Similar but Not Identical Resuscitation Trajectories of the Soil Microbial Community Based on Either DNA or RNA after Flooding

Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Experimental Setup
2.2. Soil Gas Efflux Rates
2.3. Soil Enzyme Assays
2.4. Nucleic Acid Extraction and Purification
2.5. Quantification of Bacterial and Fungal Abundance
2.6. High-Throughput Sequence Analyses
2.7. Statistical Analyses
3. Results
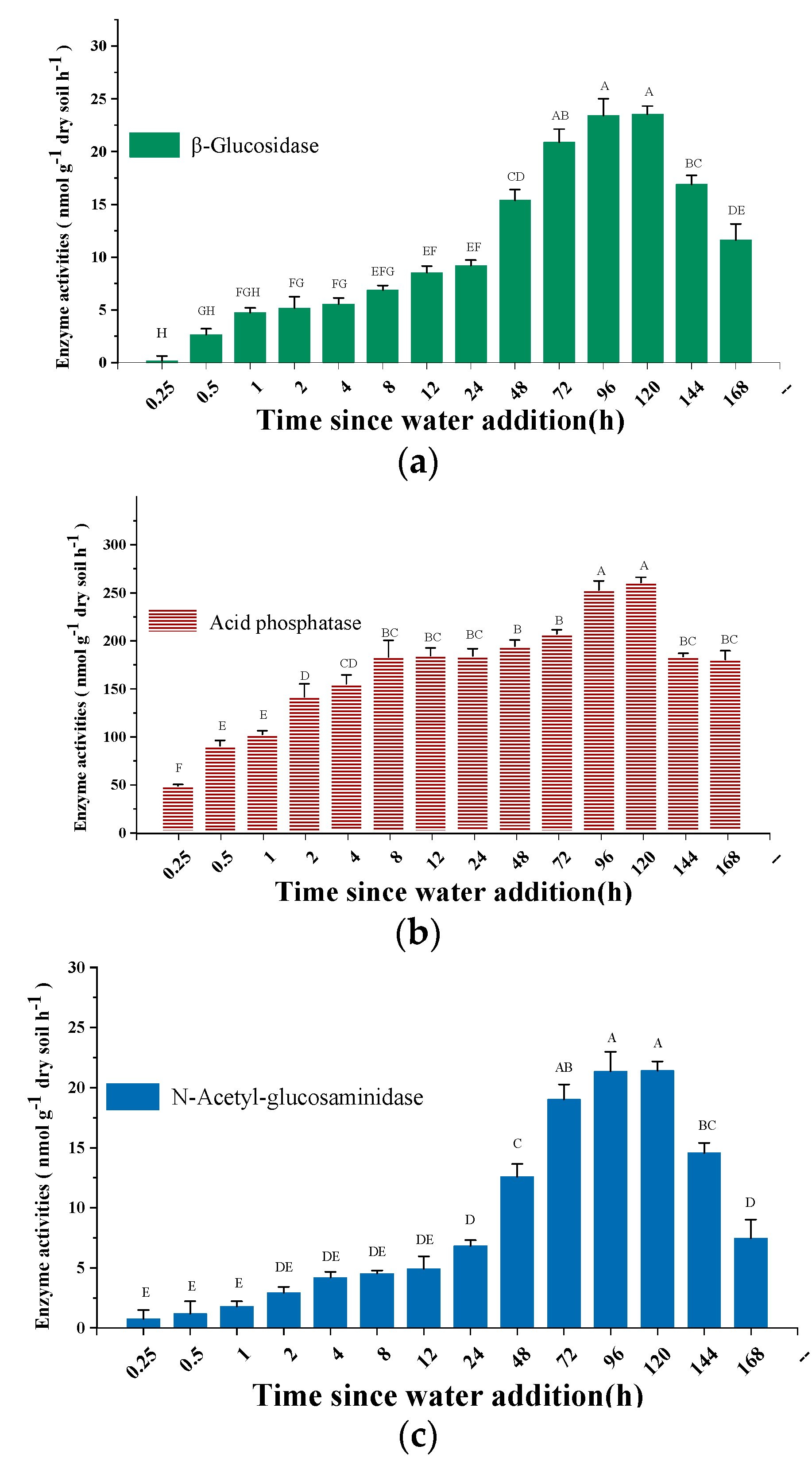
3.1. Variation in Soil Gases Efflux and Enzyme Activity
3.2. General Information of Sequencing Data
3.3. Abundance and Community Structure of Bacteria and Fungi
3.3.1. Bacterial Community
3.3.2. Fungal Community
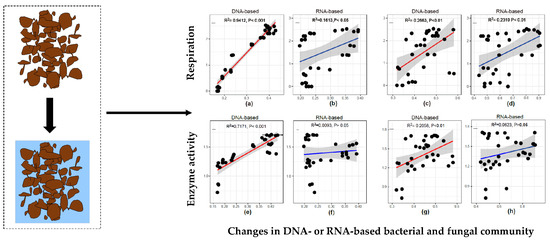
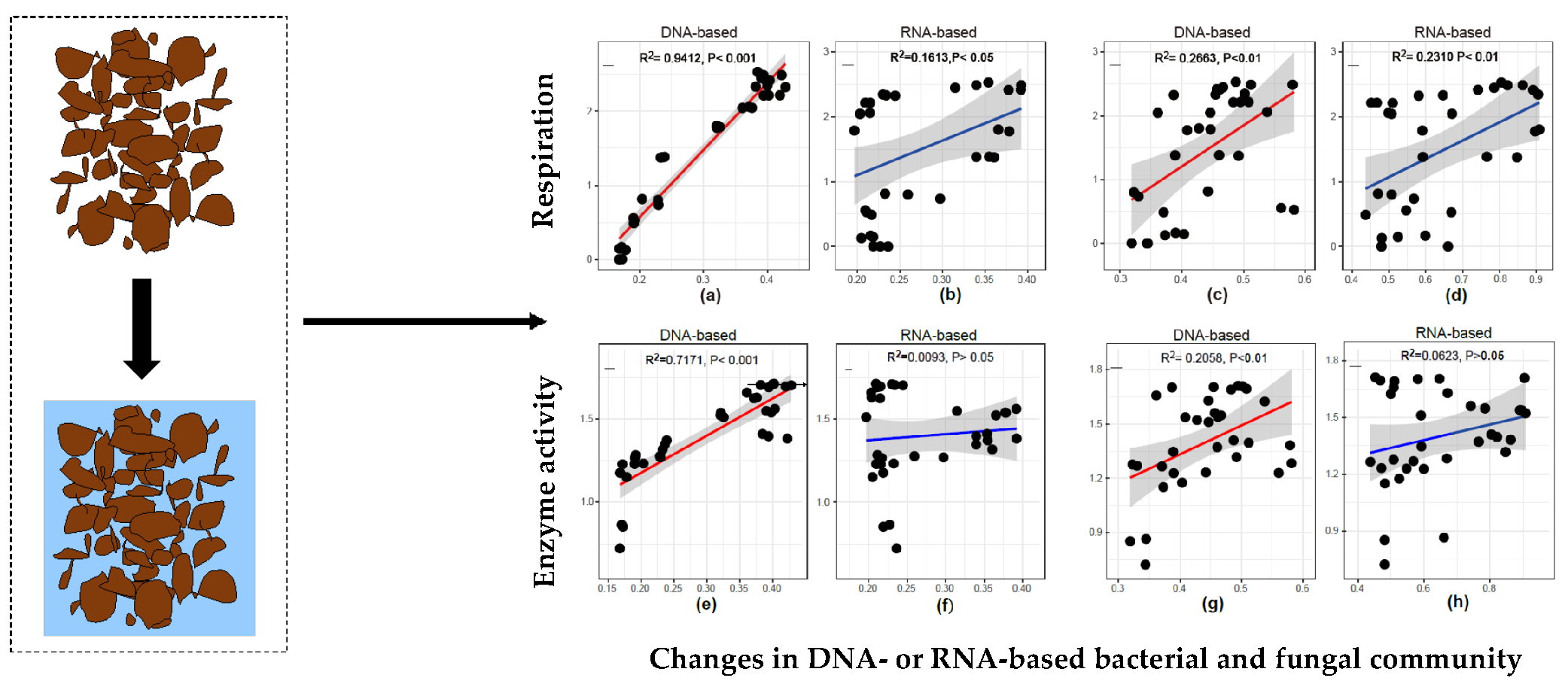
3.4. Relationship Between Microbial Activity and Community Structure
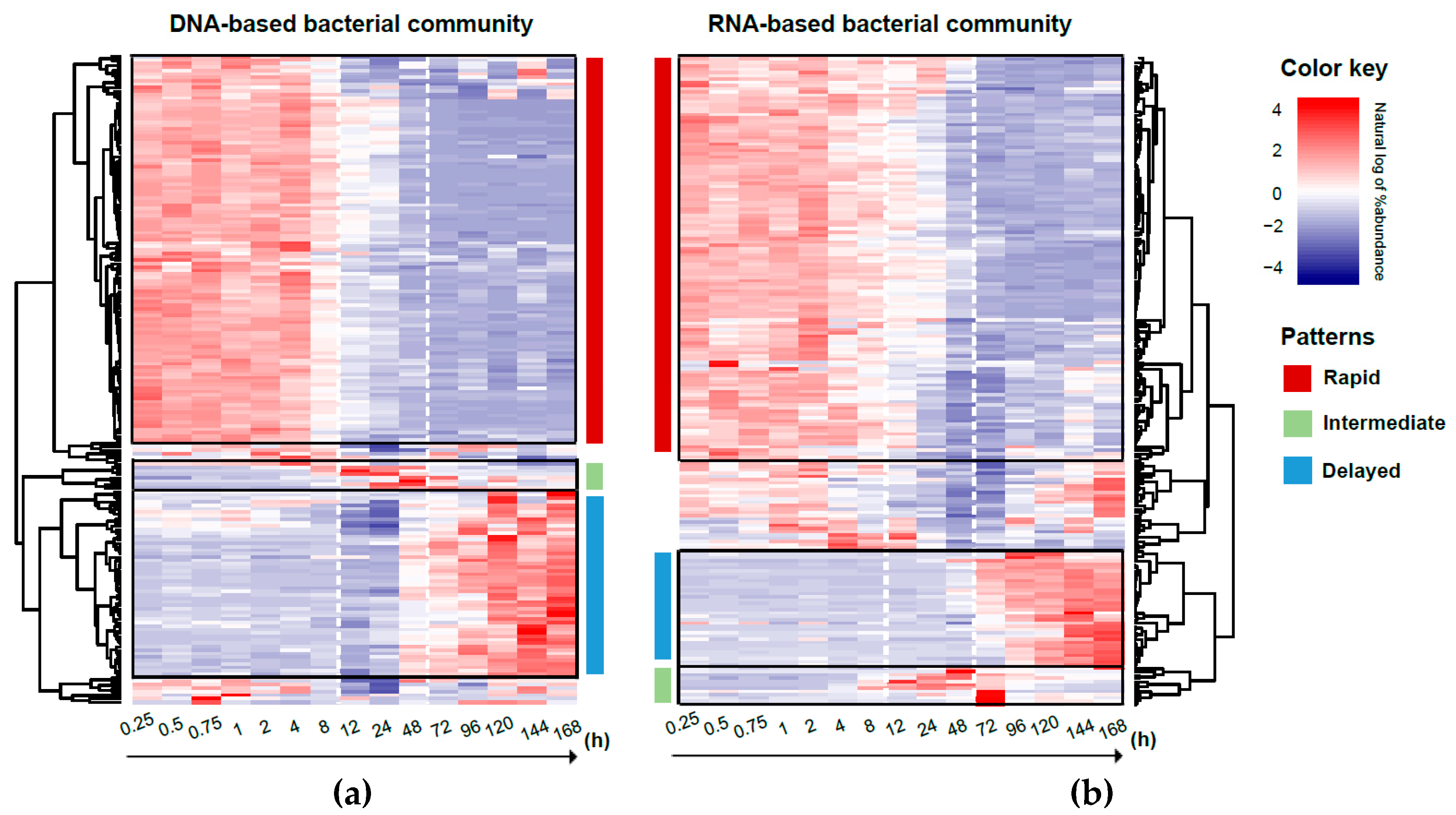
3.5. Response Trajectory of the Microbial Community to Flooding
4. Discussion
4.1. Sources of CO2 and N2O
4.2. Differences Between DNA- and RNA-Based Microbial Communities
4.3. Difference Between Bacterial and Fungal Community-level Response
4.4. Potential Role of Bacilli in the Trajectory of the Bacterial Community
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aanderud, Z.T.; Jones, S.E.; Fierer, N.; Lennon, J.T. Resuscitation of the rare boisphere contributes to pulses of ecosystem activity. Front. Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, N.; Wang, J.; Yao, H.; Qiu, Q.; Chapman, S.J. High turnover rate of free phospholipids in soil confirms the classic hypothesis of PLFA methodology. Soil Biol. Biochem. 2019, 135, 323–330. [Google Scholar] [CrossRef]
