Effect of Leptosphaeria maculans Infection on Promoter DNA Methylation of Defence Genes in Brassica napus


 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and L. maculans Isolate
2.2. Sowing and Inoculation
2.3. Sampling
2.4. DNA Extraction, Library Construction and Sequencing
2.5. Sequence Analysis
3. Results and Discussion
3.1. Disease Assessment
3.2. Differentially Methylated Promoters in the Cultivars Westar and Sturt Under Pathogen Attack
3.3. Differentially Methylated Promoters of Defence Genes in Westar and Sturt Cultivars Under Pathogen Attack
3.4. Differentially Methylated Promoters of RGAs in Westar and Sturt Cultivars Under Pathogen Attack
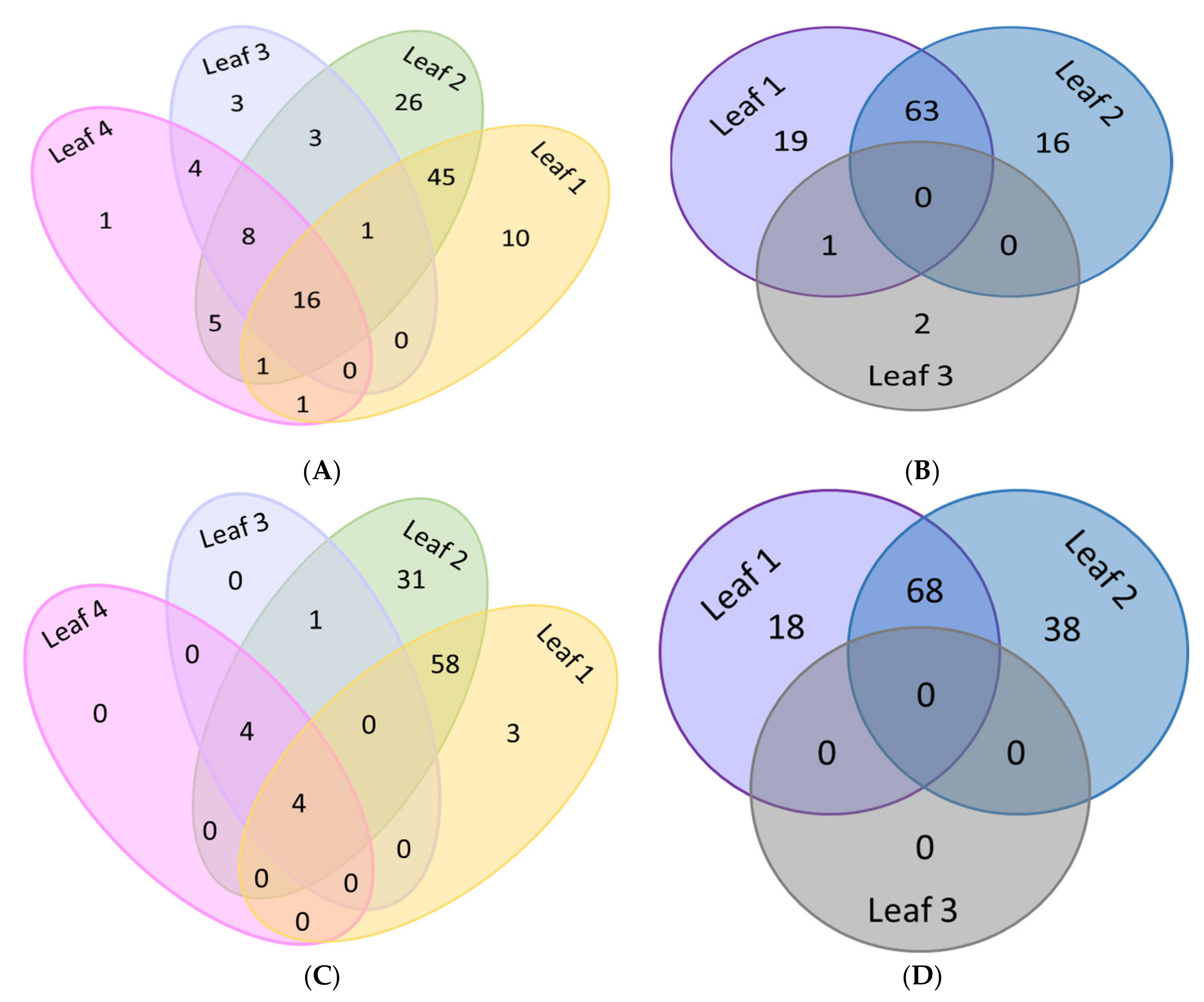
3.5. Comparative Analysis of Differentially Methylated Promoters of Defence Genes Among Leaves
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Avr gene | Avirulence gene |
| AvrLm | Avirulence to Leptosphaeria maculans |
| CC | Coiled-Coil domain |
| CpG | Cytosine-Guanine |
| CHG | Cytosine- H(Cytosine/Thymine/Adenine)- Guanine |
| CHH | Cytosine- H(Cytosine/Thymine/Adenine)- H(Cytosine/Thymine/Adenine) |
| CNL | CC-NBS-LRR |
| DMR | Differentially methylated region |
| DNA | Deoxyribonucleic acid |
| ETI | Effector-Triggered Immunity |
| GO | Gene Ontology |
| LRR | Leucine Rich Repeat |
| RLK | Receptor-Like Kinase |
| RLP | Receptor-Like Proteins |
| NBS | Nucleotide Binding Site |
| NLR | Nucleotide Binding Site- Leucine Rich Repeat |
| PAMP | Pathogen-associated molecular pattern |
| PTI | PAMP-triggered immunity |
| R gene | Resistance gene |
| RGA | Resistance Gene Analog |
| RLP | Receptor-Like Proteins |
| TE | Transposable Elements |
| TIR | Toll/Interleukin−1 Receptor domain |
| TNL | TIR-NBS-LRR |
| WGBS | Whole genome bisulfite sequencing |
References
- Kole, C. Genome Mapping and Molecular Breeding in Plants; Springer: Berlin, Germany, 2007. [Google Scholar]
- Zhang, X.; Fernando, W.D. Insights into fighting against blackleg disease of Brassica napus in Canada. Crop. Pasture Sci. 2018, 69, 40–47. [Google Scholar] [CrossRef]
- Howlett, B.J.; Idnurm, A.; Pedras, M.S.C. Leptosphaeria maculans, the causal agent of blackleg disease of Brassicas. Fungal Genet. Biol. 2001, 33, 1–14. [Google Scholar] [CrossRef]
- Sprague, S.J.; Marcroft, S.J.; Hayden, H.L.; Howlett, B.J. Major gene resistance to blackleg in Brassica napus overcome within three years of commercial production in southeastern Australia. Plant. Dis. 2006, 90, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sekhwal, M.K.; Li, P.; Lam, I.; Wang, X.; Cloutier, S.; You, F.M. Disease resistance gene analogs (RGAs) in plants. Int. J. Mol. Sci. 2015, 16, 19248–19290. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Tsuda, K.; Katagiri, F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant. Biol. 2010, 13, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Flor, H.H. Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 1971, 9, 275–296. [Google Scholar] [CrossRef]
