
Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review

Abstract
:
1. Introduction
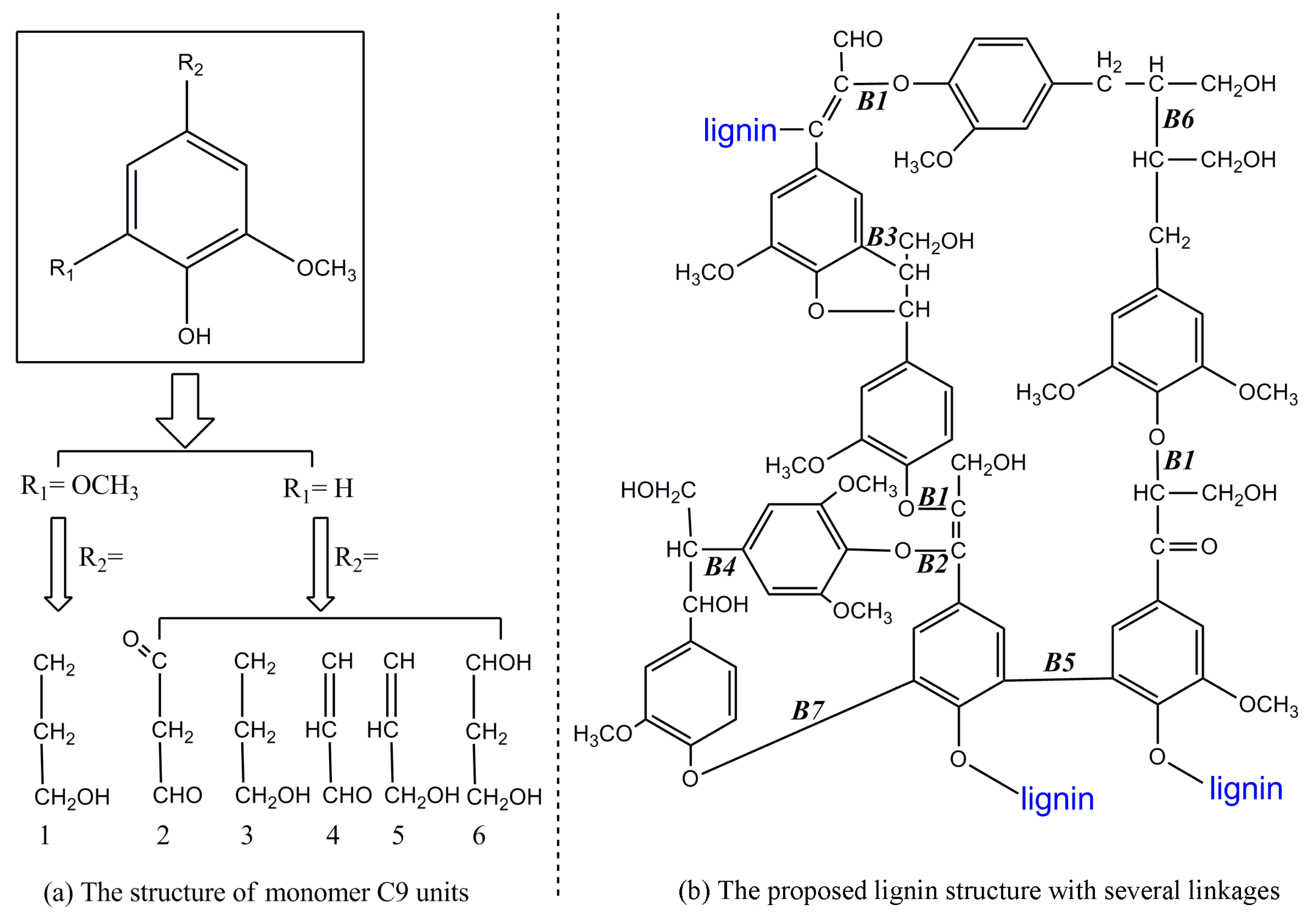
2. Chemical Structure of Lignin