- Yan, Z.; Bond-Lamberty, B.; Todd-Brown, K.E.; Bailey, V.L.; Li, S.; Liu, C.; Liu, C. A moisture function of soil heterotrophic respiration that incorporates microscale processes. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Birch, H.F. The effect of soil drying on humus decomposition and nitrogen availability. Plant Soil 1958, 10, 9–31. [Google Scholar] [CrossRef]
- Kim, D.G.; Vargas, R.; Bond-Lamberty, B.; Turetsky, M.R. Effects of soil rewetting and thawing on soil gas fluxes: A review of current literature and suggestions for future research. Biogeosciences 2012, 9, 2459–2483. [Google Scholar] [CrossRef]
- Placella, S.A.; Brodie, E.L.; Firestone, M.K. Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc. Natl. Acad. Sci. USA 2012, 109, 10931–10936. [Google Scholar] [CrossRef]
- Schimel, J.; Balser, T.C.; Wallenstein, M. Microbial stress-response physiology and its implications for ecosystem function. Ecology 2007, 88, 1386–1394. [Google Scholar] [CrossRef]
- Yang, H.; Sheng, R.; Zhang, Z.; Wang, L.; Wang, Q.; Wei, W. Responses of nitrifying and denitrifying bacteria to flooding-drying cycles in flooded rice soil. Appl. Soil Ecol. 2016, 103, 101–109. [Google Scholar] [CrossRef]
- Unger, S.; Máguas, C.; Pereira, J.S.; David, T.S.; Werner, C. The influence of precipitation pulses on soil respiration—Assessing the “Birch effect” by stable carbon isotopes. Soil Biol. Biochem. 2010, 42, 1800–1810. [Google Scholar] [CrossRef]
- Barnard, R.L.; Osborne, C.A.; Firestone, M.K. Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 2013, 7, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- More, T.T.; Yadav, J.S.S.; Yan, S.; Tyagi, R.D.; Surampalli, R.Y. Extracellular polymeric substances of bacteria and their potential environmental applications. J. Environ. Manag. 2014, 144, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Preece, C.; Verbruggen, E.; Liu, L.; Weedon, J.T.; Peñuelas, J. Effects of past and current drought on the composition and diversity of soil microbial communities. Soil Biol. Biochem. 2019, 131, 28–39. [Google Scholar] [CrossRef]
