


Genome-Wide Association Study for Non-Photochemical Quenching Traits in Oryza sativa L.


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Experimental Design and Measurement of NPQ(T)
2.3. DNA Extraction, Sequencing and Data Processing
2.4. Population Genetics Analysis
2.5. Genome-Wide Association Analyses
2.6. Analysis of Candidate Genes
3. Result
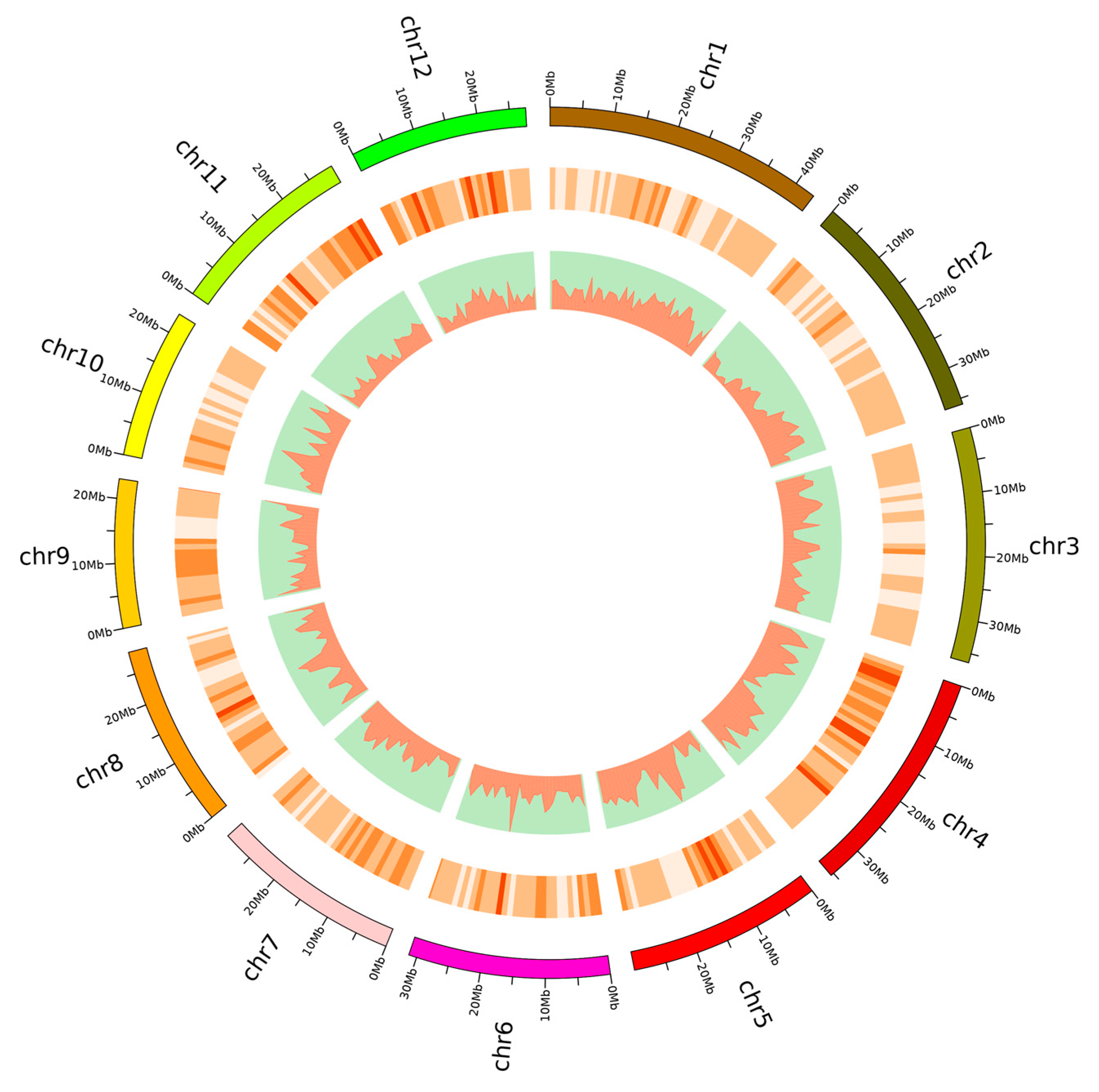
3.1. Genome Resequencing of 92 Rice Varieties
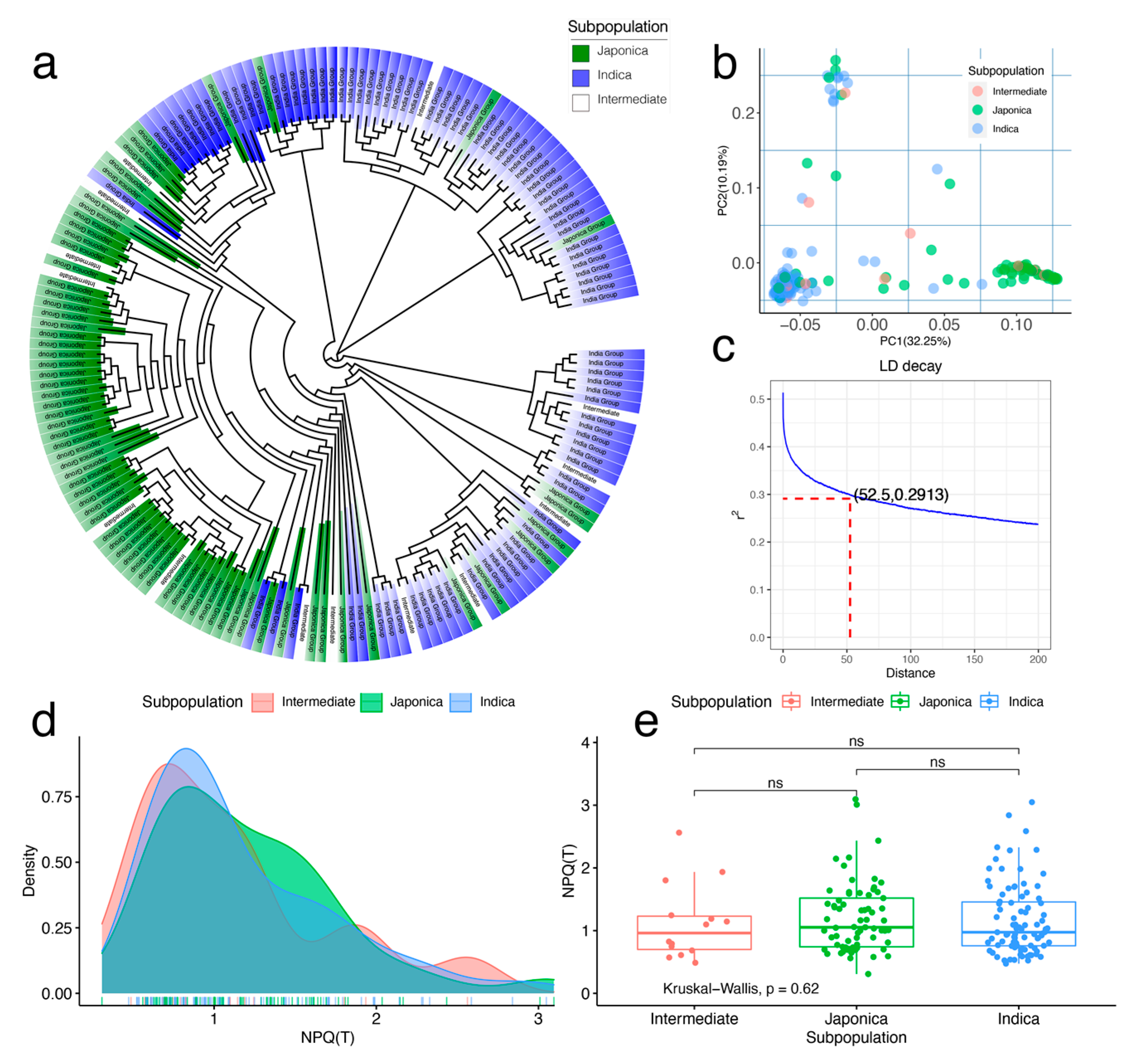
3.2. Population Structure, Linkage Disequilibrium, and Phenotypic Variation
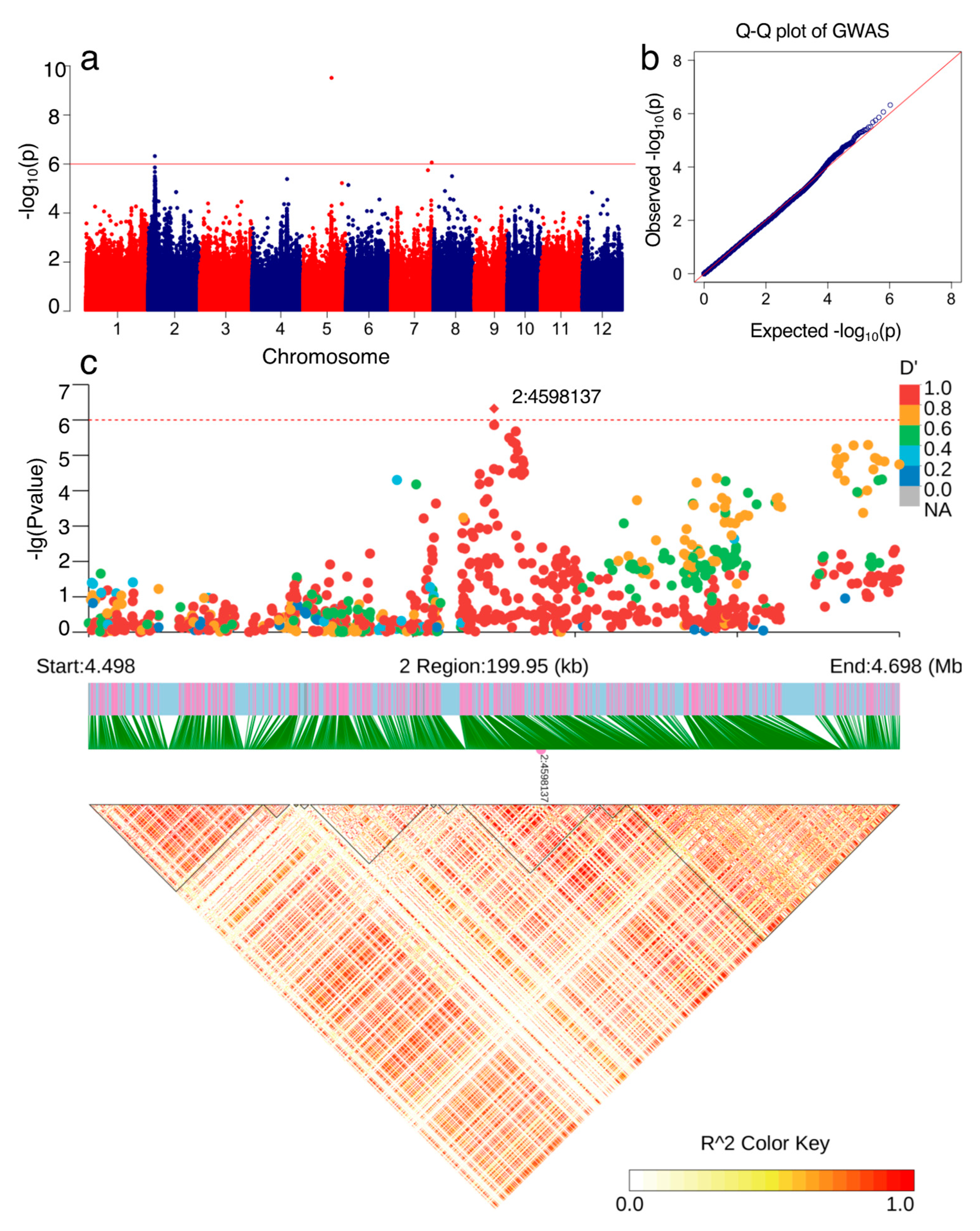
3.3. Genome-Wide Association Analysis (GWAS) for NPQ(T)
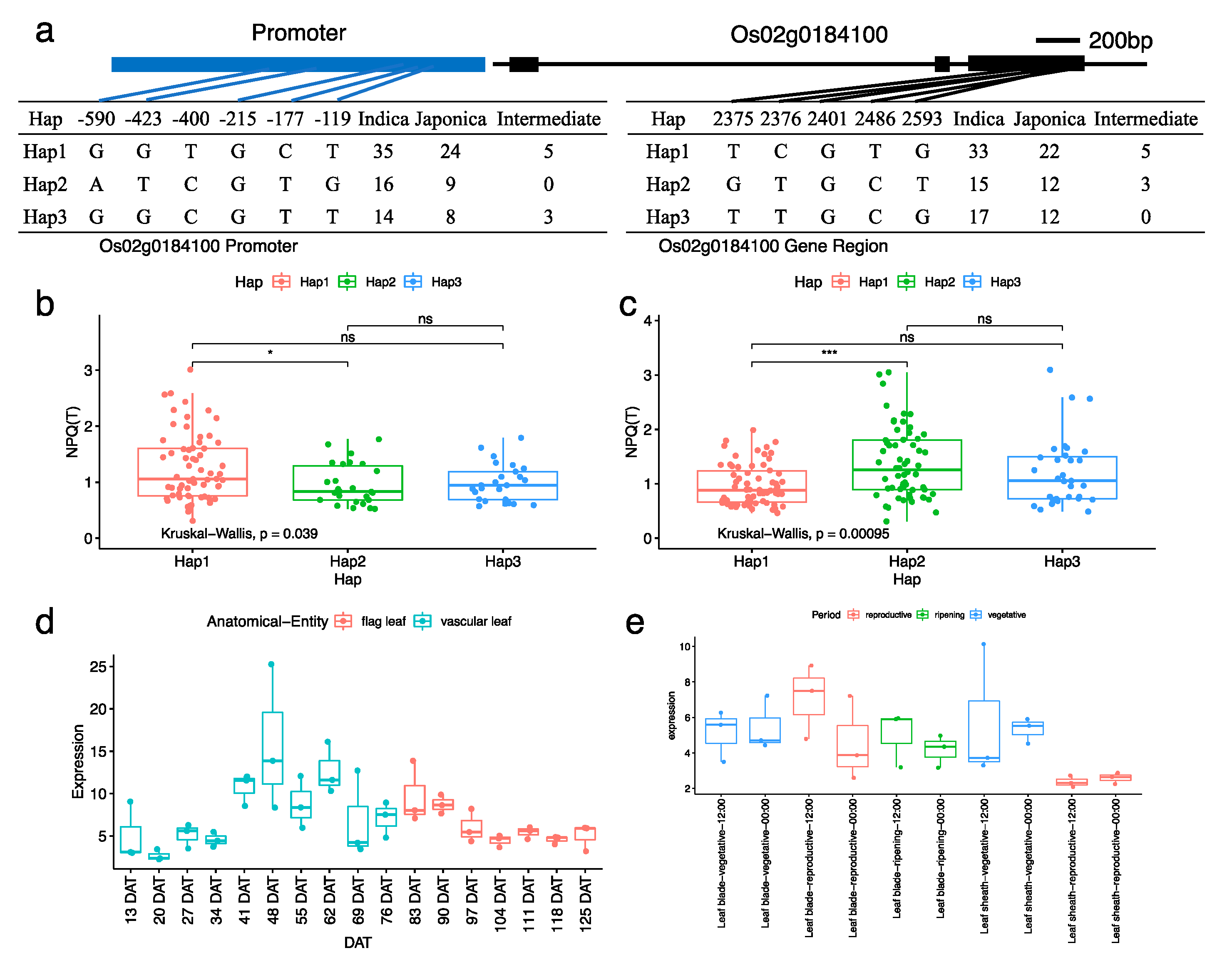
3.4. Candidate Genes Underlying the Associated Signal on Chromosome 02
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| LD | Linkage disequilibrium |
| NPQ(T)/NPQ | Non-photochemical quenching |
| SNP | Single-nucleotide polymorphism |
References
- Long, S.P.; Marshall-Colon, A.; Zhu, X.-G. Meeting the Global Food Demand of the Future by Engineering Crop Photosynthesis and Yield Potential. Cell 2015, 161, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hedden, P. The genes of the Green Revolution. Trends Genet. 2003, 19, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Long, S.P.; Zhu, X.G.; Naidu, S.L.; Ort, D.R. Can Improvement in Photosynthesis Increase Crop Yields? Plant Cell Environ. 2006, 29, 315–330. [Google Scholar] [CrossRef]