2.1. Kraft Lignin
2.2. Sulfite Lignin
2.3. Organosolv Lignin
2.4. Pyrolytic Lignin
2.5. Dilute Acid Lignin
2.6. Steam Explosion Lignin
3. Oxidative Solvolysis of Lignin
3.1. Effects of Catalysts
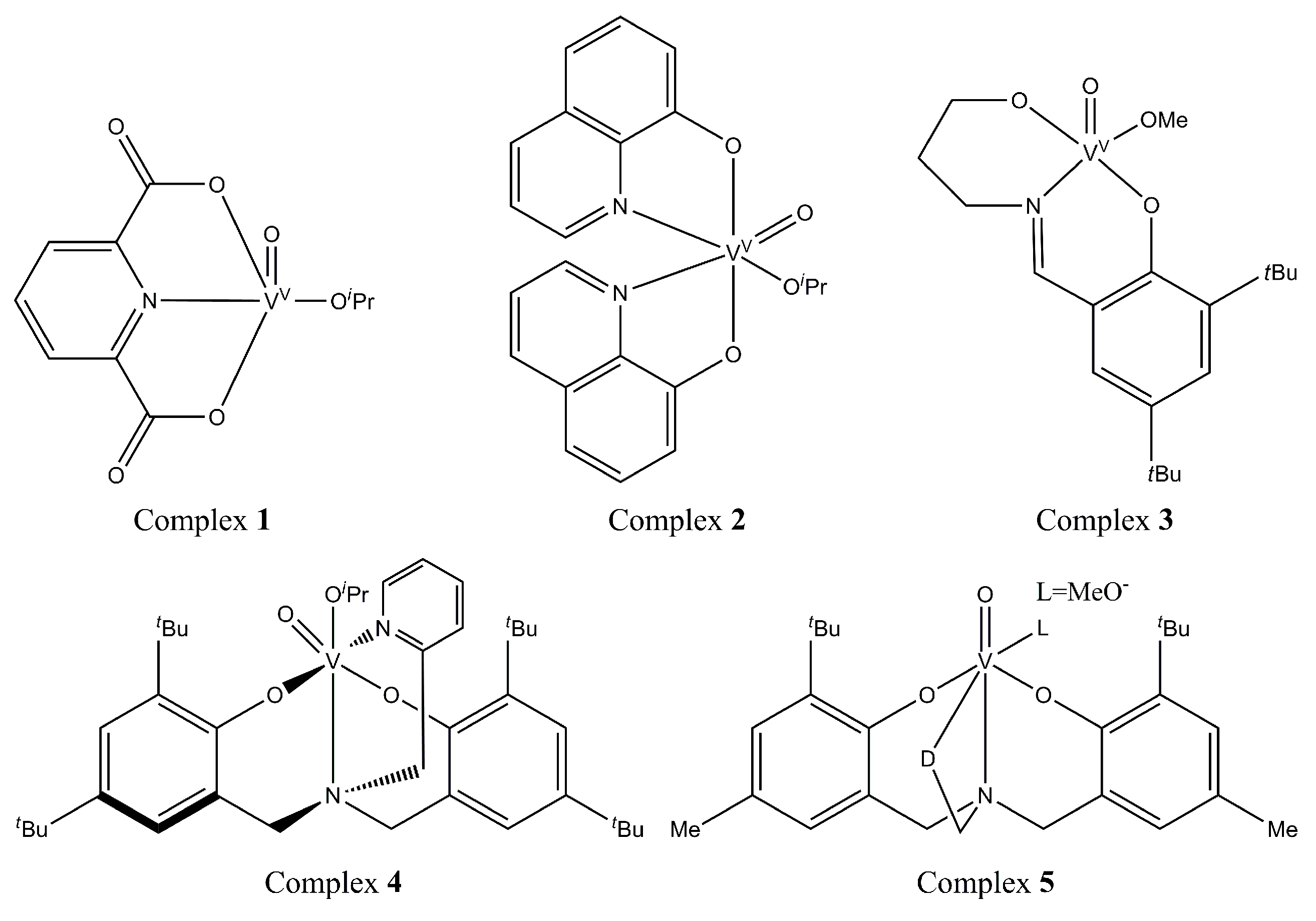
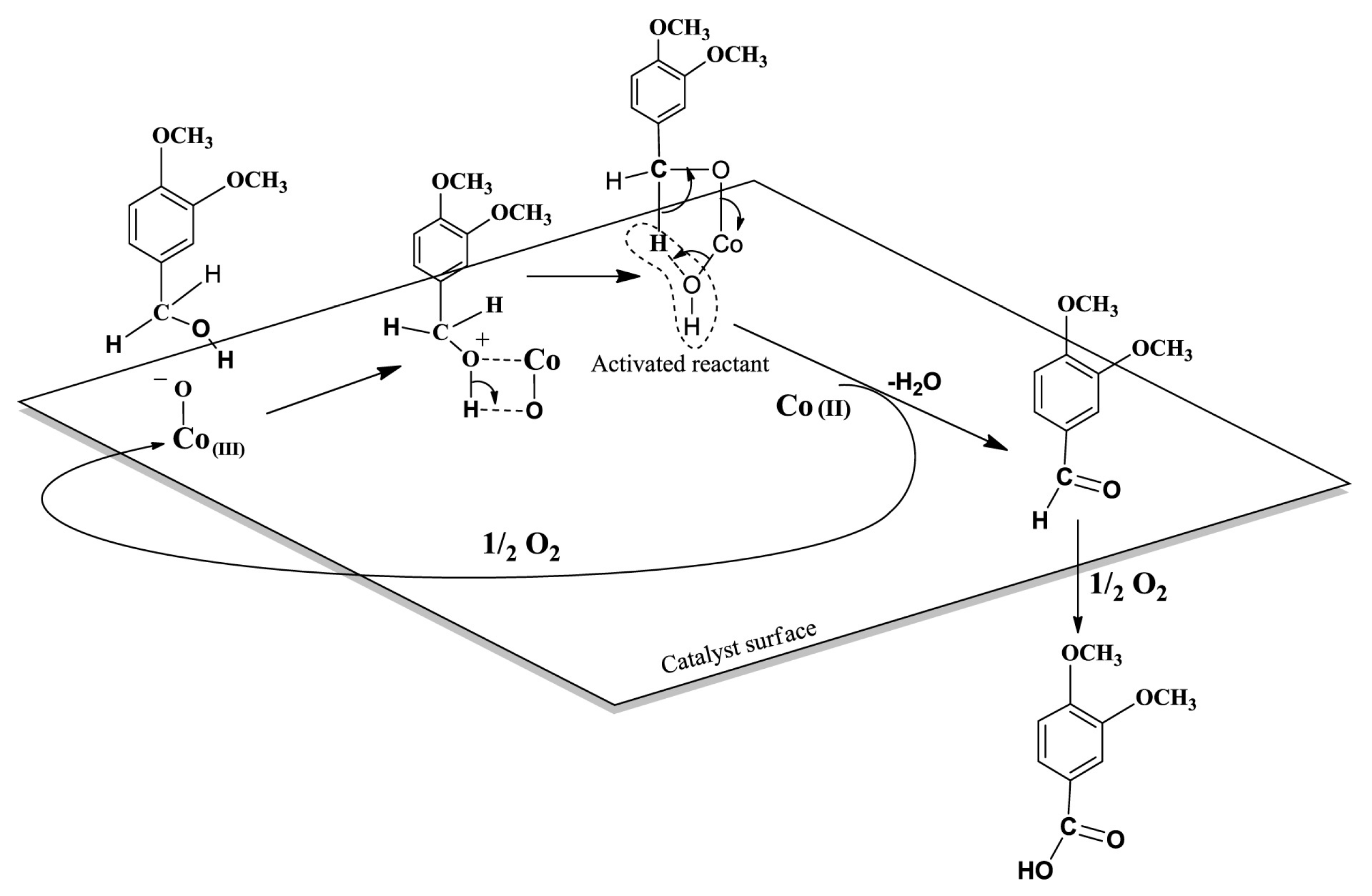
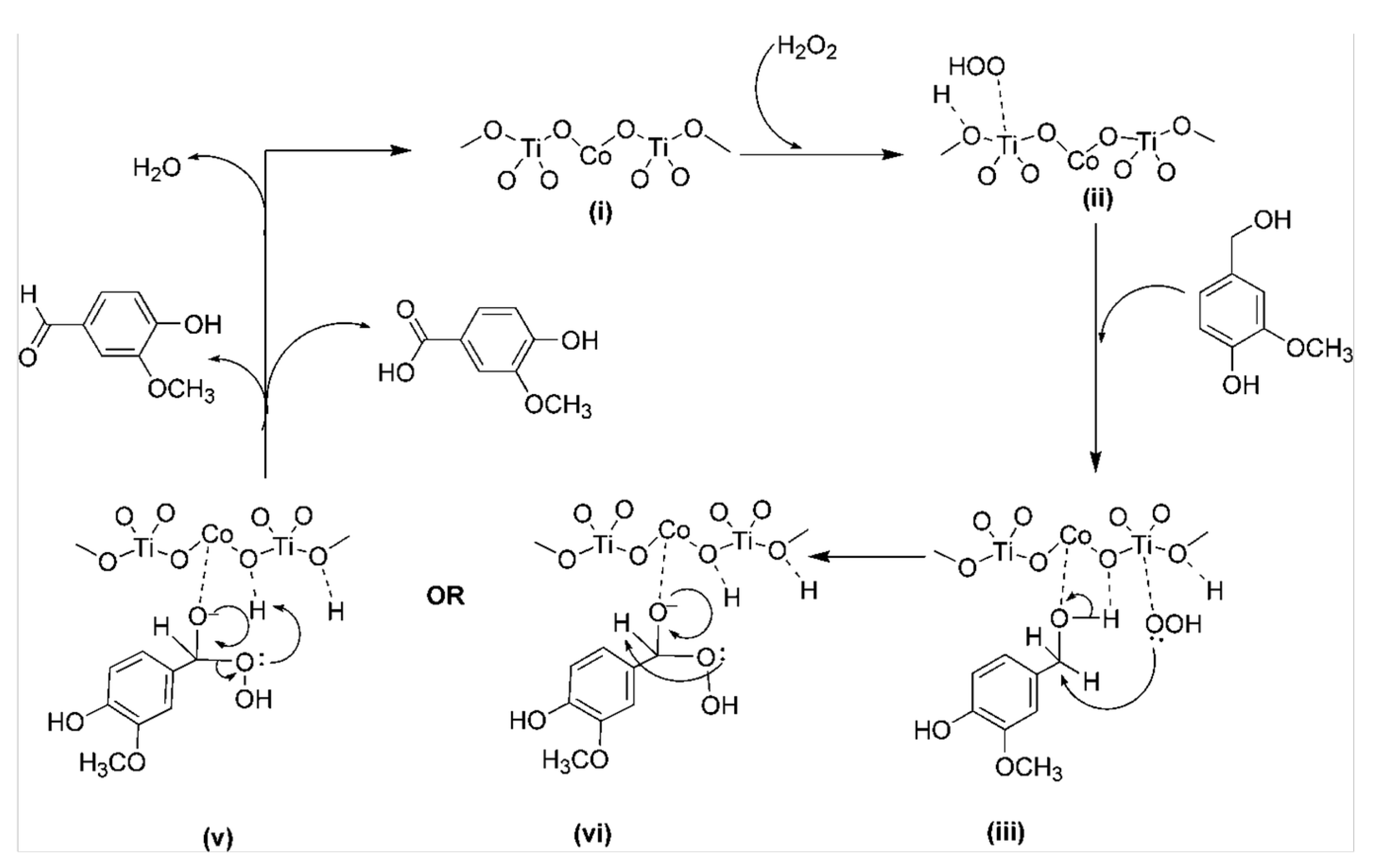