- Kaprelyants, A.S.; Gottschal, J.C.; Kell, D.B. Dormancy in Non-Sporulating Bacteria. FEMS Microbiol. Lett. 1993, 104, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Placella, S.A.; Firestone, M.K. Transcriptional Response of Nitrifying Communities to Wetting of Dry Soil. Appl. Environ. Microbiol. 2013, 79, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Wang, C.; Zhou, L.; Zhao, S.; Li, Y.; Li, F.; Jetten, M.S.M.; Lu, Y.; Schwark, L. Resuscitation of anammox bacteria after >10,000 years of dormancy. ISME J. 2019, 13, 1098–1109. [Google Scholar] [CrossRef]
- Evans, S.E.; Wallenstein, M.D. Climate change alters ecological strategies of soil bacteria. Ecol. Lett. 2014, 17, 155–164. [Google Scholar] [CrossRef]
- Meisner, A.; Jacquiod, S.; Snoek, B.L.; ten Hooven, F.C.; van der Putten, W.H. Drought Legacy Effects on the Composition of Soil Fungal and Prokaryote Communities. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Barnard, R.L.; Osborne, C.A.; Firestone, M.K. Changing precipitation pattern alters soil microbial community response to wet-up under a Mediterranean-type climate. ISME J. 2015, 9, 946–957. [Google Scholar] [CrossRef]
- Liao, H.; Chapman, S.J.; Li, Y.; Yao, H. Dynamics of microbial biomass and community composition after short-term water status change in Chinese paddy soils. Environ. Sci. Pollut. Res. 2018, 25, 2932–2941. [Google Scholar] [CrossRef]
- Kaisermann, A.; Maron, P.A.; Beaumelle, L.; Lata, J.C. Fungal communities are more sensitive indicators to non-extreme soil moisture variations than bacterial communities. Appl. Soil Ecol. 2015, 86, 158–164. [Google Scholar] [CrossRef]