- Zhu, X.-G.; Long, S.; Ort, D.R. Improving Photosynthetic Efficiency for Greater Yield. Annu. Rev. Plant Biol. 2010, 61, 235–261. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Siaca, L.G.; Coe, R.; Wang, Y.; Kromdijk, J.; Quick, W.P.; Long, S.P. Variation in photosynthetic induction between rice accessions and its potential for improving productivity. New Phytol. 2020, 227, 1097–1108. [Google Scholar] [CrossRef]
- Ambavaram, M.M.R.; Basu, S.; Krishnan, A.; Ramegowda, V.; Batlang, U.; Rahman, L.; Baisakh, N.; Pereira, A.D.C. Coordinated regulation of photosynthesis in rice increases yield and tolerance to environmental stress. Nat. Commun. 2014, 5, 5302. [Google Scholar] [CrossRef]
- Gutteridge, S. The Impact of a Changing Atmosphere on Chloroplast Function, Photosynthesis, Yield, and Food Security. Essays Biochem. 2018, 62, 1–11. [Google Scholar] [CrossRef]
- Baker, N.R. Chlorophyll fluorescence: A probe of photosynthesis in vivo. Annu. Rev. Plant Biol. 2008, 59, 89–113. [Google Scholar] [CrossRef]
- Murchie, E.; Lawson, T. Chlorophyll fluorescence analysis: A guide to good practice and understanding some new applications. J. Exp. Bot. 2013, 64, 3983–3998. [Google Scholar] [CrossRef]
- Urban, L.; Aarrouf, J.; Bidel, L.P.R. Assessing the Effects of Water Deficit on Photosynthesis Using Parameters Derived from Measurements of Leaf Gas Exchange and of Chlorophyll a Fluorescence. Front. Plant Sci. 2017, 8, 2068. [Google Scholar] [CrossRef]
