Bioinformatic Extraction of Functional Genetic Diversity from Heterogeneous Germplasm Collections for Crop Improvement


Abstract
:1. Introduction
2. Material and Methods
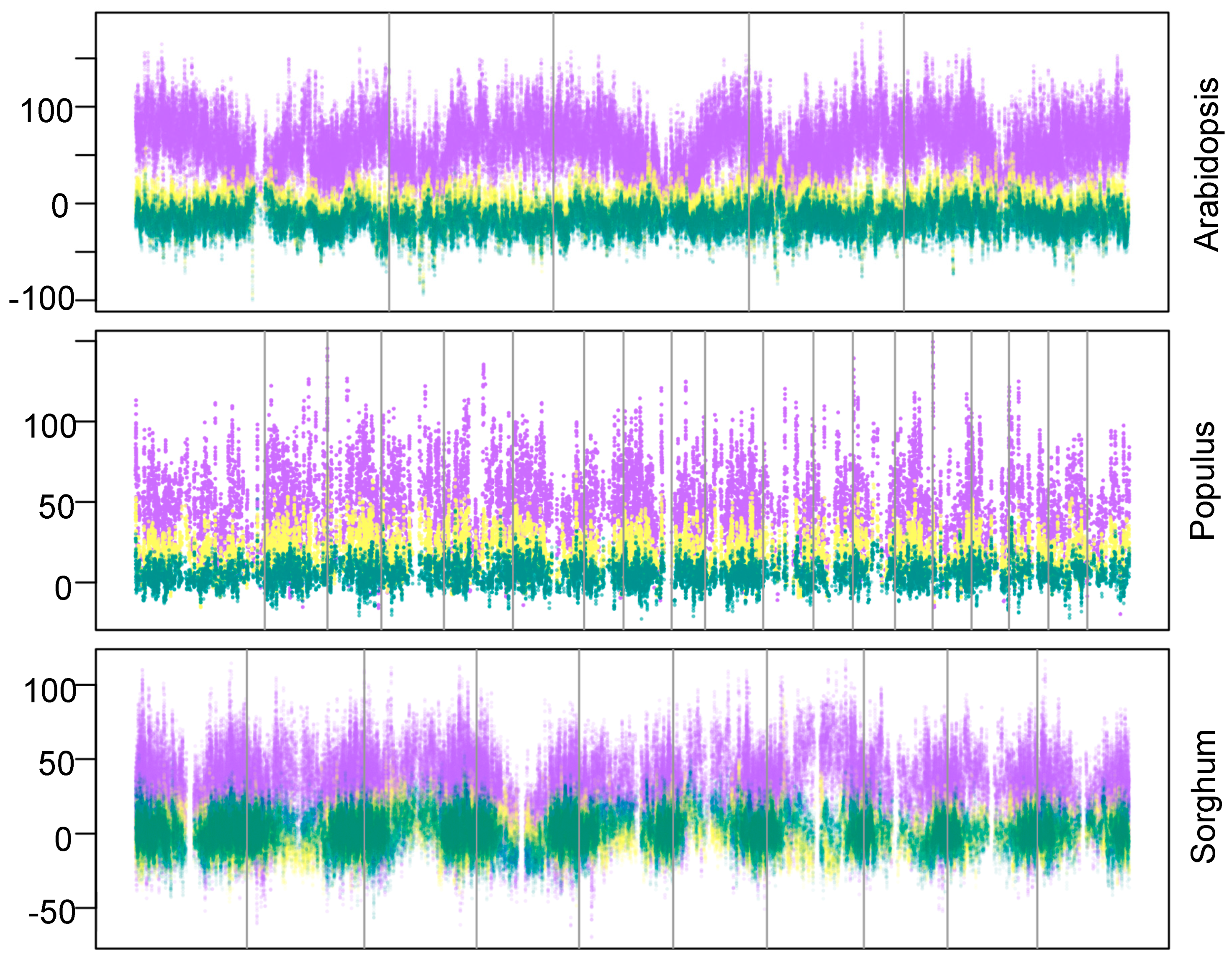
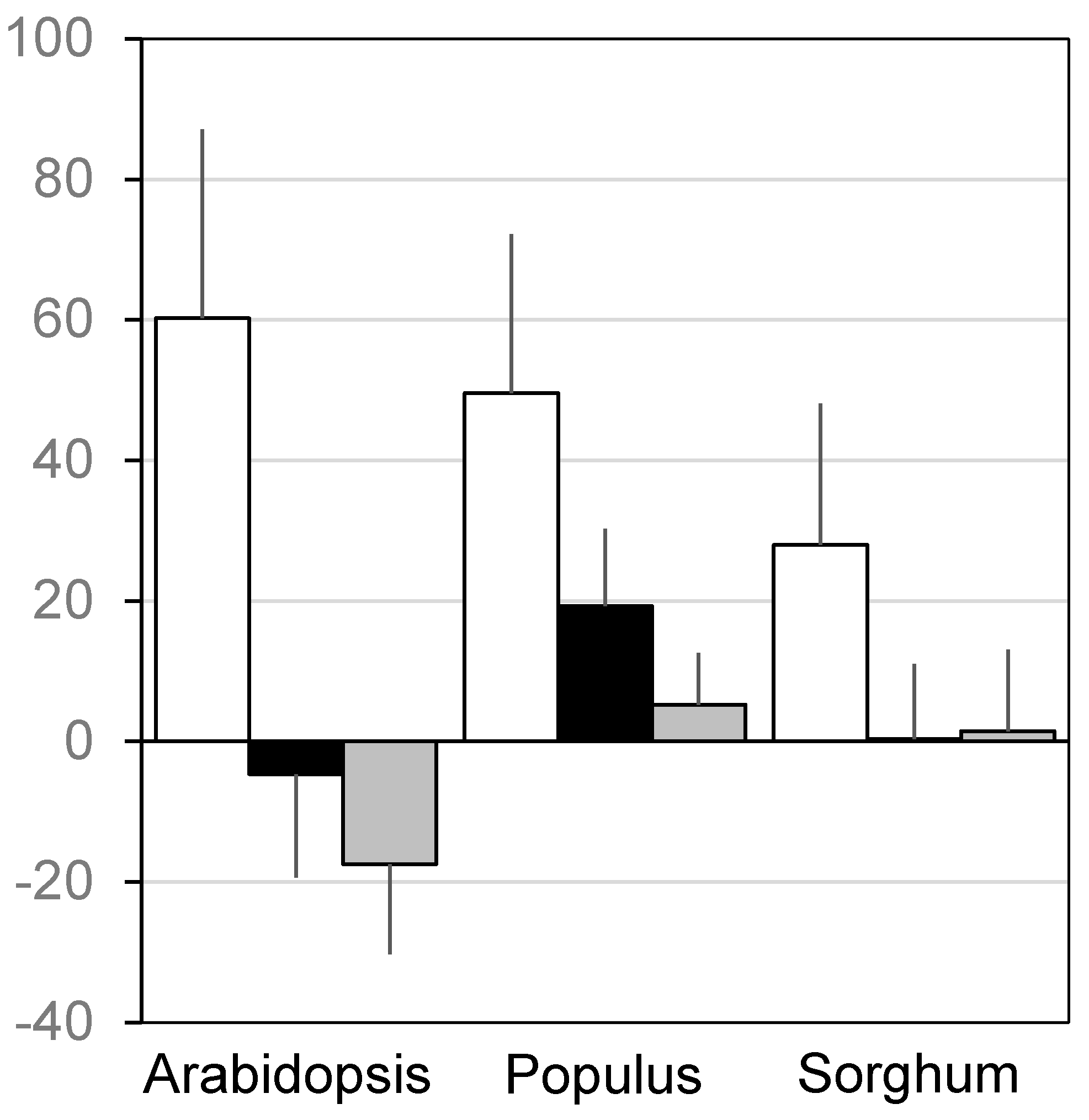
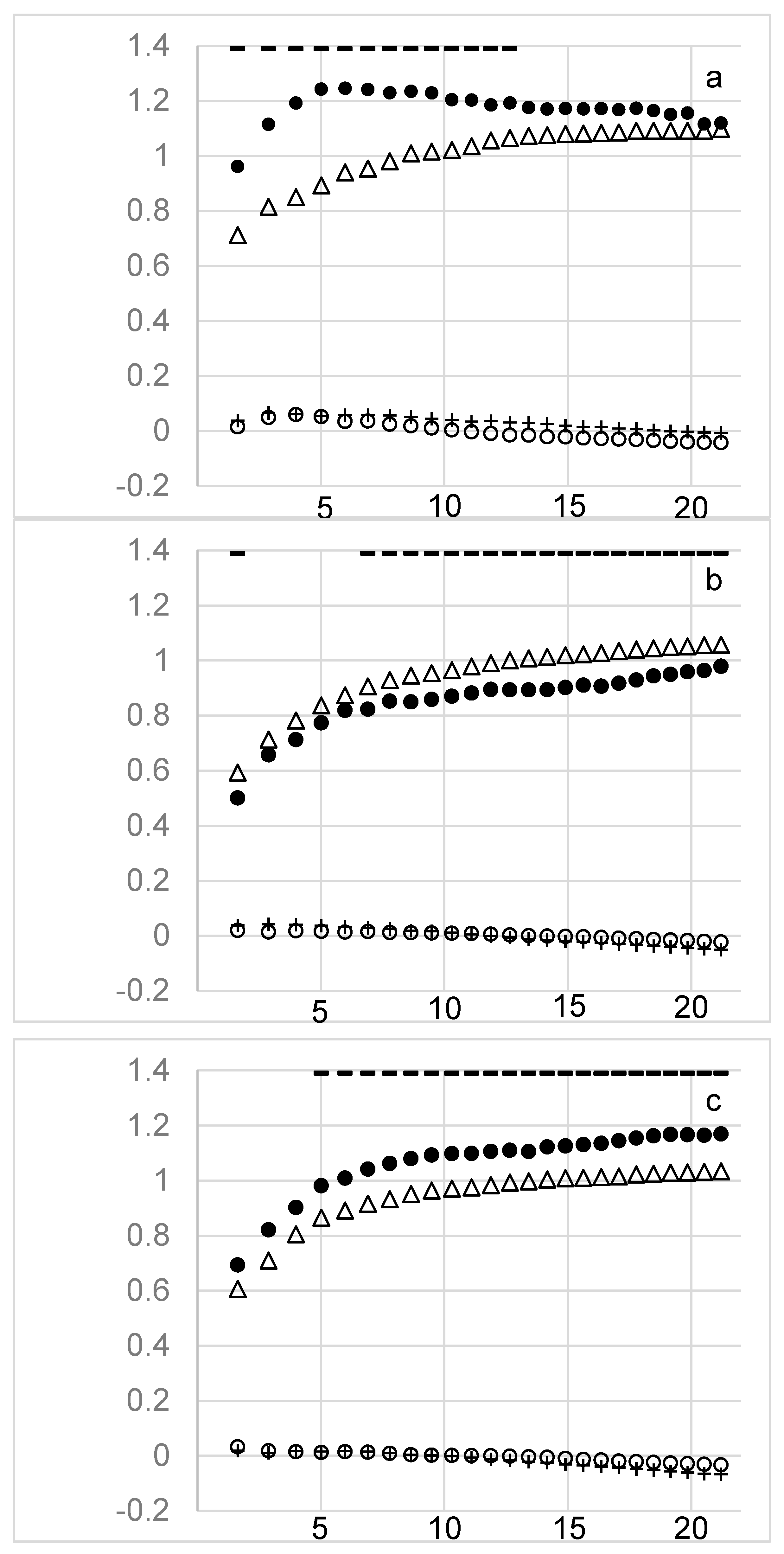
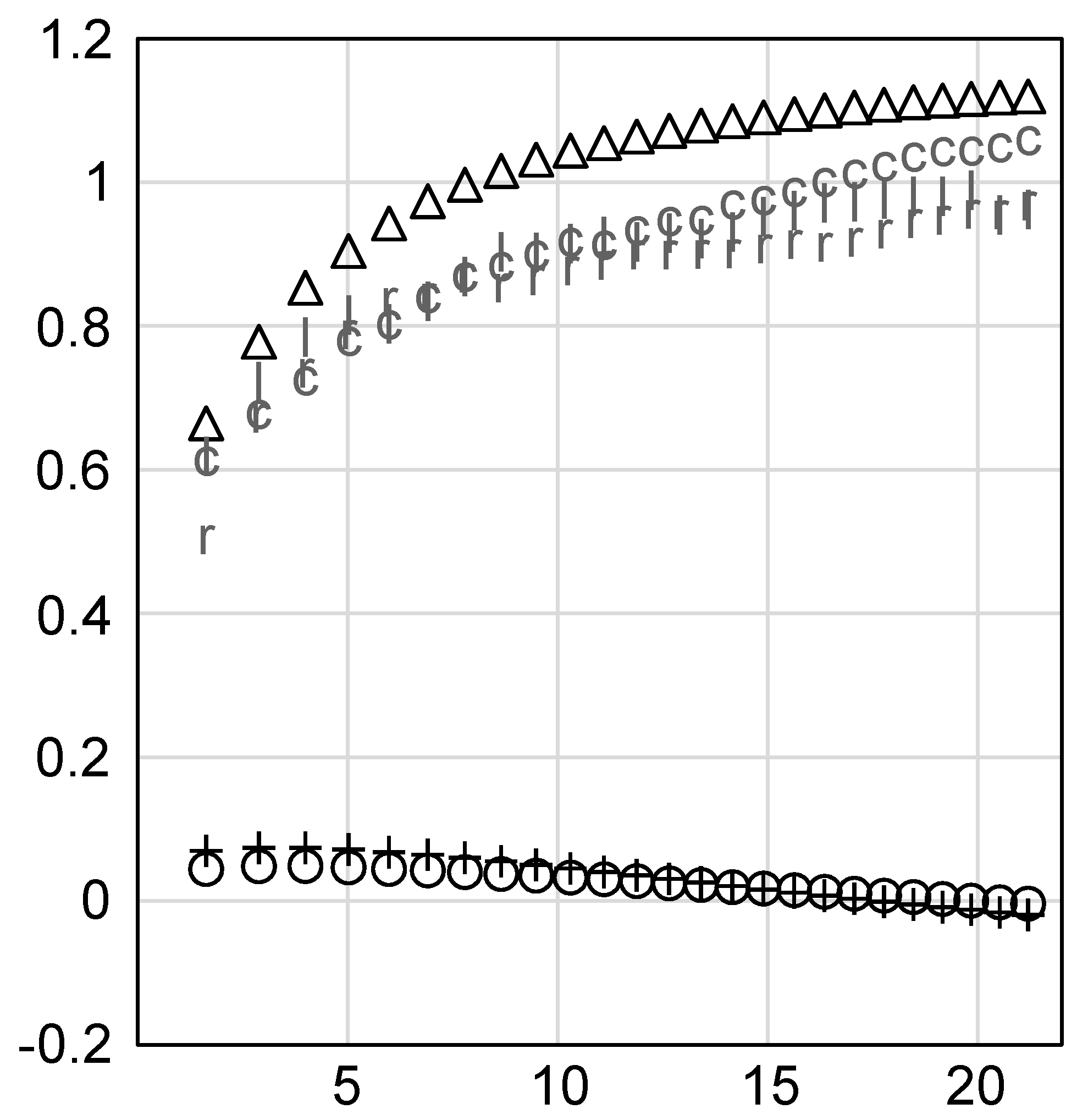
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vasudevan, K.; Vera Cruz, C.M.; Gruissem, W.; Bhullar, N.K. Large scale germplasm screening for identification of novel rice blast resistance sources. Front. Plant Sci. 2014, 5, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Sun, J.; Matthews, A.; Armas-Egas, L.; Chen, N.; Hamill, S.; Mintoff, S.; Tran-Nguyen, L.T.T.; Batley, J.; Aitken, E.A.B. Assessing variations in host resistance to Fusarium oxysporum f sp. cubense race 4 in Musa species, with a focus on the subtropical race 4. Front. Microbiol. 2019, 10, A1062. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, B.D.; Mumm, R.H. Survey of plant density tolerance in U.S. maize germplasm. Crop. Sci. 2014, 54, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, B.; Gardner, C.A.C.; Millard, M.J.; Nickson, T.; Smith, J.S.C. Global access to maize germplasm provided by the US National Plant