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Xu, B. A critical review on polyphenols and health benefits of black soybeans. Nutrients 2017, 9, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhan, J.K.; Chung, Y.C.; Chen, G.H.; Chang, C.H.; Lu, Y.C.; Hsu, C.K. Anthocyanin contents in the seed coat of black soya bean and their anti-human tyrosinase activity and antioxidative activity. Int. J. Cosmet. Sci. 2016, 38, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, J.H.; Lee, Y.S.; Yun, H.T. Antioxidant activity and extraction efficiency of anthocyanin pigments in black soybean. Korea Soybean Dig. 2006, 23, 1–9. [Google Scholar]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Ping, J.; Liu, Y.; Sun, L.; Zhao, M.; Li, Y.; She, M.; Sui, Y.; Lin, F.; Liu, X.; Tang, Z.; et al. Dt2 is a gain-of-function MADS-domain factor gene that specifies semideterminacy in soybean. Plant Cell 2014, 26, 2831–2842. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Wang, X.; Lee, R.; Li, Y.; Specht, J.E.; Nelson, R.L.; McClean, P.E.; Qiu, L.; Ma, J. Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. USA 2010, 107, 8563–8568. [Google Scholar] [CrossRef] [Green Version]
- Bernard, R.L. Two genes affecting stem termination in soybeans. Crop Sci. 1972, 12, 235–239. [Google Scholar] [CrossRef]
- Heatherly, L.G.; Smith, J.R. Effect of soybean stem growth habit on height and node number after beginning bloom in the midsouthern USA. Crop Sci. 2004, 44, 1855–1858. [Google Scholar] [CrossRef]