- Muller, P.; Li, X.-P.; Niyogi, K.K. Non-Photochemical Quenching. A Response to Excess Light Energy. Plant Physiol. 2001, 125, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Ruban, A.V.; Berera, R.; Ilioaia, C.; Van Stokkum, I.H.M.; Kennis, J.T.M.; Pascal, A.; van Amerongen, H.; Robert, B.; Horton, P.; Van Grondelle, R. Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 2007, 450, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Ruban, A.V.; Johnson, M.P.; Duffy, C.D. The photoprotective molecular switch in the photosystem II antenna. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 167–181. [Google Scholar] [CrossRef]
- Rascher, U.; Liebig, M.; Luttge, U. Evaluation of instant light-response curves of chlorophyll fluorescence parameters obtained with a portable chlorophyll fluorometer on site in the field. Plant Cell Environ. 2000, 23, 1397–1405. [Google Scholar] [CrossRef]
- Tietz, S.; Hall, C.C.; Cruz, J.A.; Kramer, D.M. Npq (T): A Chlorophyll Fluorescence Parameter for Rapid Estimation and Imaging of Non-Photochemical Quenching of Excitons in Photosystem-Ii-Associated Antenna Com-plexes. Plant Cell Environ. 2017, 40, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; Li, M.; et al. Genome-Wide Association Studies of 14 Agronomic Traits in Rice Landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 2013, 14, 483–495. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-Wide Asso-ciation Mapping Reveals a Rich Genetic Architecture of Complex Traits in Oryza Sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef]
