
Structural Variations in the Genome of Potato Varieties of the Ural Selection



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Genomic DNA Isolation and Purification
- 5 μL of RNase Cocktail™ Enzyme Mix by Invitrogen (Carlsbad, CA, USA) was mixed with 100 μL of eluate in a 2 mL tube and incubated for 1 h at 37 °C;
- At the end of incubation, 180 μL of AMPure XP by Beckman Coulter (Bray, CA, USA) was added to the eluate, mixed gently by flicking the tube, and drops were then separated by spinning, and the tubes incubated for 5 min;
- The tubes were placed on a magnetic rack until discoloration of the liquid was observed. Then the supernatant was removed;
- The precipitate was washed twice with 300 μL of freshly prepared 70% alcohol;
- Purified DNA was eluted in 100 μL of nuclease-free water by NEB (Ipswich, MA, USA), mixed gently by flicking the tube, and incubated for 5 min, after dissolving the precipitate.
2.3. Sequencing of Genomic DNA
2.4. Bioinformatic Analysis
2.4.1. Data Filtering
2.4.2. SVs Calling
2.4.3. SV-Gene Matching
3. Results
3.1. Alignment of Three Potato Varieties’ Genomes against Reference
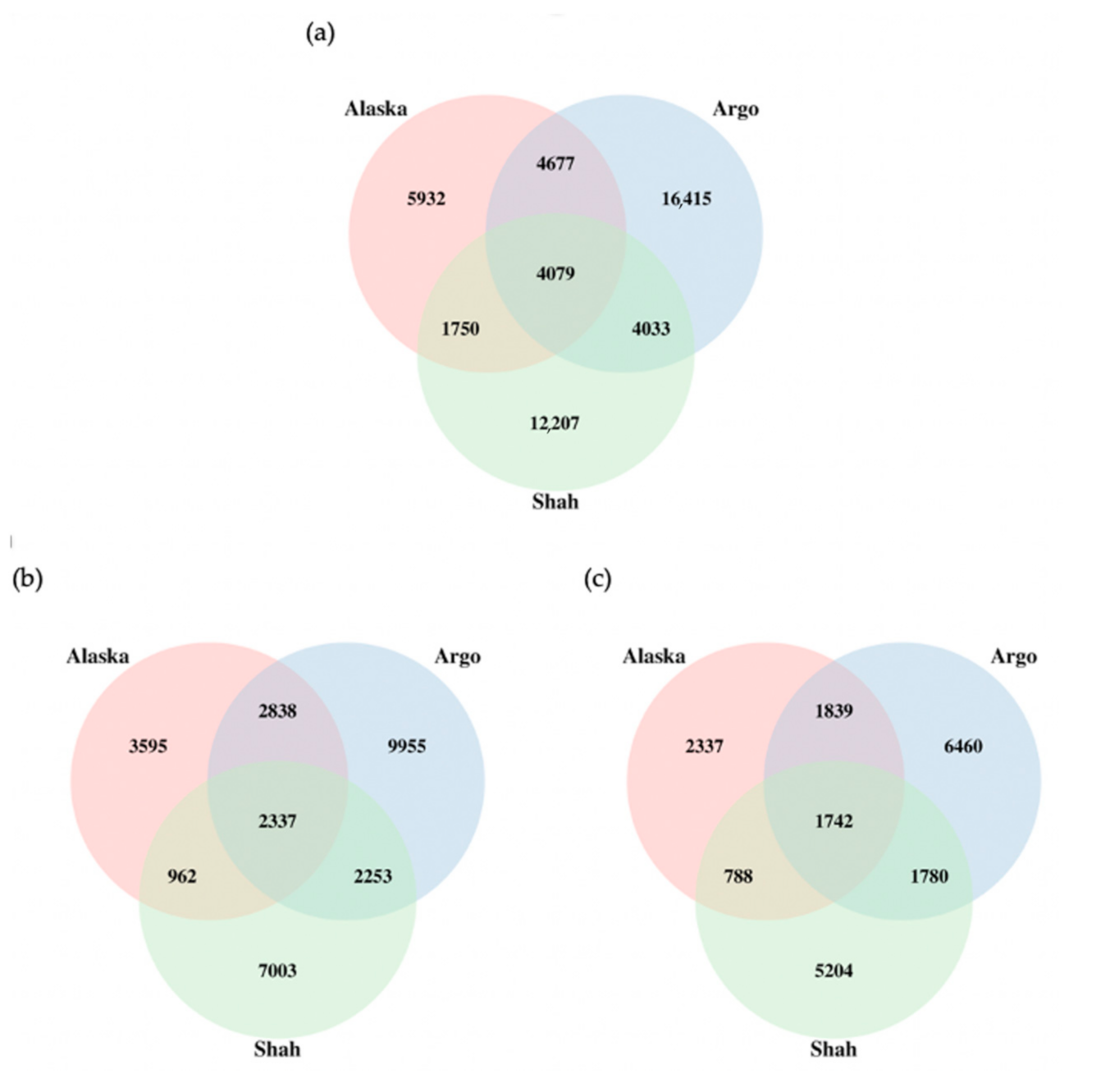
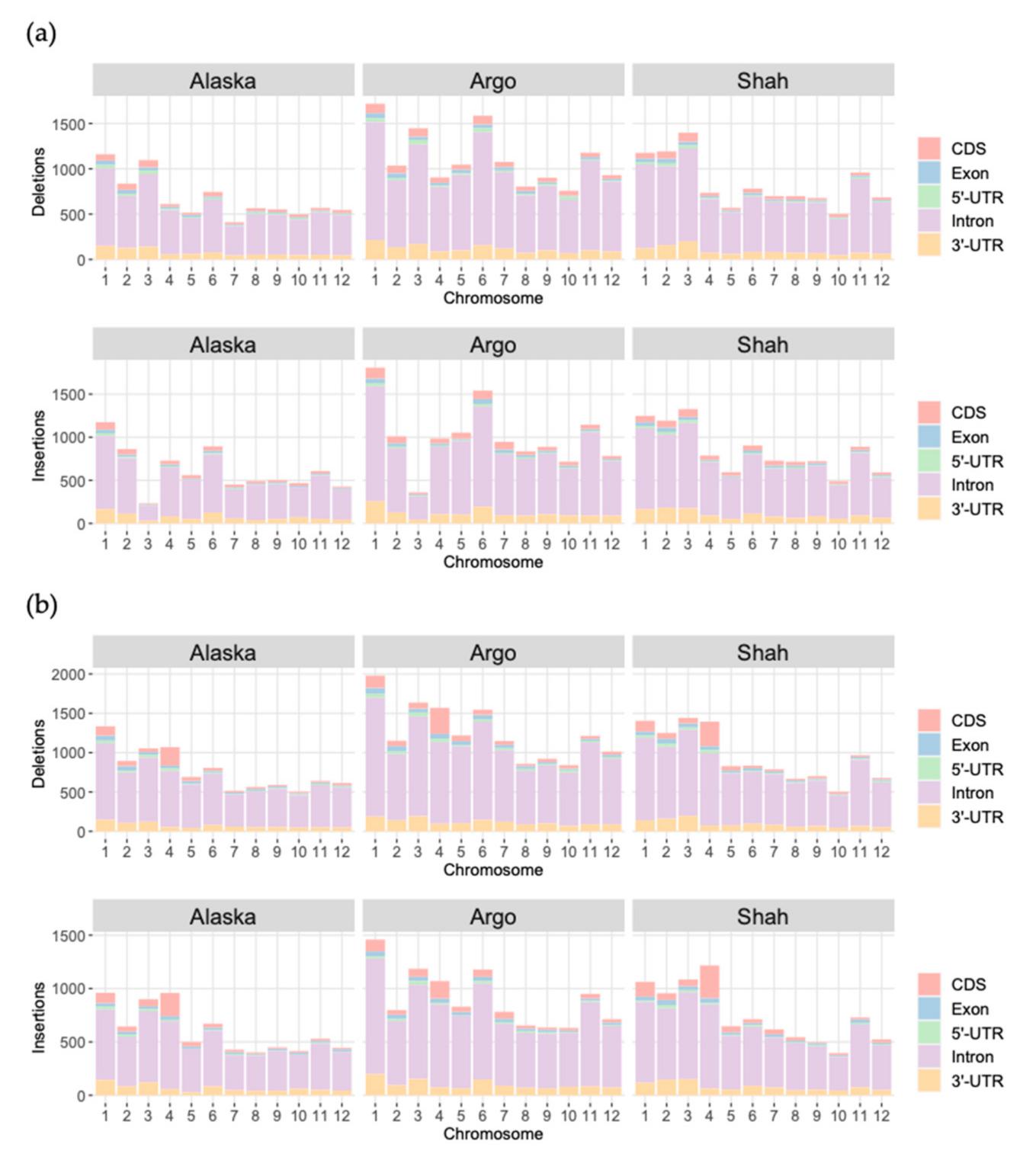
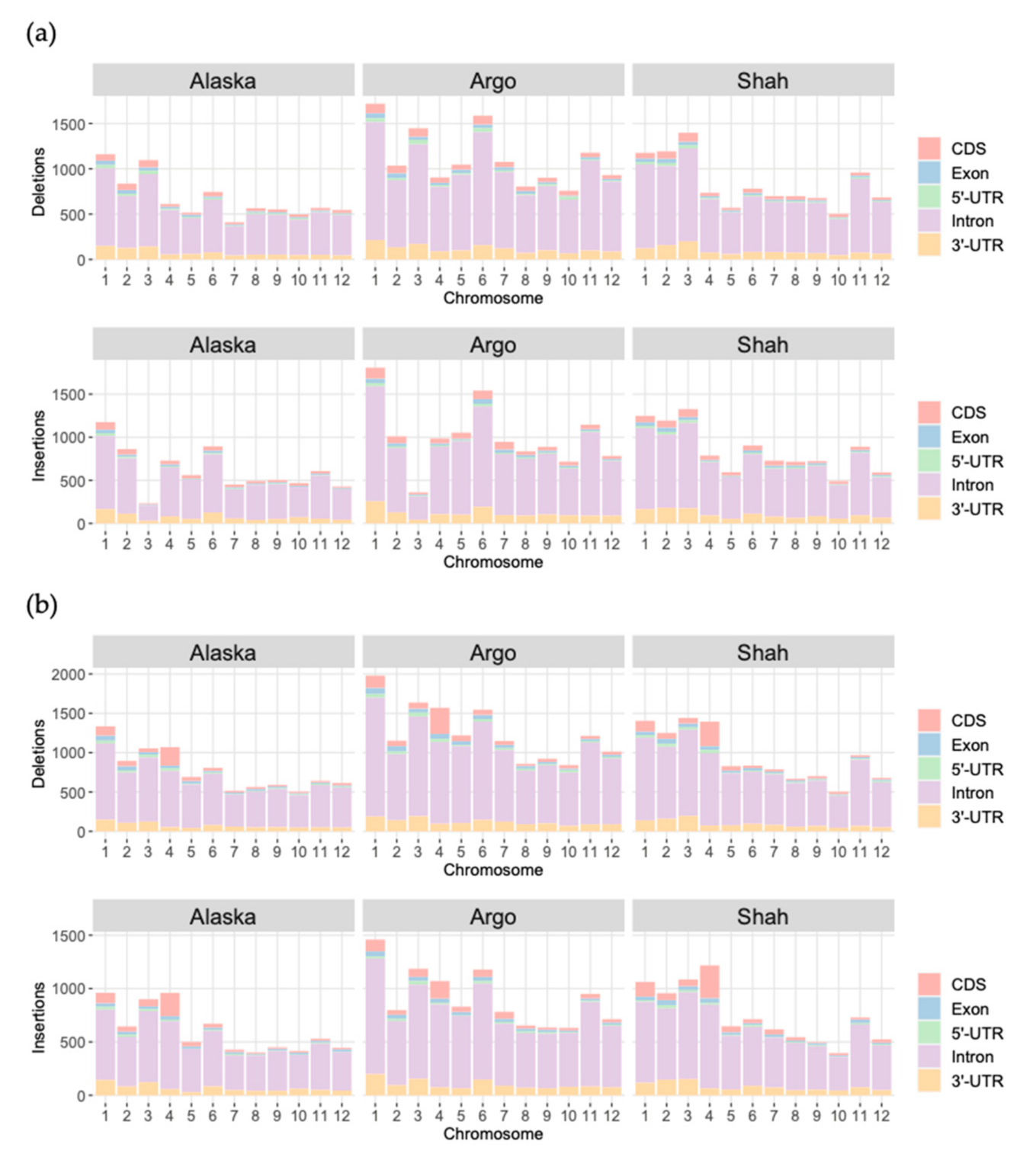
3.2. Finding Structural Variants
3.3. Structural Variants into Coding Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ghislain, M.; Douches, D.S. The genes and genomes of the potato. In The Potato Crop; Campos, H., Ortiz, O., Eds.; Springer: Cham, Switzerland, 2020; pp. 139–162. [Google Scholar]