- Specht, J.E.; Chase, K.; Macrander, M.; Graef, G.L.; Chung, J.; Markwell, J.P.; Germann, M.; Orf, J.H.; Lark, K.G. Soybean response to water: A QTL analysis of drought tolerance. Crop Sci. 2001, 41, 493–509. [Google Scholar] [CrossRef]

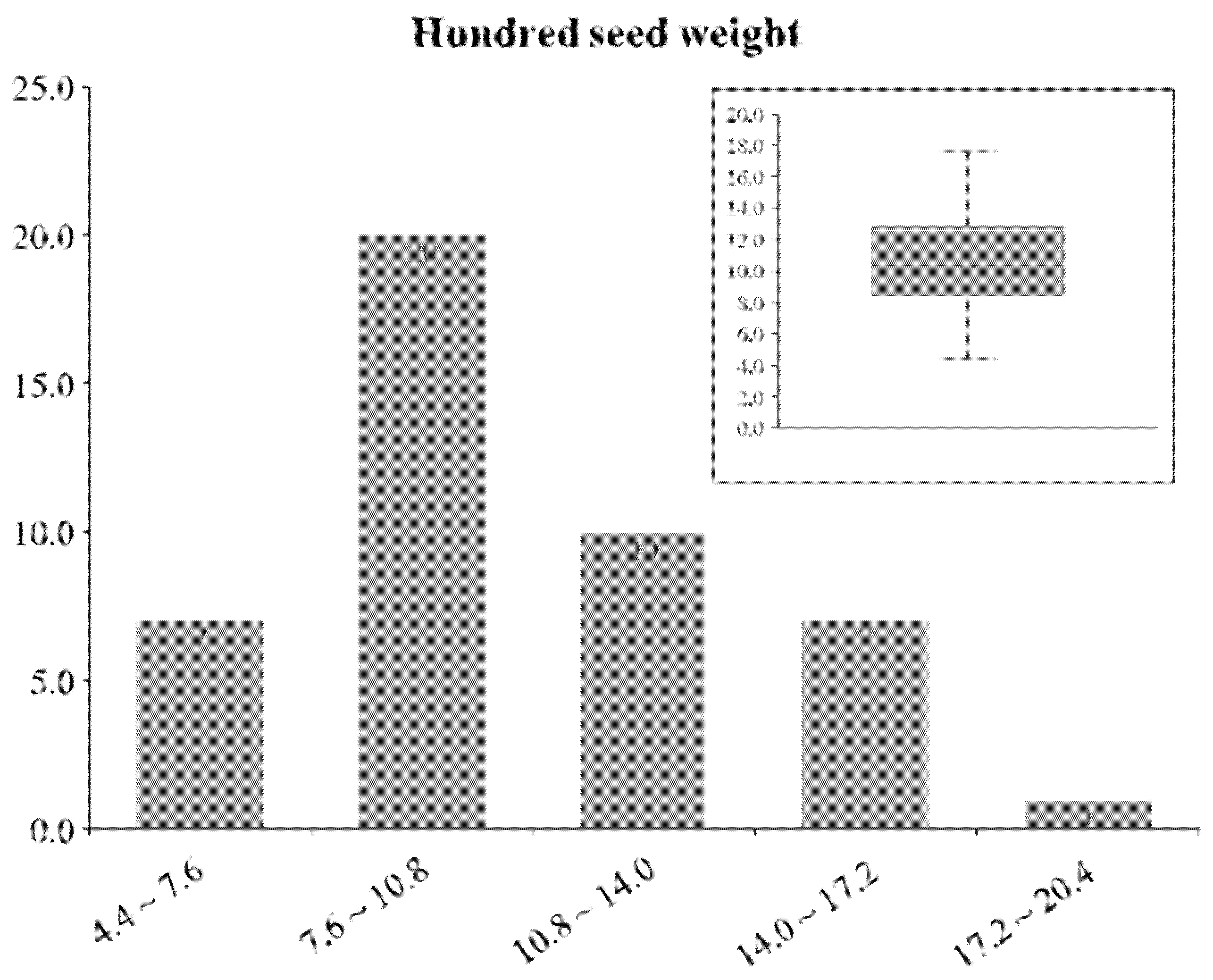

{kind=link}
{kind=link}
{kind=link}
{kind=link}
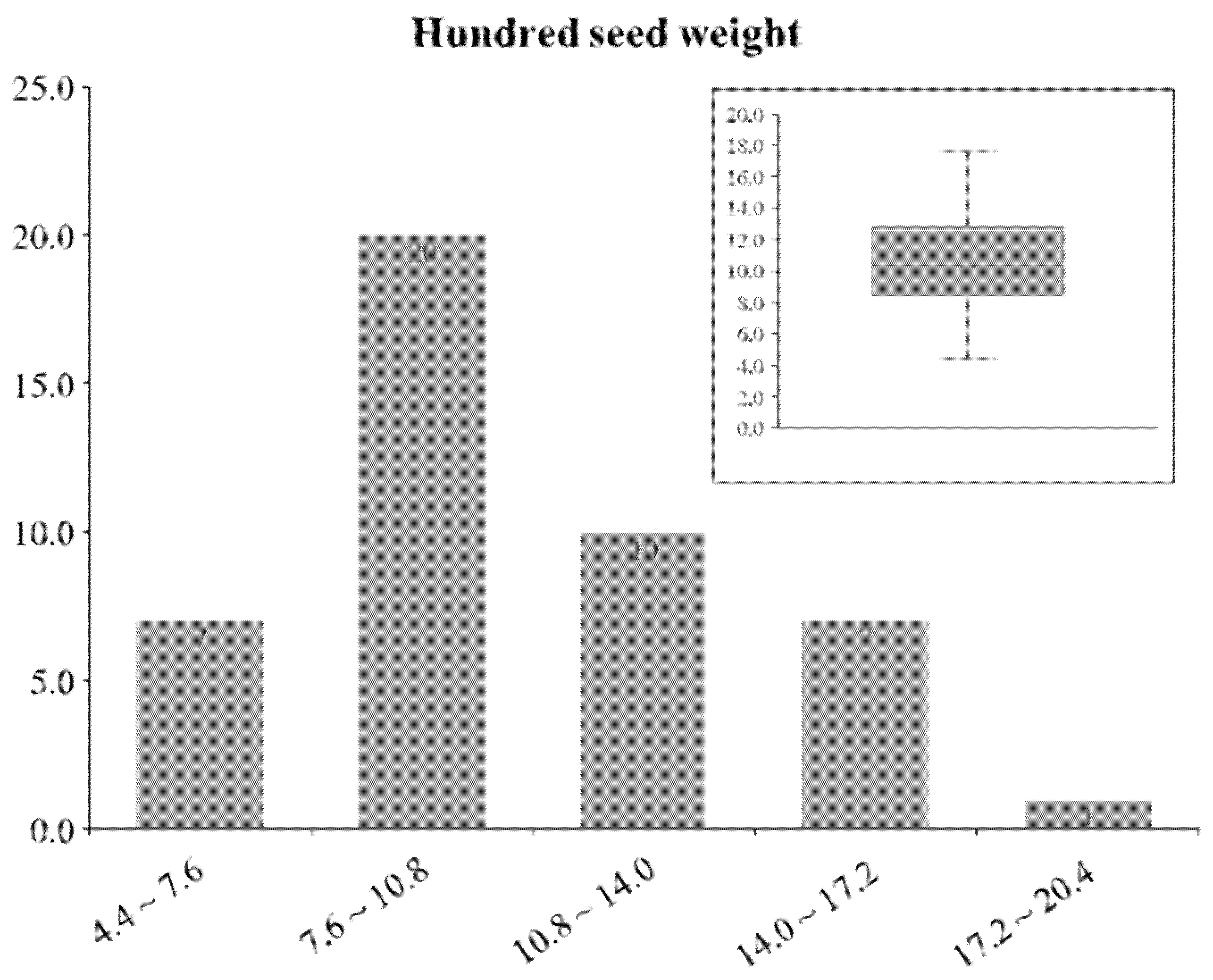{kind=link}
| Trait | SNP | Chromosome | Position | log10(p) | R2 of Model without SNP | R2 of Model without SNP | Minor Allele Frequency | Allelic Effect | LD Block |
|---|---|---|---|---|---|---|---|---|---|
| Plant height | Gm19_45000827 | 19 | 45,204,441 | 10.8 | 0.74 | 0.76 | 0.06 | −22.2 | 44,481,150–46,730,263 |
| Gm19_45441251 | 19 | 45,557,751 | 6.1 | 0.74 | 0.75 | 0.06 | −17.5 | 44,481,150–46,730,263 | |
| Number of nodes | Gm17_3252095 | 17 | 3,244,333 | 6.3 | 0.72 | 0.74 | 0.03 | −3.3 | 3,244,333–7,487,007 |
| Gm18_2284997 | 18 | 2,294,554 | 5.8 | 0.72 | 0.74 | 0.02 | −2.6 | 200,326–4,568,354 | |
| Gm19_45000827 | 19 | 45,204,441 | 9.4 | 0.72 | 0.75 | 0.06 | −2.6 | 44,481,150–46,730,263 | |
| Gm19_45441251 | 19 | 45,557,751 | 8.1 | 0.72 | 0.74 | 0.06 | −2.6 | 44,481,150–46,730,263 | |
| Gm19_45812748 | 19 | 45,930,447 | 5.9 | 0.72 | 0.74 | 0.01 | −2.7 | 44,481,150–46,730,263 | |
| Gm04_1193028 | 4 | 1,232,055 | 6.1 | 0.72 | 0.74 | 0.01 | −2.9 | 321,728–5,658,722 | |
