Comparative Transcriptome Analysis Identified Differentially Expressed Genes between Male and Female Flowers of Zanthoxylum armatum var. novemfolius

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Morphological Observation
2.3. RNA Extraction, Library Construction, and RNA-Seq
2.4. De Novo Assembly and Functional Annotation
2.5. Analysis of Differentially Expressed Genes (DEGs)
2.6. Identification of Transcription Factors (TFs)
2.7. qRT-PCR Verification
3. Results
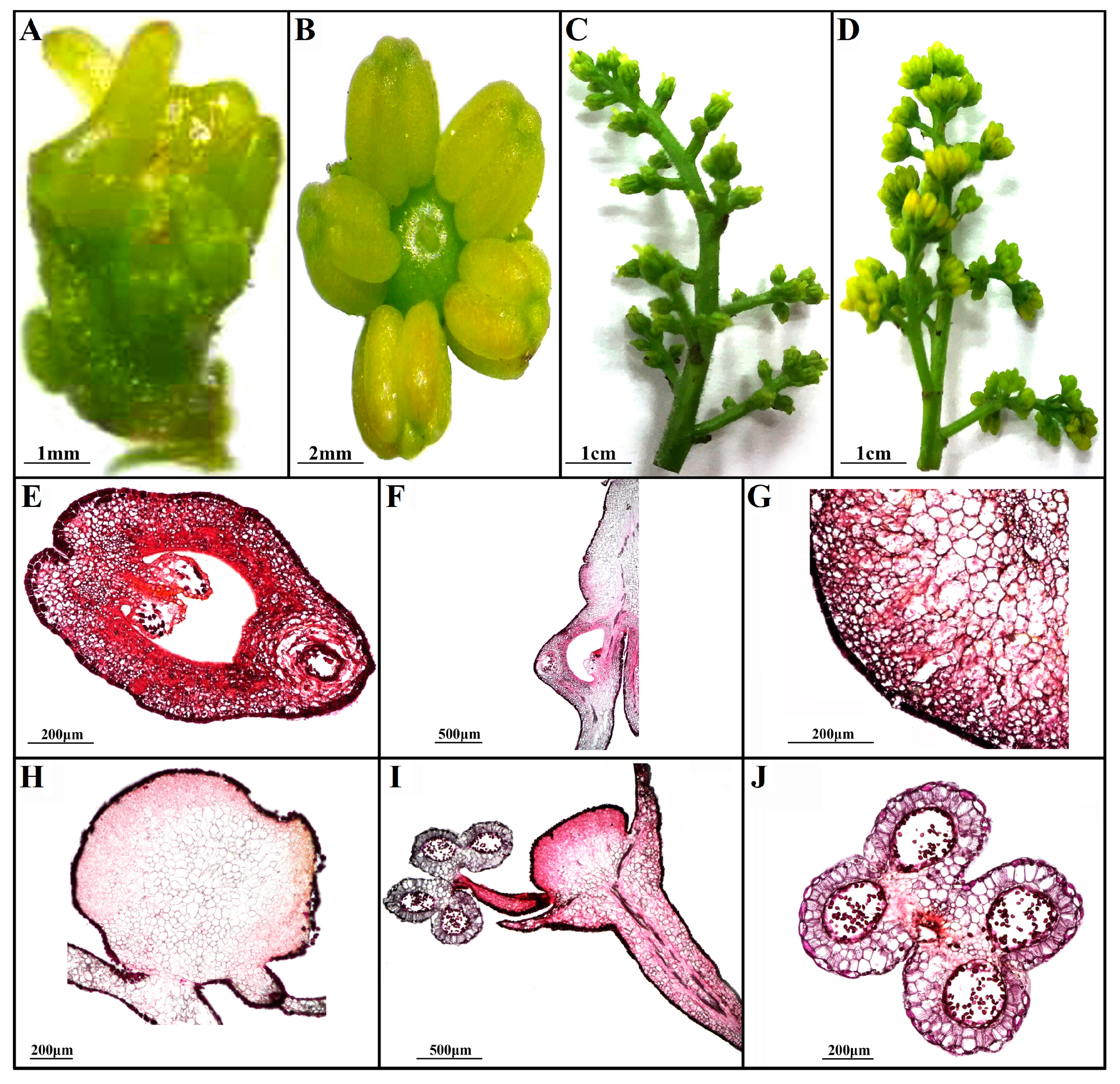
3.1. Morphological Analysis of FFS and MFS in Z. armatum
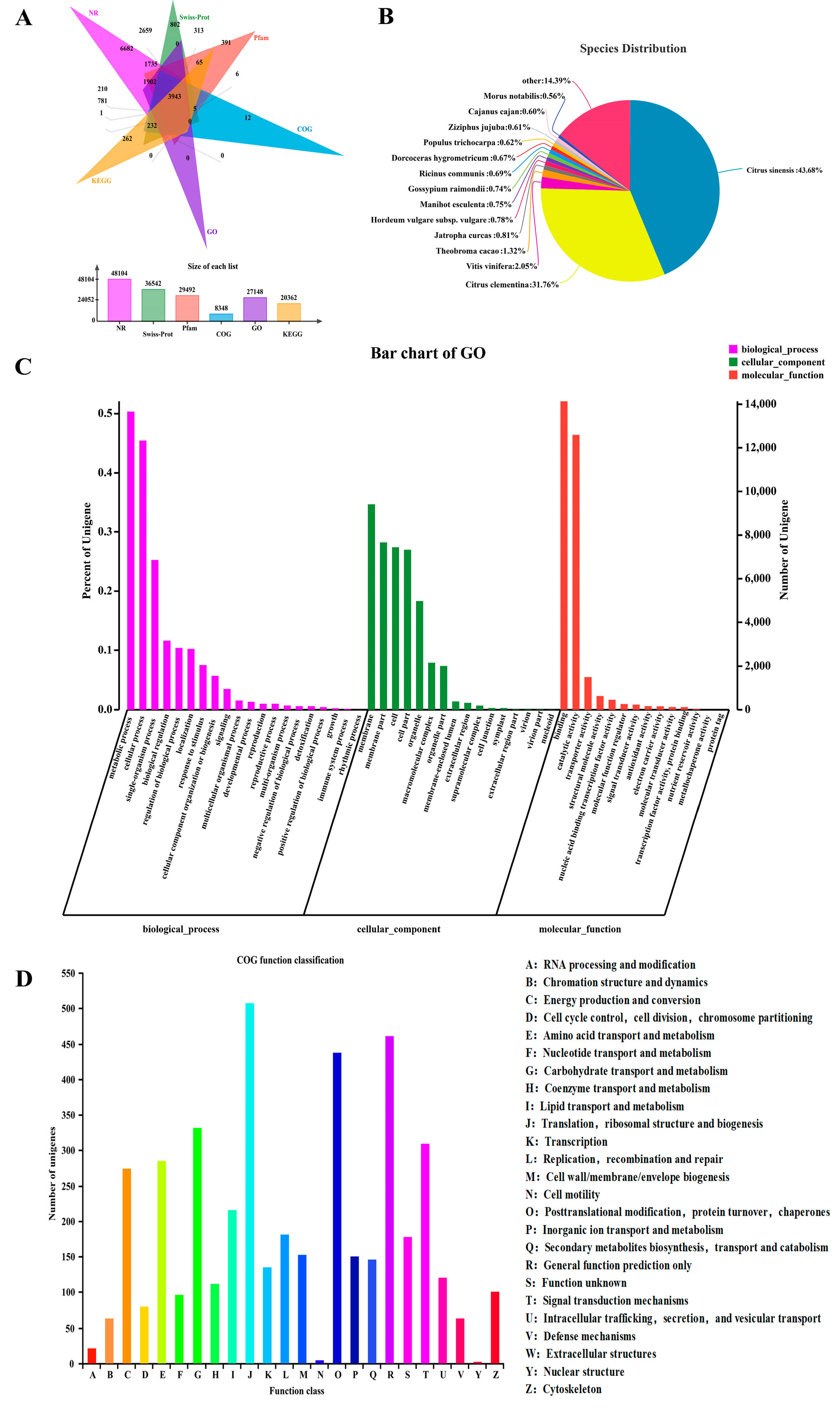
3.2. Sequencing Analysis and De Novo Assembly
3.3. Functional Classification
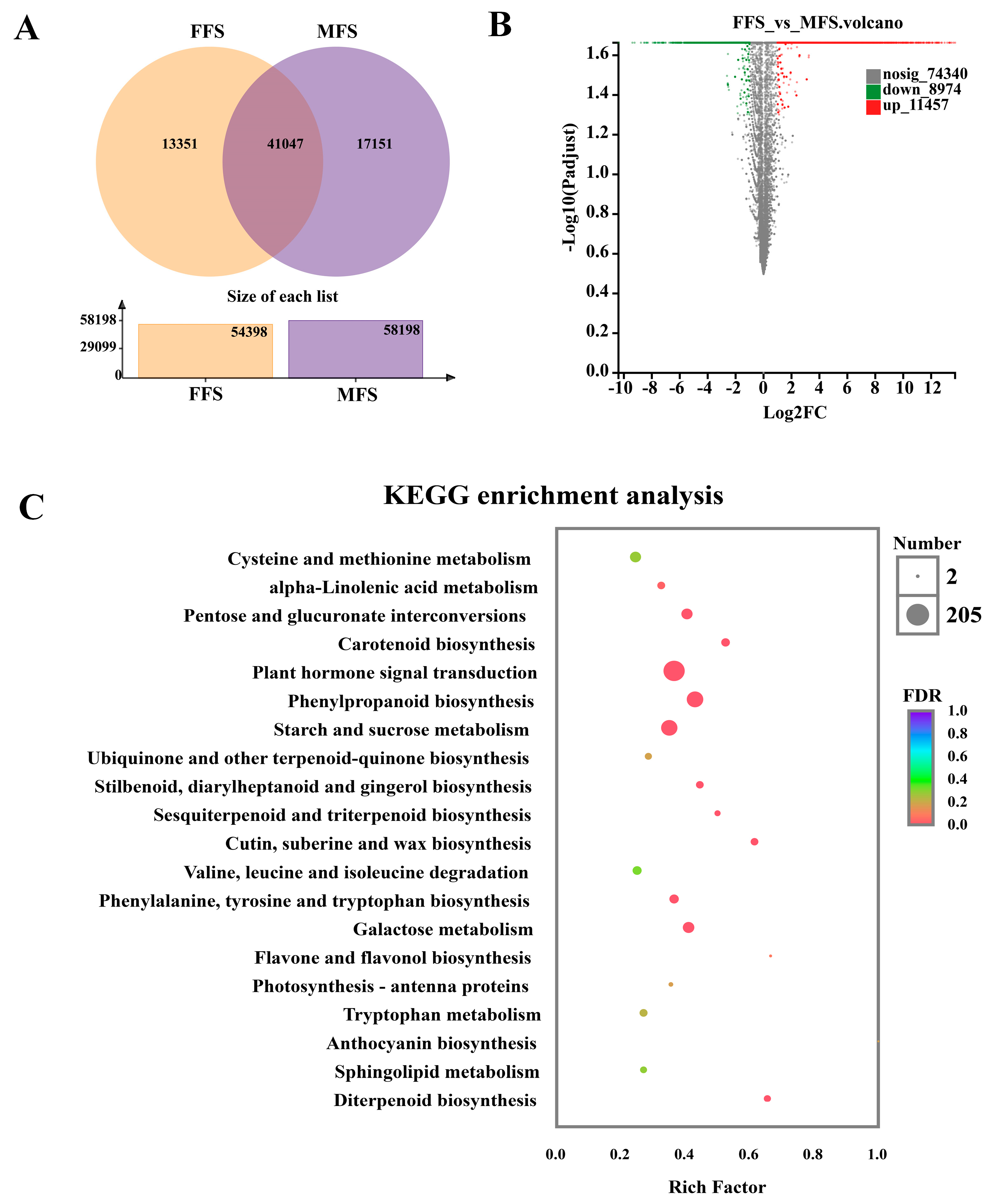
3.4. DEG Analysis
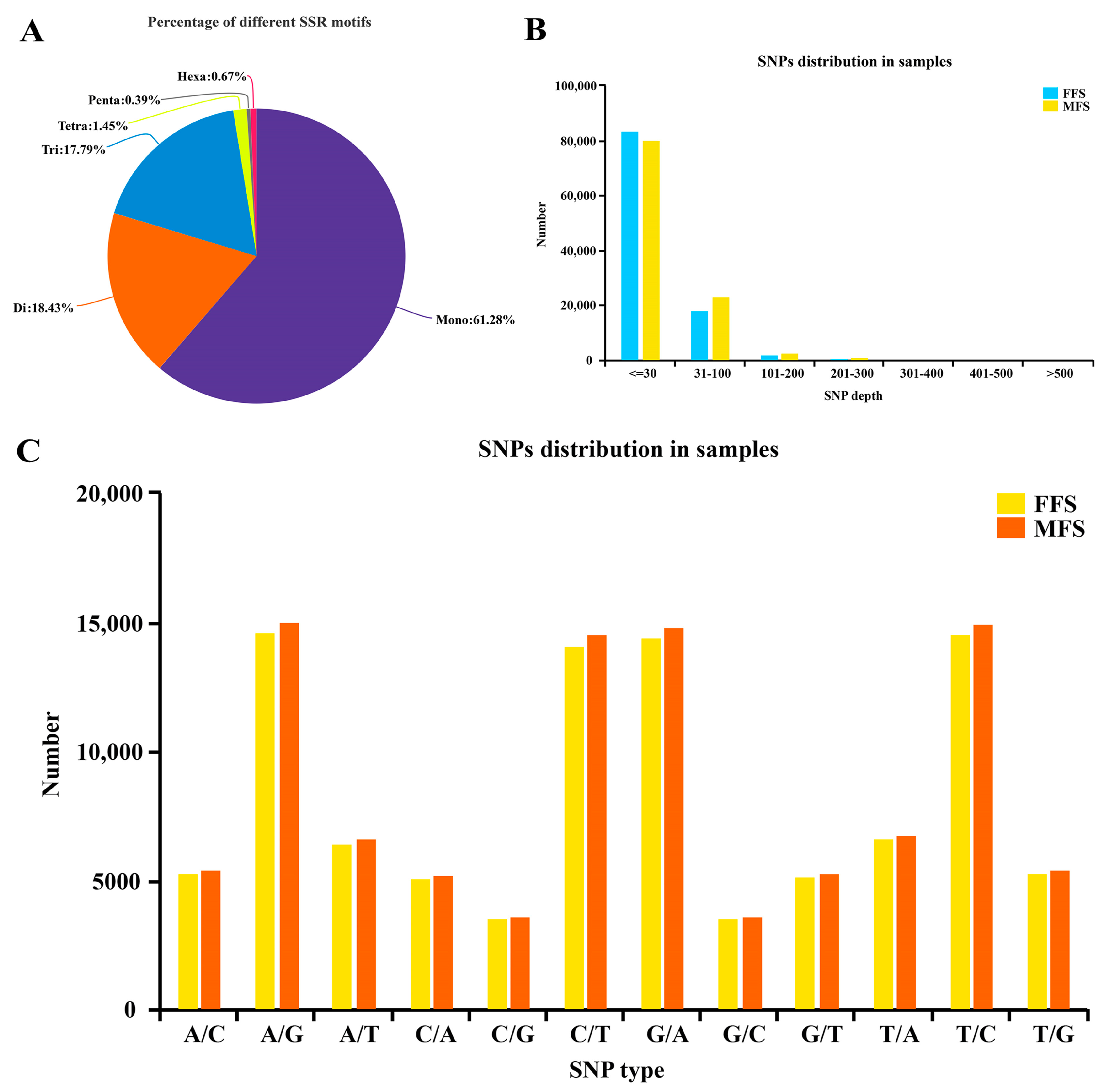
3.5. Simple Sequence Repeats (SSRs) and Single Nucleotide Polymorphisms (SNPs) Detection
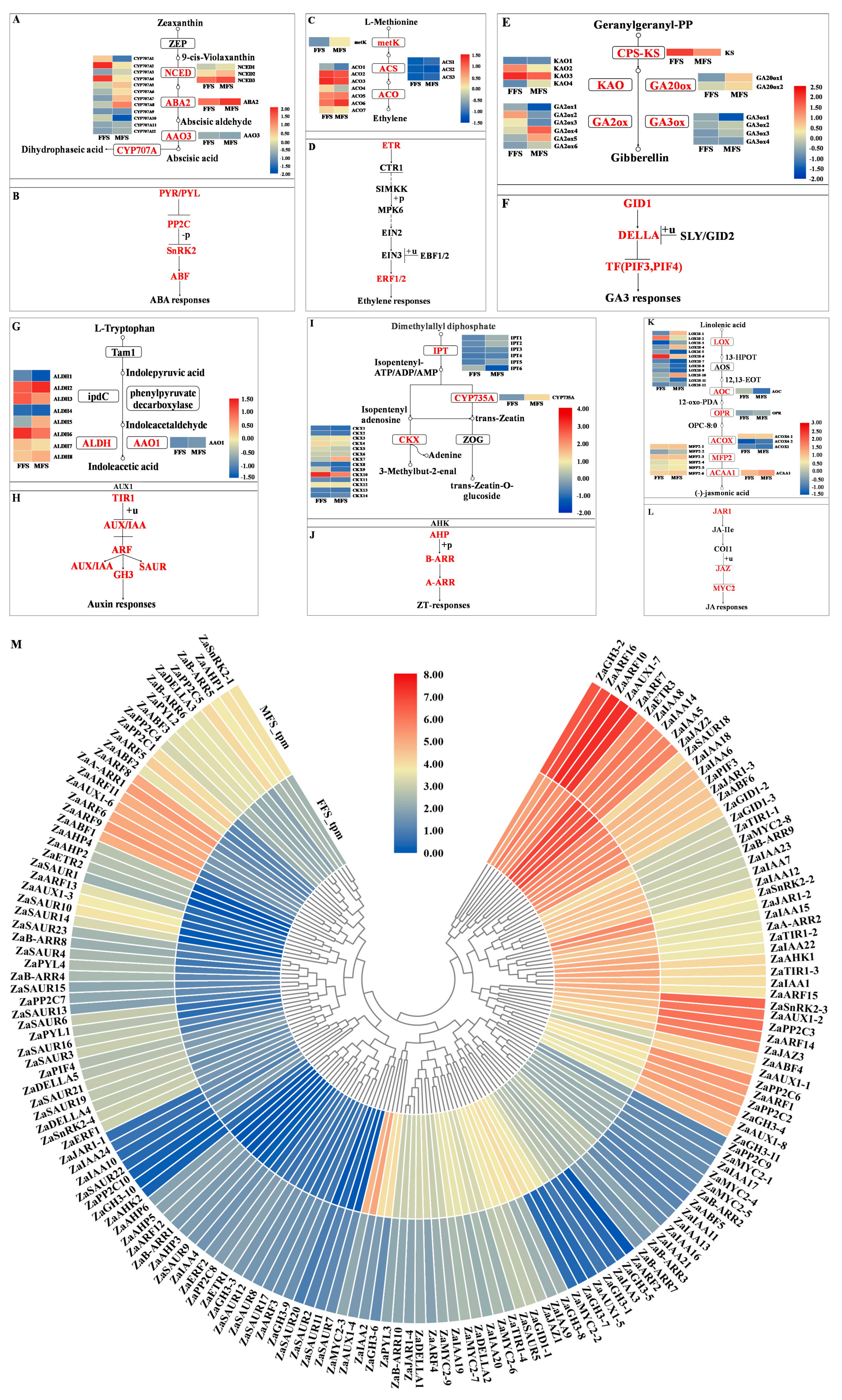
3.6. Phytohormone-Related DEGs
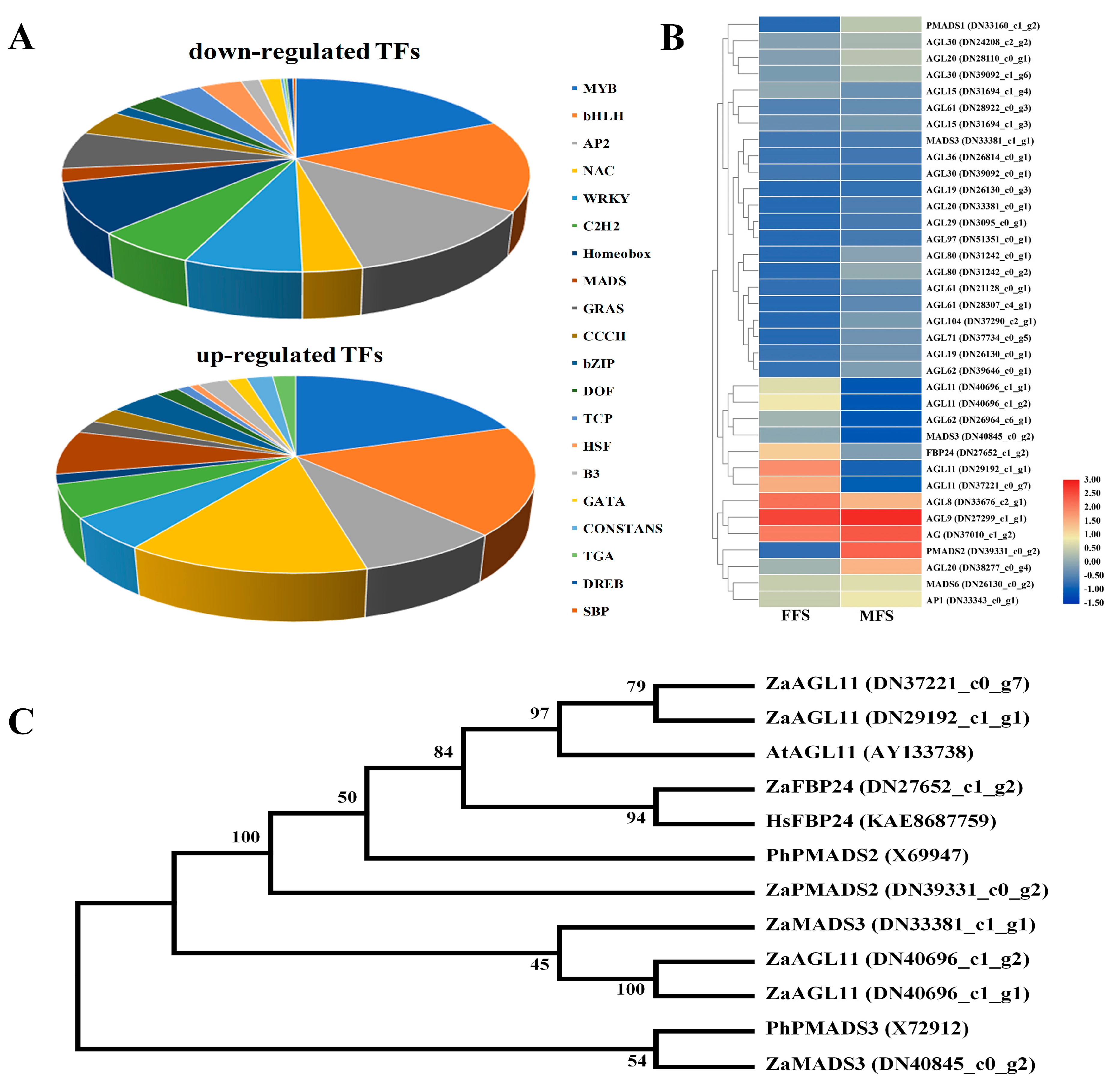
3.7. Expression Profiling of Candidate Transcription Factors
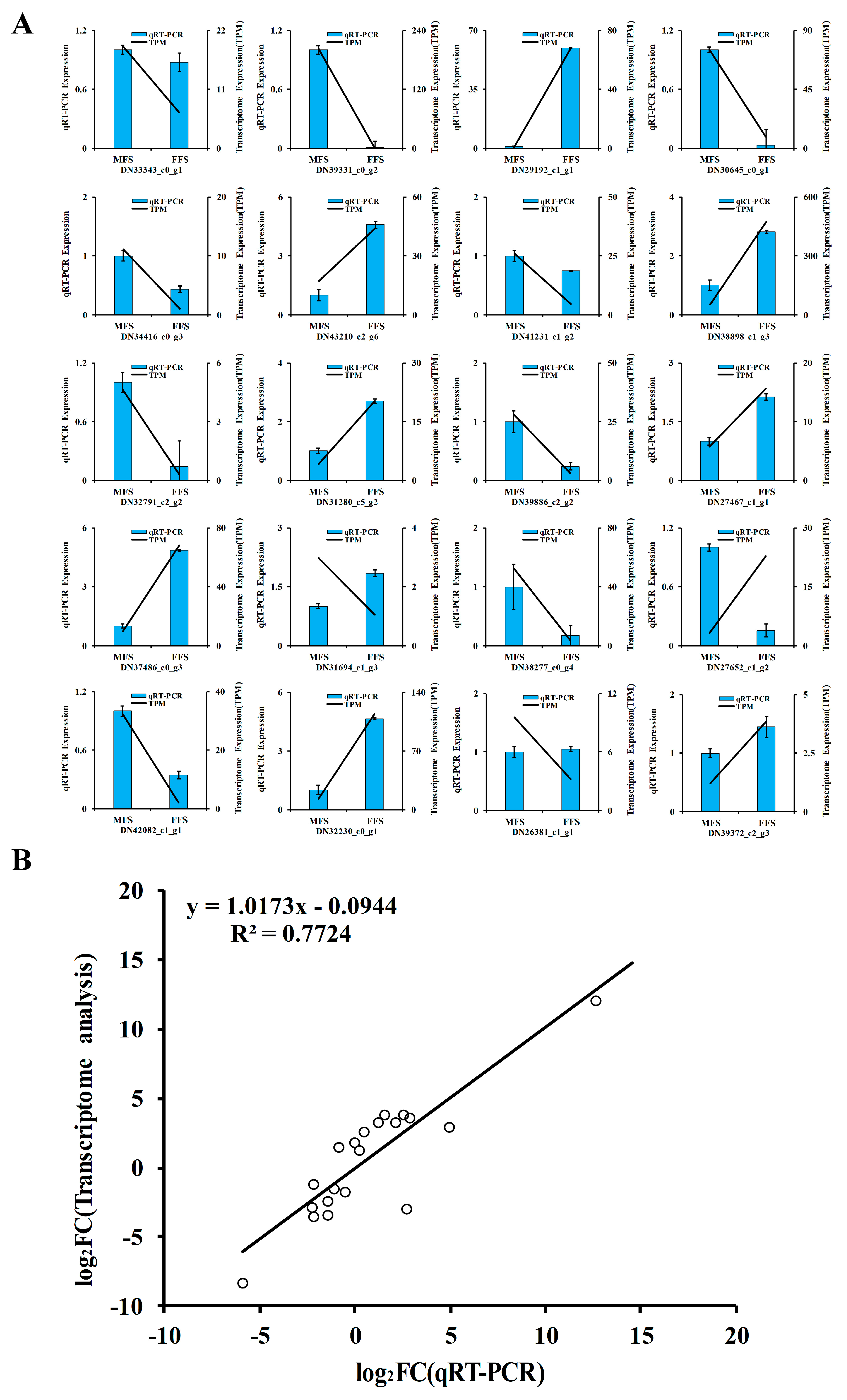
3.8. Validation of Candidate Genes by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Deng, H.P.; Xu, J.; Chen, F.; Song, Q.Z. Morphological and molecular identification on genetic diversity of Zanthoxylum armatum var. novemfolius. Acta Bot. Boreal-Occident. Sin. 2008, 28, 2103–2109. [Google Scholar]
- Qin, J.; Chen, T.; Li, Q. Simultaneous distillation and solvent extraction and GC/MS analysis of volatile oils of Zanthoxylum Bungeagum Maxim. J. Guizhou Univ. Technol. 2001, 6, 4–6. [Google Scholar]
- Ó’Maoiléidigh, D.S.; Graciet, E.; Wellmer, F. Gene networks controlling Arabidopsis thaliana flower development. New Phytol. 2014, 201, 16–30. [Google Scholar] [CrossRef]