Germplasm System and by US plant breeders. Crop. Sci. 2016, 56, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Mastrodomenico, A.T.; Hendrix, C.C.; Below, F.E. Nitrogen use efficiency and the genetic variation of maize expired Plant Variety Protection germplasm. Agriculture 2018, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.W.; Jackson, S.T. Novel climates, no analog communities, and ecological surprises. Front. Ecol. Environ. 2007, 5, 475–482. [Google Scholar] [CrossRef]
- Murdock, T.Q.; Taylor, S.W.; Flower, A.; Mehlenbacher, A.; Montenegro, A.; Zwiers, F.W.; Alfaro, R.; Spittlehouse, D.L. Pest outbreak distribution and forest management impacts in a changing climate in British Columbia. Environ. Sci. Policy 2012, 26, 75–89. [Google Scholar] [CrossRef]
- Brown, A.H.D. Core collections: A practical approach to genetic resources management. Genome 1989, 31, 818–824. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Pundir, R.P.S.; Dwivedi, S.L.; Gowda, C.L.L.; Reddy, V.G.; Singh, S. Developing a mini core collection of sorghum for diversified utilization of germplasm. Crop. Sci. 2009, 49, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 22, 1063–1066. [Google Scholar] [CrossRef] [Green Version]
- Lauter, N.; Doebley, J. Genetic variation for phenotypically invariant traits detected in teosinte: Implications for the evolution of novel forms. Genetics 2002, 160, 333–342. [Google Scholar] [PubMed]
- Le Rouzic, A.; Carlborg, Ö. Evolutionary potential of hidden genetic variation. Trends Ecol. Evolut. 2008, 23, 33–37. [Google Scholar] [CrossRef]
- Paaby, A.B.; Rockman, M.V. Cryptic genetic variation: Evolution’s hidden substrate. Nat. Rev. Genet. 2014, 15, 247–258. [Google Scholar] [CrossRef] [Green Version]
- McKhann, H.I.; Camilleri, C.; Bérard, A.; Bataillon, T.; David, J.L.; Reboud, X.; Le Corre, V.; Caloustian, C.; Gut, I.G.; Brunel, D. Nested core collections maximizing genetic diversity in Arabidopsis thaliana. Plant J. 2004, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; Dominguez-Garcia, M.C.; Atienza, S.G.; Urdíroz, N.M.; De la Rosa, R.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V.; et al. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar] [CrossRef]
- Gross, B.L.; Volk, G.M.; Richards, C.M.; Reeves, P.A.; Henk, A.D.; Forsline, P.L.; Szewc-McFadden, A.; Fazio, G.; Chao, C.T. Diversity captured in the USDA-ARS national plant germplasm system apple core collection. J. Amer. Soc. Hort. Sci. 2013, 138, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Bataillon, T.M.; David, J.L.; Schoen, D.J. Neutral genetic markers and conservation genetics: Simulated germplasm collections. Genetics 1996, 144, 409–417. [Google Scholar]
- Reeves, P.A.; Panella, L.W.; Richards, C.M. Retention of agronomically important variation in germplasm core collections: Implications for allele mining. Theor. Appl. Genet. 2012, 124, 1155–1171. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Bramel, P.J.; Singh, S. Development of a chickpea core subset using geographic distribution and quantitative traits. Crop. Sci. 2001, 41, 206–210. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Gowda, C.L.L.; Pundir, R.P.S.; Reddy, V.G.; Singh, S. Development of core subset of finger millet germplasm using geographical origin and data on 14 quantitative traits. Genet. Resour. Crop. Evol. 2006, 53, 679–685. [Google Scholar] [CrossRef]
- Yan, W.; Rutger, J.N.; Bryant, R.J.; Bockelman, H.E.; Fjellstrom, R.G.; Chen, M.H.; Tai, T.H.; McClung, A.M. Development and evaluation of core subset of the USDA rice germplasm collection. Crop. Sci. 2007, 47, 869–878. [Google Scholar] [CrossRef]
- Parra-Quijano, M.; Iriondo, J.M.; Torres, E. Improving representativeness of genebank collections through species distribution models, gap analysis and ecogeographical maps. Biodivers Conserv. 2012, 21, 79–96. [Google Scholar] [CrossRef]
- Reeves, P.A.; Richards, C.M. Capturing haplotypes in germplasm core collections using bioinformatics. Genet. Resour. Crop. Evol. 2017, 64, 1821–1828. [Google Scholar] [CrossRef]
- Reeves, P.A.; Richards, C.M. Biases induced by using geography and environment to guide ex situ conservation. Conserv. Genet. 2018, 19, 1281–1293. [Google Scholar] [CrossRef]
- Mascher, M.; Schreiber, M.; Scholz, U.; Graner, A.; Reif, J.C.; Stein, N. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 2019, 51, 1076–1081. [Google Scholar] [CrossRef]
- Lasky, J.R.; Des Marais, D.L.; McKay, J.K.; Richards, J.H.; Juenger, T.E.; Keitt, T.H. Characterizing genomic variation of Arabidopsis thaliana: The roles of geography and climate. Mol. Ecol. 2012, 21, 5512–5529. [Google Scholar] [CrossRef]
- Geraldes, A.; Farzaneh, N.; Grassa, C.J.; McKown, A.D.; Guy, R.D.; Mansfield, S.D.; Douglas, C.J.; Cronk, Q.C.B. Landscape genomics of Populus trichocarpa: The role of hybridization, limited gene flow, and natural selection in shaping patterns of population structure. Evolution 2014, 68, 3260–3280. [Google