- Engelhardt, I.C.; Blazewicz, S.J.; Barnard, R.L. Effects of contrasting precipitation patterns on the trajectory of actively growing and inactive microbial communities after rewetting. Soil Biol. Biochem. 2019. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Blazewicz, S.J.; Barnard, R.L.; Daly, R.A.; Firestone, M.K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013, 7, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Su, J.; Yang, X.; Zhu, Y. Distinct rhizosphere effect on active and total bacterial communities in paddy soils. Sci. Total Environ. 2019, 649, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhao, C.; Zheng, N.; Jia, H.; Yao, H. Interactive effects of soil texture and salinity on nitrous oxide emissions following crop residue amendment. Geoderma 2019, 337, 1146–1154. [Google Scholar] [CrossRef]
- DeForest, J.L. The influence of time, storage temperature, and substrate age on potential soil enzyme activity in acidic forest soils using MUB-linked substrates and L-DOPA. Soil Biol. Biochem. 2009, 41, 1180–1186. [Google Scholar] [CrossRef]
- Giacometti, C.; Cavani, L.; Baldoni, G.; Ciavatta, C.; Marzadori, C.; Kandeler, E. Microplate-scale fluorometric soil enzyme assays as tools to assess soil quality in a long-term agricultural field experiment. Appl. Soil Ecol. 2014, 75, 80–85. [Google Scholar] [CrossRef]
- Jing, X.; Chen, X.; Xiao, W.; Lin, L.; Wang, C.; He, J.; Zhu, B. Soil enzymatic responses to multiple environmental drivers in the Tibetan grasslands: Insights from two manipulative field experiments and a meta-analysis. Pedobiologia 2018, 71, 50–58. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Whiteley, A.S.; O’Donnell, A.G.; Bailey, M.J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 2000, 66, 5488–5491. [Google Scholar] [CrossRef]