- Sobral, R.; Silva, H.G.; Morais-Cecílio, L.; Costa, M.M. The quest for molecular regulation underlying unisexual flower development. Front. Plant Sci. 2016, 7, 160. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Ge, W.; Li, L.; Hou, D.; Ma, Y.; Liu, J.; Bai, Q.; Li, X.; Mu, S.; Gao, J. Analysis of MADS-box gene family reveals conservation in floral organ ABCDE model of Moso bamboo (Phyllostachys edulis). Front. Plant Sci. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Theissen, G.; Becker, A.; Di Rosa, A.; Kanno, A.; Kim, J.T.; Münster, T.; Winter, K.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef] [PubMed]
- Mibus, H.; Tatlioglu, T. Molecular characterization and isolation of the F/f gene for femaleness in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2004, 109, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Acosta, I.F.; Laparra, H.; Romero, S.P.; Schmelz, E.; Hamberg, M.; Mottinger, J.P.; Moreno, M.A.; Dellaporta, S.L. tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 2009, 323, 262–265. [Google Scholar] [CrossRef]
- Akagi, T.; Henry, I.M.; Ohtani, H.; Morimoto, T.; Beppu, K.; Kataoka, I.; Tao, R. A Y-encoded suppressor of feminization arose via lineage-specific duplication of a cytokinin response regulator in kiwifruit. Plant Cell 2018, 30, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, J. Suitable cryo-sectioning technique in floral organs of plants. J. Wuhan Bot. Res. 2005, 23, 285–290. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Günter, P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, D.; Li, X.; Yang, H.; Zhang, Y.; Wood, A.J. De novo transcriptome characterization and gene expression profiling of the desiccation tolerant moss Bryum argenteum following rehydration. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Niu, L.J.; Tao, Y.B.; Chen, M.S.; Fu, Q.T.; Li, C.Q. Selection of reliable reference genes for gene expression studies of a promising oilseed crop, Plukenetia volubilis, by real-time quantitative PCR. Int. J. Mol. Sci. 2015, 16, 12513–12530. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sarwar, M.B.; Ahmad, Z.; Rashid, B.; Hassan, S.; Gregersen, P.L.; Leyva, M.O.; Nagy, I.; Asp, T.; Husnain, T. De novo assembly of Agave sisalana transcriptome in response to drought stress provides insight into the tolerance mechanisms. Sci. Rep. 2019, 9, 396. [Google Scholar] [CrossRef]
- Murat, C.; Riccioni, C.; Belfiori, B.; Cichocki, N.; Labbé, J.; Morin, E.; Tisserant, E.; Paolocci, F.; Rubini, A.; Martin, F. Distribution and localization of microsatellites in the Perigord black truffle genome and identification of new molecular markers. Fungal Genet. Biol. 2011, 48, 0–601. [Google Scholar] [CrossRef]
- Liu, X.M.; Wang, X.H.; Chen, Z.X.; Ye, J.B.; Liao, Y.L.; Zhang, W.W.; Chang, J.; Xu, F. De novo assembly and comparative transcriptome analysis: Novel insights into terpenoid biosynthesis in Chamaemelum nobile L. Plant Cell Rep. 2019, 38, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, Y.; Zhang, L.; Zhuge, Q. Identification and validation of single nucleotide polymorphisms in poplar using publicly expressed sequence tags. J. Integr. Plant Biol. 2005, 12, 1493–1499. [Google Scholar] [CrossRef]