Scholar] [CrossRef]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E.; et al. Genome environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [Green Version]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, D.; Hu, J.; Zhou, X.; Ye, X.; Reichel, K.L.; Stewart, N.R.; Syrenne, R.D.; Yang, X.; Gao, P.; et al. Comparative genome analysis of lignin biosynthesis gene families across the plant kingdom. BMC Bioinformatics 2009, 10 (Suppl. S11), S3. [Google Scholar] [CrossRef] [Green Version]
- Schlenker, W.; Roberts, M.J. Nonlinear temperature effects indicate severe damages to U.S. crop yields under climate change. Proc. Nat. Acad. Sci. USA 2009, 106, 15594–15598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobell, D.B.; Roberts, M.J.; Schlenker, W.; Braun, N.; Little, B.B.; Rejesus, R.M.; Hammer, G.L. Greater sensitivity to drought accompanies maize yield increase in the U.S. Midwest. Sci. 2014, 344, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Walthall, C.L.; Hatfield, J.; Backlund, P.; Lengnick, L.; Marshall, E.; Walsh, M.; Adkins, S.; Aillery, M.; Ainsworth, E.A.; Ammann, C.; et al. Climate Change and Agriculture in the United States: Effects and Adaptation. USDA Technical Bulletin 1935; USDA: Washington, DC, USA, 2012.