- Ding, L.; Su, J.; Li, H.; Zhu, Y.; Cao, Z. Bacterial succession along a long-term chronosequence of paddy soil in the Yangtze River Delta, China. Soil Biol. Biochem. 2017, 104, 59–67. [Google Scholar] [CrossRef]
- Long, X.; Yao, H.; Huang, Y.; Wei, W.; Zhu, Y. Phosphate levels influence the utilisation of rice rhizodeposition carbon and the phosphate-solubilising microbial community in a paddy soil. Soil Biol. Biochem. 2018, 118, 103–114. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. Its Primers with Enhanced Specificity for Basidiomycetes—Application to the Identification of Mycorrhizae and Rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Li, Y.; Yao, H. Fertilization with inorganic and organic nutrients changes diazotroph community composition and N-fixation rates. J. Soils Sediments 2018, 18, 1076–1086. [Google Scholar] [CrossRef]
- Jia, Y.; Leung, M.H.Y.; Tong, X.; Wilkins, D.; Lee, P.K.H. Rare Taxa Exhibit Disproportionate Cell-Level Metabolic Activity in Enriched Anaerobic Digestion Microbial Communities. Msystems 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Ochoa-Hueso, R.; Collins, S.L.; Delgado-Baquerizo, M.; Hamonts, K.; Pockman, W.T.; Sinsabaugh, R.L.; Smith, M.D.; Knapp, A.K.; Power, S.A. Drought consistently alters the composition of soil fungal and bacterial communities in grasslands from two continents. Glob. Change Biol. 2018, 24, 2818–2827. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package, R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 September 2019).
- Kolde, R. pheatmap: Pretty Heatmaps, R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 4 April 2019).