- Golenberg, E.M.; West, N.W. Hormonal interactions and gene regulation can link monoecy and environmental plasticity to the evolution of dioecy in plants. Am. J. Bot. 2013, 100, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Leebens-Mack, J. A century of sexual determination in flowering plants. J. Hered. 2017, 108, 69–77. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, J.; Lou, H.; Wang, H.Z.; Xu, Q.J. Comparative transcriptome analysis of dioecious, unisexual floral development in Ribes diacanthum pall. Gene 2019, 699, 43–53. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, G.; Xuan, J.; Zhang, J.; Zhai, M.; Jia, X.; Guo, Z.; Li, M. Comparative transcriptome analysis of pecan female and male Inflorescences. Russ. J. Plant Physl. 2018, 65, 186–196. [Google Scholar] [CrossRef]
- Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Becker, A.; Kaufmann, K.; Freialdenhoven, A.; Vincent, C.; Li, M.A.; Saedler, H.; Theissen, G. A novel MADS-box gene subfamily with a sister-group relationship to class B floral homeotic genes. Mol. Genet. Genom. 2002, 266, 942–950. [Google Scholar] [CrossRef]
- Ito, T.; Nagata, N.; Yoshiba, Y.; Ohme-Takagi, M.; Ma, H.; Shinozaki, K. Arabidopsis MALE STERILITY1 encodes a PHD-type transcription factor and regulates pollen and tapetum development. Plant Cell 2007, 19, 3549–3562. [Google Scholar] [CrossRef]
- Baumberger, N.; Doesseger, B.; Guyot, R.; Diet, A.; Parsons, R.L.; Clark, M.A.; Simmons, M.P.; Bedinger, P.; Goff, S.A.; Ringli, C.; et al. Whole-genome comparison of leucine-rich repeat extensins in Arabidopsis and rice. A conserved family of cell wall proteins form a vegetative and a reproductive clade. Plant Physiol. 2003, 131, 1313–1326. [Google Scholar] [CrossRef]