- Deerwester, S.; Dumais, S.T.; Furnas, G.W.; Landauer, T.K.; Harshman, R. Indexing by latent semantic analysis. J. Am. Soc. Inf. Sci. 1990, 41, 391–407. [Google Scholar] [CrossRef]
- Landauer, T.K.; Foltz, P.W.; Laham, D. Introduction to latent semantic analysis. Discourse Process. 1998, 25, 259–284. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
- Hamblin, M.T.; Salas Fernandez, M.G.; Casa, A.M.; Mitchell, S.E.; Paterson, A.H.; Kresovich, S. Equilibrium processes cannot explain high levels of short- and medium-range linkage disequilibrium in the domesticated grass Sorghum bicolor. Genetics 2005, 171, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Van Treuren, R.; van Hintum, T.J.L. Marker assisted reduction of redundancy in germplasm collections: Genetic and economic aspects. Acta. Hort. 2003, 623, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Wu, S.; Raupp, W.J.; Sehgal, S.; Arora, S.; Tiwari, V.; Vikram, P.; Singh, S.; Chhuneja, P.; Gill, B.S.; et al. Efficient curation of genebanks using next generation sequencing reveals substantial duplication of germplasm accessions. Sci. Rep. 2019, 9, 650. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Olatoye, M.O.; Marla, S.; Morris, G.P. An integrated genotyping by sequencing polymorphism map for over 10,000 sorghum genotypes. Plant Genome 2019, 12, 180044. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Oksman-Caldentey, K.M.; Saito, K. Integrating genomics and metabolomics for engineering plant metabolic pathways. Curr. Opin. Biotechnol. 2005, 16, 174–179. [Google Scholar] [CrossRef]
- Jupe, F.; Witek, K.; Verweij, W.; Sliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valliyodan, B.; Nguyen, H.T. Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 2006, 9, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Loqué, D.; Scheller, H.V.; Pauly, M. Engineering of plant cell walls for enhanced biofuel production. Curr. Opin. Plant Biol. 2015, 25, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ort, D.R.; Merchant, S.S.; Alric, J.; Barkan, A.; Blankenship, R.E.; Bock, R.; Croce, R.; Hanson, M.R.; Hibberd, J.M.; Long, S.P.; et al. Redesigning photosynthesis to sustainably meet global food and bioenergy demand. Proc. Natl. Acad. Sci. USA 2015, 28, 8529–8536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.H.; Ozbun, J.L.; Munger, H.M. Physiological genetics of crop yield. Adv. Agron. 1972, 24, 97–146. [Google Scholar]
- Gur, A.; Zamir, D. Unused natural variation can lift yield barriers in plant breeding. PLoS Biol. 2004, 2, e245. [Google Scholar] [CrossRef]
- Ashraf, M.; Harris, P.J.C. Abiotic Stresses. Plant Resistance Through Breeding and Molecular Approaches; Haworth Press: New York, NY, USA, 2005. [Google Scholar]
- Das, G.; Rao, G.J.N. Molecular marker assisted gene stacking for biotic and abiotic stress resistance genes in an elite rice cultivar. Front. Plant Sci. 2015, 6, 698. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, S.; Grando, S.; Maatougui, M.; Michael, M.; Slash, M.; Haghparast, R.; Rahmanian, M.; Taheri, A.; Al-Yassin, A.; Benbelkacem, A.; et al. Plant breeding and climate changes. J. Agric. Sci. 2010, 148, 627–637. [Google Scholar] [CrossRef]
- Hanson, J.O.; Rhodes, J.R.; Riginos, C.; Fuller, R.A. Environmental and geographic variables are effective surrogates for genetic variation in conservation planning. Proc. Nat. Acad. Sci. USA 2017, 114, 12755–12760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, C.; Borlawsky, T.; Shagina, L.; Xing, H.R.; Lussier, Y.A. Bio Ontology and text: Bridging the modeling gap. Bioinformatics 2006, 22, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Sam, L.T.; Mendonça, E.A.; Li, J.; Blake, J.; Friedman, C.; Lussier, Y.A. PhenoGO: An integrated resource for the multiscale mining of clinical and biological data. BMC Bioinform. 2009, 10, S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebley, J.; Lukens, L. Transcriptional regulators and the evolution of plant form. Plant Cell 1998, 10, 1075–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.B. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 2008, 134, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Cambria, E.; White, B. Jumping NLP curves: A review of natural language processing research. IEEE Comput. Intell. Mag. 2014, 9, 48–57. [Google Scholar] [CrossRef]