- Yang, X.; Deng, F.; Ramonell, K.M. Receptor-like kinases and receptor-like proteins: Keys to pathogen recognition and defense signaling in plant innate immunity. Front. Biol. 2012, 7, 155–166. [Google Scholar] [CrossRef]
- Zhu, Q.-H.; Shan, W.-X.; Ayliffe, M.A.; Wang, M.-B. Epigenetic mechanisms: An emerging player in plant-microbe interactions. Mol. Plant.-Microbe Interact. 2016, 29, 187–196. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. PNAS USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Chan, S.W.L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Colot, V.; Rossignol, J.L. Eukaryotic DNA methylation as an evolutionary device. BioEssays 1999, 21, 402–411. [Google Scholar] [CrossRef]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Elhamamsy, A.R. DNA methylation dynamics in plants and mammals: Overview of regulation and dysregulation. Cell Biochem. Funct. 2016, 34, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Batley, J. DNA methylation: Toward crop disease resistance improvement. Trends Plant. Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Batley, J. Epigenetics: Potentials and challenges in crop breeding. Mol. Plant. 2019, 12, 1309–1311. [Google Scholar] [CrossRef]
- Wang, J.; Marowsky, N.C.; Fan, C. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS ONE 2014, 9, e110357. [Google Scholar] [CrossRef]
- Do Kim, K.; El Baidouri, M.; Abernathy, B.; Iwata-Otsubo, A.; Chavarro, C.; Gonzales, M.; Libault, M.; Grimwood, J.; Jackson, S.A. A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant. Physiol. 2015, 168, 1433–1447. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Fu, T.; Hu, L.; Xu, C.; Gong, L.; Wendel, J.F.; Liu, B. Gene-body CG methylation and divergent expression of duplicate genes in rice. Sci. Rep. 2017, 7, 2675. [Google Scholar] [CrossRef]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Q.; Kou, H.; Wang, D.; Lin, X.; Wu, Y.; Xu, C.; Xing, S.; Liu, B. Induced Pib expression and resistance to Magnaporthe grisea are compromised by cytosine demethylation at critical promoter regions in rice. J. Integr. Plant. Biol. 2011, 53, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhai, K.; Xie, Z.; Yang, D.; Zhu, X.; Liu, J.; Wang, X.; Qin, P.; Yang, Y.; Zhang, G. Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 2017, 355, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, K.; Katakami, H.; Kim, H.-J.; Ogawa, E.; Sano, C.M.; Wada, Y.; Sano, H. Epigenetic inheritance in rice plants. Ann. Bot. 2007, 100, 205–217. [Google Scholar] [CrossRef]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef]