- Nacken, W.K.; Huijser, P.; Beltran, J.P.; Saedler, H.; Sommer, H. Molecular characterization of two stamen-specific genes, tap1 and fil1, that are expressed in the wild type, but not in the deficiens mutant of Antirrhinum majus. Mol. Gen. Genet. 1991, 229, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.C.; Pearce, S.; Band, L.R.; Yang, C.; Ferjentsikova, I.; King, J.; Yuan, Z.; Zhang, D.; Wilson, Z.A. Biphasic regulation of the transcription factor ABORTED MICROSPORES (AMS) is essential for tapetum and pollen development in Arabidopsis. New Phytol. 2017, 213, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Jia, Y.; Yang, J.; Li, Z.H. Comparative transcriptome analysis of male and female conelets and development of microsatellite markers in Pinus bungeana, an endemic conifer in China. Genes 2017, 8, 393. [Google Scholar] [CrossRef] [PubMed]
- Elgizawy, A.M.; Eloksh, I.; Sharaf, A. Effect of gibberellic acid and alar on flowering and seed yield of spinach. Egypt. J. Hortic. 1992, 19, 191–200. [Google Scholar]
- Chen, X.H.; Zeng, G.G.; Cao, B.S. Relationship between endogenous plant hormones and floral sexual differentiation in cucumber (Cucumis sativus). Plant Physiol. Commun. 2002, 38, 317–320. [Google Scholar]
- Zhao, P.Q.; Yu, W.W.; Wang, Q.M. Effect of plant growth Regulator on Sexual Expression and Fruit Development of Bitter Gourd. China Veg. 2005, 1, 17–18. [Google Scholar]
- Yamasaki, S.; Fujii, N.; Matsuura, S.; Mizusawa, H.; Takahashi, H. The M locus and ethylene-controlled sex determination in andromonoecious cucumber plants. Plant Cell Physiol. 2001, 42, 608–619. [Google Scholar] [CrossRef]
- Boualem, A.; Fergany, M.; Fernandez, R.; Troadec, C.; Martin, A.; Morin, H.; Sari, M.A.; Collin, F.; Flowers, J.M.; Pitrat, M.; et al. A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science 2008, 321, 836–838. [Google Scholar] [CrossRef]