- Plaisted, R.L.; Hoopes, R.W. The past record and future prospects for the use of exotic potato germplasm. Am. Potato J. 1989, 66, 603–627. [Google Scholar] [CrossRef]
- Sood, S.; Bhardwaj, V.; Pandey, S.K.; Chakrabarti, S.K. History of potato breeding: Improvement diversification and diversity. In The Potato Genome; Xie, C., Tiwari, J.K., Eds.; Springer: Cham, Switzerland, 2017; pp. 31–72. [Google Scholar]
- Bradshaw, J.; Ramsay, G. Utilisation of the commonwealth potato collection in potato breeding. Euphytica 2005, 146, 9–19. [Google Scholar] [CrossRef]
- Simko, I.; Jansky, S.; Stephenson, S.; Spooner, D. Genetics of resistance to pests and disease. In Potato Biology and Biotechnology; Vreugdenhil, D., Bradshaw, J., Gebhardt, C., Govers, F., MacKerron, D.K.L., Taylor, M.A., Ross, H.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 117–155. [Google Scholar]
- Bradshaw, J.E. Potato breeding at the scottish plant breeding station and the scottish crop research institute: 1920–2008. Potato Res. 2009, 52, 141–172. [Google Scholar] [CrossRef]
- Finkers-Tomczak, A.; Bakker, E.; de Boer, J.; van der Vossen, E.; Achenbach, U.; Golas, T.; Suryaningrat, S.; Smant, G.; Bakker, J.; Goverse, A. Comparative sequence analysis of the potato cyst nematode resistance locus H1 reveals a major lack of co-linearity between three haplotypes in potato (Solanum tuberosum ssp.). Theor. Appl. Genet. 2011, 122, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, J.E. Review and analysis of limitations in ways to improve conventional potato breeding. Potato Res. 2017, 60, 171–193. [Google Scholar] [CrossRef]