- Niyogi, K.K.; Grossman, A.R.; Bjorkman, O. Arabidopsis Mutants Define a Central Role for the Xanthophyll Cycle in the Regulation of Photosynthetic Energy Conversion. Plant Cell 1998, 10, 1121. [Google Scholar] [CrossRef]
- Rungrat, T.; Almonte, A.A.; Cheng, R.; Gollan, P.J.; Stuart, T.; Aro, E.M.; Borevitz, J.O.; Pogson, B.; Wilson, P.B. A Ge-nome-Wide Association Study of Non-Photochemical Quenching in Response to Local Seasonal Climates in Arabidopsis Tha-liana. Plant Direct. 2019, 5, e00138. [Google Scholar]
- Shamim, M.J.; Kaga, A.; Tanaka, Y.; Yamatani, H.; Shiraiwa, T. Analysis of Physiological Variations and Genetic Archi-tecture for Photosynthetic Capacity of Japanese Soybean Germplasm. Front. Plant Sci. 2022, 13, 910527. [Google Scholar] [CrossRef] [PubMed]
- Kasajima, I.; Ebana, K.; Yamamoto, T.; Takahara, K.; Yano, M.; Kawai-Yamada, M.; Uchimiya, H. Molecular Distinction in Genetic Regulation of Nonphotochemical Quenching in Rice. Proc. Natl. Acad. Sci. USA 2011, 33, 13835–13840. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Z.; Li, Z.; Zhang, Q.; Hu, J.; Xiao, Y.; Cai, D.; Wu, J.; King, G.J.; Li, H.; et al. Dissection of the genetic architecture of three seed-quality traits and consequences for breeding in Brassica napus. Plant Biotechnol. J. 2017, 16, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, C.; Li, Y.; Liao, S.; Cheng, H.; Tu, Y.; Zhu, X.; Chen, K.; He, Y.; Wang, G. The Coordination of Osbzip72 and Osmybs2 with Reverse Roles Regulates the Transcription Ofospsbs1in Rice. New Phytol. 2021, 229, 370–387. [Google Scholar] [CrossRef] [PubMed]