- Yan, L.W.; Chen, Y.Y.; Liu, Z.C. Effects of ethephon on activities of oxidase enzymes and sexual differentiation of bitter gourd. J. South. Agric. 2017, 48, 1844–1848. [Google Scholar]
- Liu, Y.; Zhong, Z.C.; Wang, X.X.; Xie, J.; Yang, W.Y. Comparison in contents of endogenous hormones and polyamines in female and male plants of Trichosanthes Kirilowii. Acta Hortic. Sin. 2010, 37, 1645–1650. [Google Scholar]
- Zhang, H.J.; Wang, X.Z.; Gao, P.; Gao, M.L.; Luan, F.S. Progress of study on sex differentiation in melon. Acta Hortic. Sin. 2012, 39, 1773–1780. [Google Scholar]
- Staswick, P.E. Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell. 2015, 17, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Liscum, E.; Reed, J.W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 2002, 49, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Schauer, N.; Usadel, B.; Frasse, P.; Zouine, M.; Hernould, M.; Latché, A.; Pech, J.C.; Fernie, A.R.; Bouzayen, M. Regulatory features underlying pollination-dependent and -independent tomato fruit set revealed by transcript and primary metabolite profiling. Plant Cell 2009, 21, 1428–1452. [Google Scholar] [CrossRef]
- Pagnussat, G.C.; Alandete-Saez, M.; Bowman, J.L.; Sundaresan, V. Auxin-dependent patterning and gamete specification in the Arabidopsis female gametophyte. Science 2009, 324, 1684–1689. [Google Scholar] [CrossRef]
- Ai, J.; Li, A.; Li, C.; Wang, J.; Shen, Y. The effect of cytokinin on sex conversion of male plants of Vitis amurensis. Acta Hortic. Sin. 2002, 29, 163–164. [Google Scholar]
- Yan, Y.; Christensen, S.; Isakeit, T.; Engelberth, J.; Meeley, R.; Hayward, A.; Kolomiets, M.V. Disruption of OPR7 and OPR8 reveals the versatile functions of jasmonic acid in maize development and defense. Plant Cell 2012, 24, 1420–1436. [Google Scholar] [CrossRef]