- Jansky, S.H.; Spooner, D.M. The evolution of potato breeding. Plant Breed. Rev. 2017, 41, 169–211. [Google Scholar]
- Gebhardt, C. Bridging the gap between genome analysis and precision breeding in potato. Trends Genet. 2013, 29, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bhardwaj, V.; Dalamu, D.; Kaushik, S.K.; Singh, B.P.; Sharma, S.K.; Umamaheshwari, R.; Baswaraj, R.; Kumar, V.; Gebhardt, C. Identification of elite potato genotypes possessing multiple disease resistance genes through molecular approaches. Sci. Hortic. 2014, 179, 204–211. [Google Scholar] [CrossRef]
- Kasai, K.; Morikawa, Y.; Sorri, V.A.; Valkonen, J.P.; Gebhardt, C.; Watanabe, K.N. Development of SCAR markers to the PVY resistance gene Ryadg based on a common feature of plant disease resistance genes. Genome 2000, 43, 1–8. [Google Scholar] [CrossRef]
- Mori, K.; Sakamoto, Y.; Mukojima, N.; Tamiya, S.; Nakao, T.; Ishii, T.; Hosaka, K. Development of a multiplex PCR method for simultaneous detection of diagnostic DNA markers of five disease and pest resistance genes in potato. Euphytica 2011, 180, 347–355. [Google Scholar] [CrossRef]
- Song, Y.S.; Hepting, L.; Schweize, G.; Hartl, L.; Wenzel, G.; Schwarzfischer, A. Mapping of extreme resistance to PVY (Ry (sto)) on chromosome XII using anther-culture-derived primary dihaploid potato lines. Theor. Appl. Genet. 2005, 111, 879–887. [Google Scholar] [CrossRef]