- Kembel, S.W.; Coman, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Webb, C.O.; Ackerly, D.D.; McPeek, M.A.; Donoghue, M.J. Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 2002, 33, 475–505. [Google Scholar] [CrossRef]
- Liu, S.; Schloter, M.; Bruggemann, N. Accumulation of NO2- during periods of drying stimulates soil N2O emissions during subsequent rewetting. Eur. J. Soil Sci. 2018, 69, 936–946. [Google Scholar] [CrossRef]
- Kaiser, M.; Kleber, M.; Berhe, A.A. How air-drying and rewetting modify soil organic matter characteristics: An assessment to improve data interpretation and inference. Soil Biol. Biochem. 2015, 80, 324–340. [Google Scholar] [CrossRef]
- Moyano, F.E.; Manzoni, S.; Chenu, C. Responses of soil heterotrophic respiration to moisture availability: An exploration of processes and models. Soil Biol. Biochem. 2013, 59, 72–85. [Google Scholar] [CrossRef]
- Inglima, I.; Alberti, G.; Bertolini, T.; Vaccari, F.P.; Gioli, B.; Miglietta, F.; Cotrufo, M.F.; Peressotti, A. Precipitation pulses enhance respiration of Mediterranean ecosystems: The balance between organic and inorganic components of increased soil CO2 efflux. Glob. Change Biol. 2009, 15, 1289–1301. [Google Scholar] [CrossRef]
- Heil, J.; Vereecken, H.; Bruggemann, N. A review of chemical reactions of nitrification intermediates and their role in nitrogen cycling and nitrogen trace gas formation in soil. Eur. J. Soil Sci. 2016, 67, 23–39. [Google Scholar] [CrossRef]
- Li, Y.; Chapman, S.J.; Nicol, G.W.; Yao, H. Nitrification and nitrifiers in acidic soils. Soil Biol. Biochem. 2018, 116, 290–301. [Google Scholar] [CrossRef]
- Blagodatskaya, E.; Kuzyakov, Y. Active microorganisms in soil: Critical review of estimation criteria and approaches. Soil Biol. Biochem. 2013, 67, 192–211. [Google Scholar] [CrossRef]
- Musat, N.; Foster, R.; Vagner, T.; Adam, B.; Kuypers, M.M.M. Detecting metabolic activities in single cells, with emphasis on nanoSIMS. FEMS Microbiol. Rev. 2012, 36, 486–511. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, I.C.; Welty, A.; Blazewicz, S.J.; Bru, D.; Rouard, N.; Breuil, M.C.; Gessler, A.; Galiano, L.; Miranda, J.C.; Spor, A.; et al. Depth matters: Effects of precipitation regime on soil microbial activity upon rewetting of a plant-soil system. ISME J. 2018, 12, 1061–1071. [Google Scholar] [CrossRef]
- De Vries, F.T.; Liiri, M.E.; Bjørnlund, L.; Bowker, M.A.; Christensen, S.; Setälä, H.M.; Bardgett, R.D. Land use alters the resistance and resilience of soil food webs to drought. Nat. Clim. Change 2012, 2, 276. [Google Scholar] [CrossRef]
- Ludemann, H.; Arth, I.; Liesack, W. Spatial changes in the bacterial community structure along a vertical oxygen gradient in flooded paddy soil cores. Appl. Environ. Microbiol. 2000, 66, 754–762. [Google Scholar] [CrossRef]
- Noll, M.; Matthies, D.; Frenzel, P.; Derakshani, M.; Liesack, W. Succession of bacterial community structure and diversity in a paddy soil oxygen gradient. Environ. Microbiol. 2005, 7, 382–395. [Google Scholar] [CrossRef]