| Hundred seed weight | Gm02_8807595 | 2 | 8,896,955 | 7.3 | 0.83 | 0.84 | 0.05 | 5.3 | 8,487,794–12,414,985 |
| Gm02_8730196 | 2 | 8,819,494 | 6.8 | 0.83 | 0.84 | 0.04 | 5.1 | 8,487,794–12,414,985 | |
| Gm02_10302121 | 2 | 10,381,395 | 5.7 | 0.83 | 0.83 | 0.04 | 4.0 | 8,487,794–12,414,985 | |
| Gm16_31454423 | 16 | 31,822,897 | 5.8 | 0.83 | 0.84 | 0.05 | 3.8 | 31,454,423–32,838,190 | |
| Gm16_31710278 | 16 | 32,078,578 | 5.7 | 0.83 | 0.83 | 0.06 | 4.0 | 31,454,423–32,838,190 | |
| Gm16_32200441 | 16 | 32,698,542 | 5.6 | 0.83 | 0.83 | 0.18 | 3.0 | 31,454,423–32,838,190 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, H.; Lee, J.Y.; Lee, J.-D. Genome-Wide Association Mapping for Seed Weight in Soybean with Black Seed Coats and Green Cotyledons. Agronomy 2022, 12, 250. https://doi.org/10.3390/agronomy12020250
Jo H, Lee JY, Lee J-D. Genome-Wide Association Mapping for Seed Weight in Soybean with Black Seed Coats and Green Cotyledons. Agronomy. 2022; 12(2):250. https://doi.org/10.3390/agronomy12020250
Chicago/Turabian StyleJo, Hyun, Ji Yun Lee, and Jeong-Dong Lee. 2022. "Genome-Wide Association Mapping for Seed Weight in Soybean with Black Seed Coats and Green Cotyledons" Agronomy 12, no. 2: 250. https://doi.org/10.3390/agronomy12020250
APA StyleJo, H., Lee, J. Y., & Lee, J.-D. (2022). Genome-Wide Association Mapping for Seed Weight in Soybean with Black Seed Coats and Green Cotyledons. Agronomy, 12(2), 250. https://doi.org/10.3390/agronomy12020250