- Ohbayashi, K.; Nakata, N.; Chaya, M.; Komura, K. Development of a detection method of resistance to potato disease and pest using DNA markers. 1. Detection methods of resistance to potato virus X, potato cyst nematode and late blight. Bull. Nagasaki Agri. Fore. Technol. Dev. Cen. 2010, 1, 1–26. [Google Scholar]
- Asano, K.; Kobayashi, A.; Tsuda, S.; Nishinaka, M.; Tamiya, S. DNA marker-assisted evaluation of potato genotypes for potential resistance to potato cyst nematode pathotypes not yet invading into Japan. Breed. Sci. 2012, 62, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.; Lopez-Vizcon, C. Application of molecular marker-assisted selection (MAS) for disease resistance in a practical potato breeding programme. Potato Res. 2012, 55, 1–13. [Google Scholar] [CrossRef]
- Wang, M.; Allefs, S.; van den Berg, R.G.; Vleeshouwers, V.G.; van der Vossen, E.A.; Vosman, B. Allele mining in Solanum: Conserved homologues of Rpi-blb1 are identified in Solanum stoloniferum. Theor. Appl. Genet. 2008, 116, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, Y.; Vossen, J.H.; Visser, R.G.F.; Jacobsen, E. Functional stacking of three resistance genes against Phytophthora infestans in potato. Transgenic Res. 2012, 21, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Bellin, D.; Henselewski, H.; Lehmann, W.; Schwarzfischer, J.; Valkonen, J.P. Marker-assisted combination of major genes for pathogen resistance in potato. Theor. Appl. Genet. 2006, 112, 1458–1464. [Google Scholar] [CrossRef]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195.