- Bemer, M.; Heijmans, K.; Airoldi, C.; Heijmans, K.; Airoldi, C.; Davies, B.; Angenent, G.C. An atlas of type I MADS Box gene expression during female gametophyte and seed development in Arabidopsis. Plant Physiol. 2010, 154, 287–300. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X. Identification of the sex-biased gene expression and putative sex-associated genes in Eucommia ulmoides Oliver using comparative transcriptome analyses. Molecules 2017, 22, 2255. [Google Scholar] [CrossRef]
- Tsuchimoto, S.; van der Krol, A.R.; Chua, N.H. Ectopic expression of pMADS3 in transgenic petunia phenocopies the petunia blind mutant. Plant Cell 1993, 5, 843–853. [Google Scholar]
- Folter, S.D.; Shchennikova, A.V.; Franken, J.; Busscher, M.; Baskar, R.; Grossniklaus, U.; Angenent, G.C.; Immink, R.G. A Bsister MADS-box gene involved in ovule and seed development in petunia and Arabidopsis. Plant J. 2006, 47, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Zluvova, J.; Nicolas, M.; Berger, A.; Negrutiu, I.; Monéger, F. Premature arrest of the male flower meristem precedes sexual dimorphism in the dioecious plant Silene latifolia. Proc. Natl. Acad. Sci. USA 2006, 103, 18854–18859. [Google Scholar] [CrossRef]
- Makkena, S.; Lee, E.; Sack, F.D.; Lamb, R.S. The R2R3 MYB transcription factors FOUR LIPS and MYB88 regulate female reproductive development. J. Exp. Bot. 2012, 63, 5545–5558. [Google Scholar] [CrossRef]
- Gonzalez, A.; Mendenhall, J.; Huo, Y.; Lloyd, A. TTG1 complex MYBs, MYB5 and TT2, control outer seed coat differentiation. Dev. Biol. 2009, 325, 412–421. [Google Scholar] [CrossRef]