- Lennon, J.T.; Aanderud, Z.T.; Lehmkuhl, B.K.; Schoolmaster, D.R. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 2012, 93, 1867–1879. [Google Scholar] [CrossRef]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Schimel, J.P. Life in Dry Soils: Effects of Drought on Soil Microbial Communities and Processes. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 409–432. [Google Scholar] [CrossRef]
- Filippidou, S.; Wunderlin, T.; Junier, T.; Jeanneret, N.; Dorador, C.; Molina, V.; Johnson, D.R.; Junier, P.A. Combination of Extreme Environmental Conditions Favor the Prevalence of Endospore-Forming Firmicutes. Front. Microbiol. 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Sharipova, M.R.; Balaban, N.P.; Gabdrakhmanova, L.A.; Shilova, M.A.; Kadyrova, Y.M.; Rudenskaya, G.N.; Leshchinskaya, I.B. Hydrolytic enzymes and sporulation in Bacillus intermedius. Microbiology 2002, 71, 420–424. [Google Scholar] [CrossRef]
- Feng, M.; Adams, J.M.; Fan, K.; Shi, Y.; Sun, R.; Wang, D.; Guo, X.; Chu, H. Long-term fertilization influences community assembly processes of soil diazotrophs. Soil Biol. Biochem. 2018, 126, 151–158. [Google Scholar] [CrossRef]
- Li, Y.; Pan, F.; Yao, H. Response of symbiotic and asymbiotic nitrogen-fixing microorganisms to nitrogen fertilizer application. J. Soil Sediment. 2019, 19, 1948–1958. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Kou, Y.; Wang, J.; Tu, B.; Li, H.; Li, X.; Wang, C.; Yao, M. Soil pH is a major driver of soil diazotrophic community assembly in Qinghai-Tibet alpine meadows. Soil Biol. Biochem. 2017, 115, 547–555. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA-Based | RNA-Based | ||||||
|---|---|---|---|---|---|---|---|
| Time | Richness | Shannon | Evenness | Richness | Shannon | Evenness | |
| Bacteria | 0.25 h | 4927 ± 46 a | 10.61 ± 0.02 a | 0.996 ± 0.000 a | 4357 ± 87 ab | 9.34 ± 0.14 ab | 0.977 ± 0.003 a |
| 0.5 h | 4961 ± 38 a | 10.61 ± 0.02 a | 0.996 ± 0.000 a | 4246 ± 115 ab | 9.08 ± 0.18 abc | 0.972 ± 0.004 a | |
| 0.75 h | 4967 ± 18 a | 10.65 ± 0.01 a | 0.996 ± 0.000 a | 4242 ± 70 ab | 9.17 ± 0.13 abc | 0.975 ± 0.002 a | |
| 1 h | 4901 ± 11 a | 10.50 ± 0.04 a | 0.996 ± 0.000 a | 4294 ± 33 ab | 9.22 ± 0.07 abc | 0.975 ± 0.002 a | |
| 2 h | 4923 ± 20 a | 10.54 ± 0.01 a | 0.996 ± 0.000 a | 4433 ± 36 a | 9.46 ± 0.05 a | 0.981 ± 0.001 a | |
| 4 h | 4889 ± 27 a | 10.5 ± 0.04 b | 0.995 ± 0.000 a | 4037 ± 22 abcd | 8.83 ± 0.07 bcd | 0.971 ± 0.003 a | |
| 8 h | 4541 ± 19 b | 9.54 ± 0.02 b | 0.981 ± 0.000 a | 4006 ± 24 bcd | 8.73 ± 0.03 cd | 0.973 ± 0.001 a | |
| 12 h | 3938 ± 84 c | 7.97 ± 0.15 d | 0.929 ± 0.005 a | 3864 ± 57 bcd | 8.34 ± 0.11 de | 0.964 ± 0.002 a | |
| 24 h | 3742 ± 25 cd | 8.07 ± 0.05 d | 0.962 ± 0.002 a | 3743 ± 41 de | 7.86 ± 0.11 ef | 0.94 ± 0.009 b | |
| 48 h | 3491 ± 67 de | 8.94 ± 0.12 c | 0.988 ± 0.001 a | 3104 ± 88 fg | 7.06 ± 0.11 g | 0.924 ± 0.006 b | |
| 72 h | 3197 ± 75 f | 8.80 ± 0.09 c | 0.986 ± 0.000 a | 2798 ± 169 g | 7.58 ± 0.19 fg | 0.966 ± 0.003 a | |