- Kuhlgert, S.; Austic, G.; Zegarac, R.; Osei-Bonsu, I.; Hoh, D.; Chilvers, M.I.; Roth, M.G.; Bi, K.; TerAvest, D.; Weebadde, P.; et al. MultispeQ Beta: A tool for large-scale plant phenotyping connected to the open PhotosynQ network. R. Soc. Open Sci. 2016, 3, 160592. [Google Scholar] [CrossRef]
- Uzunova, M.; Ecke, W.; Weissleder, K.; Röbbelen, G. Mapping the genome of rapeseed (Brassica napus L.). I. Construction of an RFLP linkage map and localization of QTLs for seed glucosinolate content. Theor. Appl. Genet. 1995, 90, 194–204. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A Mapreduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. Ensemblcompara Genetrees: Complete, Duplica-tion-Aware Phylogenetic Trees in Vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007; 81, 559–575. [Google Scholar]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Murchie, E.H.; Ruban, A.V. Dynamic Non-Photochemical Quenching in Plants: From Molecular Mechanism to Produc-tivity. Plant J. 2020, 101, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.; Lepetit, B. Biodiversity of NPQ. J. Plant Physiol. 2015, 172, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Ruban, A.V. Nonphotochemical Chlorophyll Fluorescence Quenching: Mechanism and Effectiveness in Protecting Plants from Photodamage. Plant Physiol. 2016, 170, 1903–1916. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, H.; Jiang, J.; Xu, J.; Xie, W.; Fu, X.; Liu, C.; He, Y.; Wang, G. Genetic Architecture of Natural Variation in Rice Nonphotochemical Quenching Capacity Revealed by Genome-Wide Association Study. Front. Plant Sci. 2017, 8, 1773. [Google Scholar] [CrossRef]
- Bender-Machado, L.; Bäuerlein, M.; Carrari, F.; Schauer, N.; Lytovchenko, A.; Gibon, Y.; Müller-röber, B. Expression of a Yeast Acetyl Coa Hydrolase in the Mitochondrion of Tobacco Plants Inhibits Growth and Restricts Photosynthesis. Plant Mol. Biol. 2004, 55, 645–662. [Google Scholar] [CrossRef]
- Shimizu, R.; Dempo, Y.; Nakayama, Y.; Nakamura, S.; Bamba, T.; Fukusaki, E.; Fukui, T. New Insight into the Role of the Calvin Cycle: Reutilization of Co2 Emitted through Sugar Degradation (Vol 5, 11617, 2015). Sci. Rep. 2016, 6, 27961. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, F.; Wang, Y.; Xue, H.; Jiang, Q.; Shi, J.; Dai, H.; Zhang, Z.; Li, L.; He, P.; et al. MdHAL3, a 4′-phosphopantothenoylcysteine decarboxylase, is involved in the salt tolerance of autotetraploid apple. Plant Cell Rep. 2020, 39, 1479–1491. [Google Scholar] [CrossRef]
- Miloslavina, Y.; de Bianchi, S.; Dall’Osto, L.; Bassi, R.; Holzwarth, A.R. Quenching in Arabidopsis thaliana Mutants Lacking Monomeric Antenna Proteins of Photosystem II. J. Biol. Chem. 2011, 286, 36830–36840. [Google Scholar] [CrossRef] [PubMed]