- Song, Y.; Ma, K.; Ci, D.; Zhang, Z.; Zhang, D. Sexual dimorphism floral microRNA profiling and target gene expression in andromonoecious poplar (Populus tomentosa). PLoS ONE 2013, 8. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Reads | Clean Bases | Q30 (%) | N50 | Average Length (nt) | Total Unigenes (num) |
|---|---|---|---|---|---|---|
| Female flowers (FFS) | 65,533,834 | 9,602,149,462 | 93.23 | 1151 | 826.88 | 94,771 |
| Male flowers (MFS) | 77,155,592 | 11,300,693,240 | 93.55 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Tang, N.; Liu, X.; Ye, J.; Zhang, J.; Chen, Z.; Xu, F.; Zhang, W.; Liao, Y. Comparative Transcriptome Analysis Identified Differentially Expressed Genes between Male and Female Flowers of Zanthoxylum armatum var. novemfolius. Agronomy 2020, 10, 283. https://doi.org/10.3390/agronomy10020283
Zhang X, Tang N, Liu X, Ye J, Zhang J, Chen Z, Xu F, Zhang W, Liao Y. Comparative Transcriptome Analysis Identified Differentially Expressed Genes between Male and Female Flowers of Zanthoxylum armatum var. novemfolius. Agronomy. 2020; 10(2):283. https://doi.org/10.3390/agronomy10020283
Chicago/Turabian StyleZhang, Xian, Ning Tang, Xiaomeng Liu, Jiabao Ye, Jingyi Zhang, Zexiong Chen, Feng Xu, Weiwei Zhang, and Yongling Liao. 2020. "Comparative Transcriptome Analysis Identified Differentially Expressed Genes between Male and Female Flowers of Zanthoxylum armatum var. novemfolius" Agronomy 10, no. 2: 283. https://doi.org/10.3390/agronomy10020283
APA StyleZhang, X., Tang, N., Liu, X., Ye, J., Zhang, J., Chen, Z., Xu, F., Zhang, W., & Liao, Y. (2020). Comparative Transcriptome Analysis Identified Differentially Expressed Genes between Male and Female Flowers of Zanthoxylum armatum var. novemfolius. Agronomy, 10(2), 283. https://doi.org/10.3390/agronomy10020283