- Pham, G.M.; Hamilton, J.P.; Wood, J.C.; Burke, J.T.; Zhao, H.; Vaillancourt, B.; Ou, S.; Jiang, J.; Buell, C.R. Construction of a chromosome-scale long-read reference genome assembly for potato. Gigascience 2020, 9, giaa100. [Google Scholar] [CrossRef]
- Hardigan, M.A.; Crisovan, E.; Hamilton, J.P.; Kim, J.; Laimbeer, P.; Leisner, C.P.; Manrique-Carpintero, N.C.; Newton, L.; Pham, G.M.; Vaillancourt, B.; et al. Genome reduction uncovers a large dispensable genome and adaptive role for copy number variation in asexually propagated Solanum tuberosum. Plant Cell 2016, 28, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Tang, D.; Huang, W.; Yang, Z.; Zhang, Y.; Hamilton, J.P.; Visser, R.G.F.; Bachem, C.W.B.; Robin Buell, C.; Zhang, Z.; et al. Haplotype-resolved genome analyses of a heterozygous diploid potato. Nat. Genet. 2020, 52, 1018–1023. [Google Scholar] [CrossRef]
- Kyriakidou, M.; Achakkagari, S.R.; Gálvez López, J.H.; Zhu, X.; Tang, C.Y.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Structural genome analysis in cultivated potato taxa. Theor. Appl. Genet. 2020, 133, 951–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, A.; Halterman, D. Structural variation within the potato Ve gene locus and correlation with molecular marker analysis. Crop Sci. 2016, 56, 3133–3142. [Google Scholar] [CrossRef]
- Shanina, E.P.; Sergeeva, L.B.; Stafeeva, M.A.; Kurkin, E.M. The using of DNA markers to assess the original breeding material of potatoes. Achiev. Sci. Technol. AIC 2018 32, 47–49.
- Available online: https://community.nanoporetech.com/posts/gupp.y-v4-4-2-patch-release (accessed on 25 February 2021).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Fidel, R.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Heller, D.; Vingron, M. SVIM: Structural variant identification using mapped long reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://solanaceae.plantbiology.msu.edu/dm_v6_1_download.shtml\ (accessed on 15 June 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Knaus, B.J.; Grunwald, N.J. VCFR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, K.; Ludidi, N. Drought and exogenous abscisic acid alter hydrogen peroxide accumulation and differentially regulate the expression of two maize RD22-like genes. Sci. Rep. 2017, 7, 8821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hasegawa, Y.; Lu, Y.; Sato, T. Ubiquitin related enzymes and plant-specific ubiquitin ligase ATL family in tomato plants. Plant Biotechnol. 2017, 34, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Kalunke, R.M.; Tundo, S.; Benedetti, M.; Cervone, F.; De Lorenzo, G.; D’Ovidio, R. An update on polygalacturonase-inhibiting protein (PGIP), a leucine-rich repeat protein that protects crop plants against pathogens. Front. Plant Sci. 2015, 6, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Y.; Song, B.; Liu, X.; Xie, C.; Li, M.; Lin, Y.; Zhang, H.; Liu, J. Promoter regions of potato vacuolar invertase gene in response to sugars and hormones. Plant Physiol. Biochem. 2013, 69, 9–16. [Google Scholar] [CrossRef]