- Liu, G.; Xia, Y.; Liu, T.; Dai, S.; Hou, X. The DNA methylome and association of differentially methylated regions with differential gene expression during heat stress in Brassica rapa. Int. J. Mol. Sci. 2018, 19, 1414. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Genetic and epigenetic effects of plant–pathogen interactions: An evolutionary perspective. Mol. Plant. 2011, 4, 1014–1023. [Google Scholar] [CrossRef]
- Purwantara, A.; Salisbury, P.A.; Burton, W.A.; Howlett, B.J. Reaction of Brassica juncea (Indian Mustard) Lines to Australian Isolates of Leptosphaeria maculans under Glasshouse and Field Conditions. Eur. J. Plant. Pathol. 1998, 104, 895–902. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.-K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A.P.; et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant. Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef]
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y. Assembly and comparison of two closely related Brassica napus genomes. Plant. Biotechnol. J. 2017, 15, 1602–1610. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Quan, X.; Jia, G.; Xiao, J.; Cloutier, S.; You, F.M. RGAugury: A pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 2016, 17, 852. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Bayer, P.; Inturrisi, F.; Neik, T.; Yang, H.; Dolatabadian, A.; Zhang, F.; Severn-Ellis, A.; Patel, D.; Pradhan, A.; et al. Resistance gene analogs in the Brassicaceae: Identification, characterization, distribution and evolution. Plant. Physiol. 2020, in press. [Google Scholar]
- Yaish, M.W.; Al-Lawati, A.; Al-Harrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukushima, N.; Osabe, K.; Itabashi, E.; Shimizu, M.; Miyaji, N.; Takasaki-Yasuda, T.; Suzuki, Y.; Seki, M.; Fujimoto, R. Identification of DNA methylated regions by using methylated DNA immunoprecipitation sequencing in Brassica rapa. Crop. Pasture Sci. 2018, 69, 107–120. [Google Scholar] [CrossRef]
- Gyetvai, G.; Sønderkær, M.; Göbel, U.; Basekow, R.; Ballvora, A.; Imhoff, M.; Kersten, B.; Nielsen, K.-L.; Gebhardt, C. The transcriptome of compatible and incompatible interactions of potato (Solanum tuberosum) with Phytophthora infestans revealed by DeepSAGE analysis. PLoS ONE 2012, 7, e31526. [Google Scholar] [CrossRef]
- Alvarez, M.E.; Nota, F.; Cambiagno, D.A. Epigenetic control of plant immunity. Mol. Plant. Pathol. 2010, 11, 563–576. [Google Scholar] [CrossRef]
- Yaish, M.W. DNA methylation-associated epigenetic changes in stress tolerance of plants. In Molecular Stress Physiology of Plants; Rout, G.R., Das, A.B., Eds.; Springer: New Delhi, India, 2013; pp. 427–440. [Google Scholar]
- Stokes, T.L.; Kunkel, B.N.; Richards, E.J. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 2002, 16, 171–182. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, M.; He, Y. DNA methylation analysis of the Citrullus lanatus response to Cucumber Green Mottle Mosaic Virus infection by whole-genome bisulfite sequencing. Genes 2019, 10, 344. [Google Scholar] [CrossRef]