- Rutkauskas, D.; Chmeliov, J.; Johnson, M.; Ruban, A.; Valkunas, L. Exciton annihilation as a probe of the light-harvesting antenna transition into the photoprotective mode. Chem. Phys. 2012, 404, 123–128. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subpopulation | No. of Line | Mean | SD | Median | Max. | Min. |
|---|---|---|---|---|---|---|
| Indica | 90 | 1.16 | 0.56 | 0.97 | 3.05 | 0.47 |
| Intermediate | 14 | 1.12 | 0.55 | 1.05 | 3.1 | 0.31 |
| Japonica | 69 | 1.21 | 0.6 | 0.96 | 2.56 | 0.49 |
| All | 173 | 1.18 | 0.56 | 1.02 | 3.1 | 0.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Liu, S.; Xiong, D.; Xiong, Z.; Zhang, Z.; Wang, F.; Huang, J. Genome-Wide Association Study for Non-Photochemical Quenching Traits in Oryza sativa L. Agronomy 2022, 12, 3216. https://doi.org/10.3390/agronomy12123216
Wei Y, Liu S, Xiong D, Xiong Z, Zhang Z, Wang F, Huang J. Genome-Wide Association Study for Non-Photochemical Quenching Traits in Oryza sativa L. Agronomy. 2022; 12(12):3216. https://doi.org/10.3390/agronomy12123216
Chicago/Turabian StyleWei, Youbo, Sicheng Liu, Dongliang Xiong, Zhuang Xiong, Zuolin Zhang, Fei Wang, and Jianliang Huang. 2022. "Genome-Wide Association Study for Non-Photochemical Quenching Traits in Oryza sativa L." Agronomy 12, no. 12: 3216. https://doi.org/10.3390/agronomy12123216
APA StyleWei, Y., Liu, S., Xiong, D., Xiong, Z., Zhang, Z., Wang, F., & Huang, J. (2022). Genome-Wide Association Study for Non-Photochemical Quenching Traits in Oryza sativa L. Agronomy, 12(12), 3216. https://doi.org/10.3390/agronomy12123216