| 96 h | 3178 ± 64 f | 8.85 ± 0.04 c | 0.984 ± 0.000 a | 3108 ± 6f g | 8.34 ± 0.02 de | 0.976 ± 0.000 a | |
| 120 h | 3234 ± 62 ef | 9.31 ± 0.06 b | 0.992 ± 0.000 a | 3143 ± 62 fg | 8.45 ± 0.10 d | 0.976 ± 0.002 a | |
| 144 h | 3408 ± 22 ef | 9.29 ± 0.03 b | 0.986 ± 0.000 a | 3256 ± 80 f | 8.71 ± 0.04 cd | 0.974 ± 0.000 a | |
| 168 h | 3324 ± 74 ef | 9.41 ± 0.07 b | 0.992 ± 0.000 a | 3366 ± 137 ef | 8.84 ± 0.10 bcd | 0.977 ± 0.001 a | |
| Fungi | 0.25 h | 365 ± 15 a | 4.02 ± 0.43 a | 0.784 ± 0.064 a | 209 ± 11 ab | 4.94 ± 0.07 a | 0.925 ± 0.007 a |
| 0.5 h | 349 ± 14 a | 3.33 ± 0.12a | 0.689 ± 0.04 a | 228 ± 46 ab | 4.79 ± 0.73 a | 0.875 ± 0.065 a | |
| 0.75 h | 412 ± 8 a | 4.68 ± 0.28a | 0.895 ± 0.031 a | 202 ± 25 ab | 4.22 ± 0.93 a | 0.812 ± 0.122 a | |
| 1 h | 386 ± 6 a | 3.99 ± 0.48a | 0.809 ± 0.044 a | 228 ± 10 ab | 4.01 ± 0.42 a | 0.8 ± 0.066 a | |
| 2 h | 400 ± 38a | 4.58 ± 0.18a | 0.901 ± 0.004 a | 233 ± 32 ab | 4.81 ± 0.35 a | 0.897 ± 0.027 a | |
| 4 h | 381 ± 28a | 4.28 ± 0.38a | 0.867 ± 0.036 a | 183 ± 9 ab | 4.05 ± 0.77 a | 0.787 ± 0.121 a | |
| 8 h | 356 ± 48a | 3.72 ± 0.41 a | 0.765 ± 0.056 a | 214 ± 39 ab | 3.70 ± 0.77 a | 0.802 ± 0.083 a | |
| 12 h | 377 ± 23a | 4.17 ± 0.27 a | 0.85 ± 0.026 a | 252 ± 34 a | 4.42 ± 0.91 a | 0.791 ± 0.139 a | |
| 24 h | 420 ± 30a | 4.16 ± 0.45 a | 0.809 ± 0.07 a | 204 ± 35 ab | 5 ± 0.32 a | 0.909 ± 0.029 a | |
| 48 h | 440 ± 20 a | 4.19 ± 0.18 a | 0.806 ± 0.023 a | 143 ± 13 ab | 4.3 ± 0.57 a | 0.811 ± 0.066 a | |
| 72 h | 397 ± 40 a | 4.17 ± 0.47 a | 0.814 ± 0.072 a | 136 ± 6 ab | 4.35 ± 1.049 a | 0.812 ± 0.136 a | |
| 96 h | 427 ± 40 a | 4.59 ± 0.23 a | 0.887 ± 0.016 a | 162 ± 50 ab | 3.21 ± 0.87 a | 0.636 ± 0.174 a | |
| 120 h | 320 ± 17 a | 3.81 ± 0.26 a | 0.795 ± 0.044 a | 114 ± 28 ab | 4.45 ± 0.79 a | 0.824 ± 0.068 a | |
| 144 h | 431 ± 10 a | 4.47 ± 0.27 a | 0.874 ± 0.03 a | 88 ± 26 b | 3.24 ± 0.47 a | 0.708 ± 0.089 a | |
| 168 h | 320 ± 16 a | 3.4 ± 0.29 a | 0.704 ± 0.06 a | 116 ± 44 b | 3.01 ± 0.21 a | 0.673 ± 0.043 a | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Li, Y.; Zheng, N.; Chapman, S.J.; Yao, H. Similar but Not Identical Resuscitation Trajectories of the Soil Microbial Community Based on Either DNA or RNA after Flooding. Agronomy 2020, 10, 502. https://doi.org/10.3390/agronomy10040502
Zhu Y, Li Y, Zheng N, Chapman SJ, Yao H. Similar but Not Identical Resuscitation Trajectories of the Soil Microbial Community Based on Either DNA or RNA after Flooding. Agronomy. 2020; 10(4):502. https://doi.org/10.3390/agronomy10040502
Chicago/Turabian StyleZhu, Yizu, Yaying Li, Ningguo Zheng, Stephen James Chapman, and Huaiying Yao. 2020. "Similar but Not Identical Resuscitation Trajectories of the Soil Microbial Community Based on Either DNA or RNA after Flooding" Agronomy 10, no. 4: 502. https://doi.org/10.3390/agronomy10040502
APA StyleZhu, Y., Li, Y., Zheng, N., Chapman, S. J., & Yao, H. (2020). Similar but Not Identical Resuscitation Trajectories of the Soil Microbial Community Based on Either DNA or RNA after Flooding. Agronomy, 10(4), 502. https://doi.org/10.3390/agronomy10040502