- Rodríguez-Leal, D.; Lemmon, Z.H.; Man, J.; Bartlett, M.E.; Lippman, Z.B. Engineering quantitative trait variation for crop improvement by genome editing. Cell 2017, 171, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, Z.H.; Reem, N.T.; Dalrymple, J.; Soyk, S.; Swartwood, K.E.; Rodriguez-Leal, D.; Van Eck, J.; Lippman, Z.B. Rapid improvement of domestication traits in an orphan crop by genome editing. Nat. Plants 2018, 4, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and exploiting pan-genomics for crop improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Term | ID | Term |
|---|---|---|---|
| GO:0010114 | response to red light | GO:0019740 | nitrogen utilization |
| GO:0010018 | far-red light signaling pathway | GO:0071481 | cellular response to X-ray |
| GO:0009408 | response to heat | GO:0016209 | antioxidant activity |
| GO:0015979 | photosynthesis | GO:0045087 | innate immune response |
| GO:0009409 | response to cold | GO:0080167 | response to karrikin |
| GO:0031990 | mRNA export from nucleus in response to heat stress | GO:0071480 | cellular response to gamma radiation |
| GO:0009640 | photomorphogenesis | GO:0019915 | lipid storage |
| GO:0009651 | response to salt stress | GO:0048316 | seed development |
| GO:0009555 | pollen development | GO:0006281 | DNA repair |
| GO:0006979 | response to oxidative stress | GO:0009411 | response to UV |
| GO:0034599 | cellular response to oxidative stress | GO:0032502 | developmental process |
| GO:0009266 | response to temperature stimulus | GO:0009646 | response to absence of light |
| GO:0042538 | hyperosmotic salinity response | GO:0043207 | response to external biotic stimulus |
| GO:0009631 | cold acclimation | GO:0006974 | cellular response to DNA damage stimulus |
| GO:0071470 | cellular response to osmotic stress | GO:0030332 | cyclin binding |
| GO:0006338 | chromatin remodeling | GO:0019760 | glucosinolate metabolic process |
| GO:0070483 | detection of hypoxia | GO:0009414 | response to water deprivation |
| GO:0050826 | response to freezing | GO:0019684 | photosynthesis, light reaction |
| GO:0048577 | negative regulation of short-day photoperiodism, flowering | GO:0016132 | brassinosteroid biosynthetic process |
| GO:0048364 | root development | GO:0016131 | brassinosteroid metabolic process |
| GO:0019748 | secondary metabolic process | GO:0015250 | water channel activity |
| GO:0043044 | ATP-dependent chromatin remodeling | GO:0006995 | cellular response to nitrogen starvation |
| GO:0042276 | error-prone translesion synthesis | GO:0009611 | response to wounding |
| GO:0030154 | cell differentiation | GO:0009793 | embryo development ending in seed dormancy |
| GO:0009908 | flower development | GO:0010014 | meristem initiation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reeves, P.A.; Tetreault, H.M.; Richards, C.M. Bioinformatic Extraction of Functional Genetic Diversity from Heterogeneous Germplasm Collections for Crop Improvement. Agronomy 2020, 10, 593. https://doi.org/10.3390/agronomy10040593
Reeves PA, Tetreault HM, Richards CM. Bioinformatic Extraction of Functional Genetic Diversity from Heterogeneous Germplasm Collections for Crop Improvement. Agronomy. 2020; 10(4):593. https://doi.org/10.3390/agronomy10040593
Chicago/Turabian StyleReeves, Patrick A., Hannah M. Tetreault, and Christopher M. Richards. 2020. "Bioinformatic Extraction of Functional Genetic Diversity from Heterogeneous Germplasm Collections for Crop Improvement" Agronomy 10, no. 4: 593. https://doi.org/10.3390/agronomy10040593
APA StyleReeves, P. A., Tetreault, H. M., & Richards, C. M. (2020). Bioinformatic Extraction of Functional Genetic Diversity from Heterogeneous Germplasm Collections for Crop Improvement. Agronomy, 10(4), 593. https://doi.org/10.3390/agronomy10040593