- Bayer, P.E.; Golicz, A.A.; Tirnaz, S.; Chan, C.K.K.; Edwards, D.; Batley, J. Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant. Biotechnol. J. 2019, 17, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.G.F.; Soares, M.R.; Gimenez, D.F.J.; Fonseca, L.F.S.; Torrieri, E.; Ramos, E.S.; Giuliatti, S. Detection of differentially methylated regions of irradiated fig tree selections. Sci. Agric. 2017, 74, 285–293. [Google Scholar] [CrossRef]
- Yang, S.; Tang, F.; Caixeta, E.T.; Zhu, H. Epigenetic regulation of a powdery mildew resistance gene in Medicago truncatula. Mol. Plant. 2013, 6, 2000–2003. [Google Scholar] [CrossRef][Green Version]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.-L.; Penterman, J.; Fischer, R.L.; Voinnet, O. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. PNAS USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Alamery, S.; Tirnaz, S.; Bayer, P.; Tollenaere, R.; Chaloub, B.; Edwards, D.; Batley, J. Genome-wide identification and comparative analysis of NBS-LRR resistance genes in Brassica napus. Crop. Pasture Sci. 2018, 69, 72–93. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sturt_Leaf 1 | Sturt_Leaf 2 | Sturt_Leaf 3 | Sturt_Leaf 4 | Westar_Leaf 1 | Westar_Leaf 2 | Westar_Leaf 3 | |
|---|---|---|---|---|---|---|---|
| All differentially methylated promoters | |||||||
| CpG% | 93 | 91 | 92 | 92 | 90 | 90 | 90 |
| CHG% | 5 | 7 | 6 | 6 | 7 | 7 | 7 |
| CHH% | 2 | 2 | 2 | 2 | 3 | 3 | 3 |
| Differentially methylated promoters of defence genes | |||||||
| CpG% | 96 | 96 | 96 | 96 | 94 | 94 | 94 |
| CHG% | 2 | 3 | 3 | 3 | 4 | 5 | 5 |
| CHH% | 2 | 1 | 1 | 1 | 2 | 1 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirnaz, S.; Merce, C.; Bayer, P.E.; Severn-Ellis, A.A.; Edwards, D.; Batley, J. Effect of Leptosphaeria maculans Infection on Promoter DNA Methylation of Defence Genes in Brassica napus. Agronomy 2020, 10, 1072. https://doi.org/10.3390/agronomy10081072
Tirnaz S, Merce C, Bayer PE, Severn-Ellis AA, Edwards D, Batley J. Effect of Leptosphaeria maculans Infection on Promoter DNA Methylation of Defence Genes in Brassica napus. Agronomy. 2020; 10(8):1072. https://doi.org/10.3390/agronomy10081072
Chicago/Turabian StyleTirnaz, Soodeh, Clementine Merce, Philipp E. Bayer, Anita A. Severn-Ellis, David Edwards, and Jacqueline Batley. 2020. "Effect of Leptosphaeria maculans Infection on Promoter DNA Methylation of Defence Genes in Brassica napus" Agronomy 10, no. 8: 1072. https://doi.org/10.3390/agronomy10081072
APA StyleTirnaz, S., Merce, C., Bayer, P. E., Severn-Ellis, A. A., Edwards, D., & Batley, J. (2020). Effect of Leptosphaeria maculans Infection on Promoter DNA Methylation of Defence Genes in Brassica napus. Agronomy, 10(8), 1072. https://doi.org/10.3390/agronomy10081072