- Tweneboah, S.; Oh, S.-K. Biological roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in solanaceous crops. J. Plant Biotechnol. Korean Soc. Plant Biotechnol. 2017, 44, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Iovene, M.; Zhang, T.; Lou, Q.; Buell, C.R.; Jiang, J. Copy number variation in potato—An asexually propagated autotetraploid species. Plant J. 2013, 75, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Pham, G.M.; Newton, L.; Wiegert-Rininger, K.; Vaillancourt, B.; Douches, D.S.; Buell, C.R. Extensive genome heterogeneity leads to preferential allele expression and copy number-dependent expression in cultivated potato. Plant J. 2017, 92, 624–637. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Parameter | SVIM | Sniffles | ||
|---|---|---|---|---|
| Option | Value | Option | Value | |
| Minimum SV length | --min_sv_size | 3 | −l | 3 |
| Maximum SV length | --max_sv_size | 100,000,000 | — | — |
| Minimum reads number for SV determination | --minimum_depth | 20 | –s | 10 |
| Minimum quality | --min_mapq | 40 | –q | 40 |
| Maximum distance to group SVs together | --segment_gap_tolerance | 5 | –d | 5 |
| Variety | Number of Reads | Total Reads Length, Gbp | Mean Read Length, bp | Max Read Length, bp | Mean Read Quality | Coverage 1 |
|---|---|---|---|---|---|---|
| Alaska | 7,009,345 | 42 | 5992 | 138,417 | 22,5 | 42 |
| Argo | 7,916,456 | 47 | 5937 | 142,819 | 21,3 | 46 |
| Shah | 7,841,739 | 44 | 5611 | 119,045 | 20,8 | 44 |
| Variety | SVIM | Sniffles |
|---|---|---|
| Short SVs | ||
| Alaska | 17,809/16,686/-/- | 20,472/15,000/32/6 |
| Argo | 30,161/27,393/4/- | 33,934/22,806/50/9 |
| Shah | 22,391/22,433/7/- | 25,708/18,674/32/5 |
| Medium SVs | ||
| Alaska | 28/-/-/- | 207/1/9/8 |
| Argo | 55/-/-/- | 315/-/14/11 |
| Shah | 42/-/-/- | 220/-/9/7 |
| Large SVs | ||
| Alaska | -/-/-/- | 4/-/2/22 |
| Argo | 1/-/-/- | 6/-/2/24 |
| Shah | 3/-/-/- | 3/-/4/12 |
| Variety | SVIM | Sniffles | SVIM–Sniffles |
|---|---|---|---|
| Alaska | 8106/7398/-/- | 9274/7302/9/1 | 4747/3410/-/- |
| Argo | 13,381/12,069/-/- | 15,082/10,886/21/1 | 8236/5884/-/- |
| Shah | 10,070/10,188/3/- | 11,451/8987/12/1 | 6000/4857/-/- |
| Variety | Number of Genes | Number of SVs | ||||
|---|---|---|---|---|---|---|
| Total | >100 kbpSVs | <100 kbpSVs | Total | >100 kbpSVs | <100 kbpSVs | |
| Alaska | 2594/170 | 2540/135 | 54/35 | 29/10 | 3/2 | 26/8 |
| Argo | 1498/731 | 1435/680 | 63/51 | 41/17 | 5/2 | 36/15 |
| Shah | 926/1336 | 892/1305 | 34/31 | 26/13 | 3/2 | 23/11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lihodeevskiy, G.A.; Shanina, E.P. Structural Variations in the Genome of Potato Varieties of the Ural Selection. Agronomy 2021, 11, 1703. https://doi.org/10.3390/agronomy11091703
Lihodeevskiy GA, Shanina EP. Structural Variations in the Genome of Potato Varieties of the Ural Selection. Agronomy. 2021; 11(9):1703. https://doi.org/10.3390/agronomy11091703
Chicago/Turabian StyleLihodeevskiy, Georgiy A., and Elena P. Shanina. 2021. "Structural Variations in the Genome of Potato Varieties of the Ural Selection" Agronomy 11, no. 9: 1703. https://doi.org/10.3390/agronomy11091703
APA StyleLihodeevskiy, G. A., & Shanina, E. P. (2021). Structural Variations in the Genome of Potato Varieties of the Ural Selection. Agronomy, 11(9), 1703. https://doi.org/10.3390/agronomy11091703