3.1.1. Organometallic Catalysts
3.1.2. Metal-Free-Organic Catalysts
3.1.3. Acid/Base Catalysts
3.1.4. Metal Salt Catalysts
3.1.5. Heterogeneous Catalyst
3.2. Effects of Solvent System
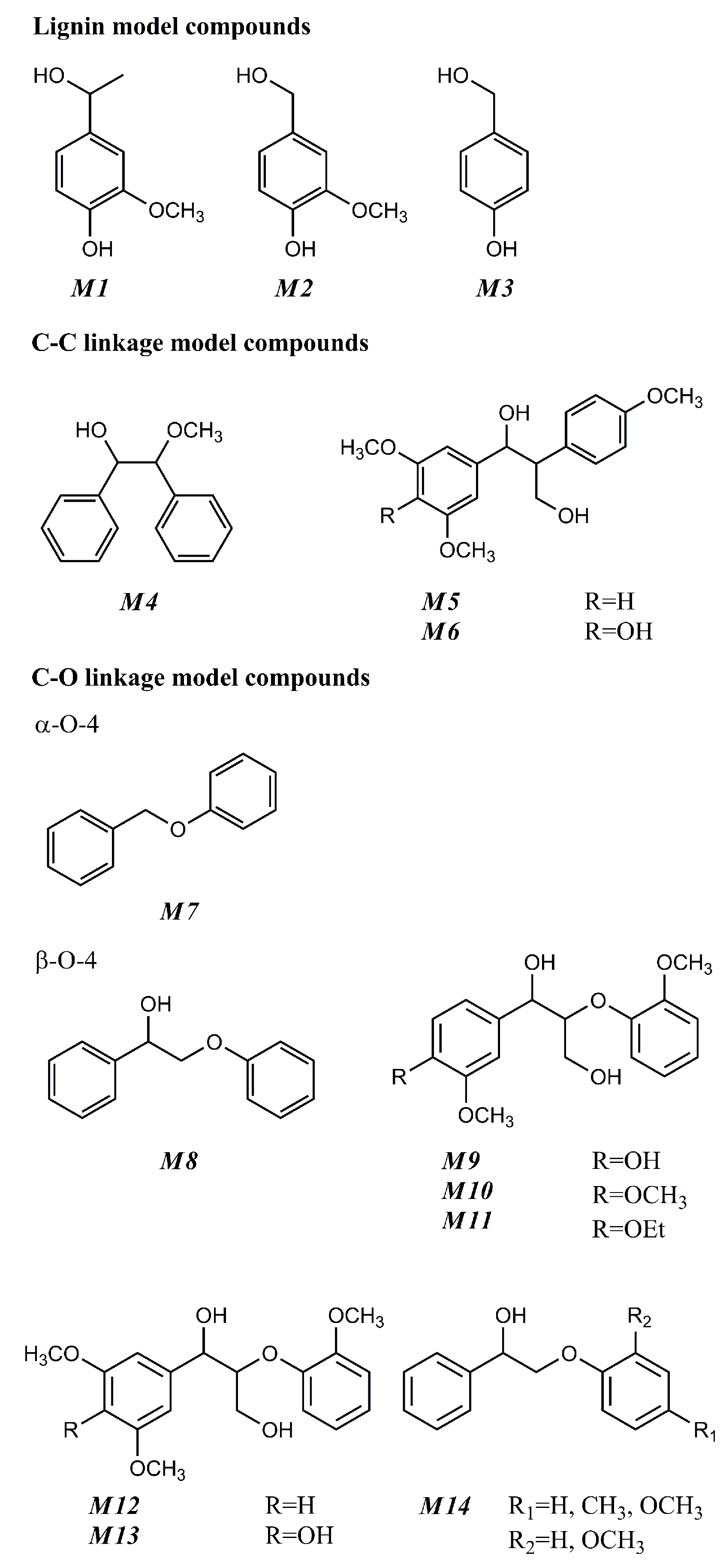
4. Oxidative Solvolysis of Lignin-Related Model Compounds
4.1. Oxidative Reforming of Lignin-Derived Monomers
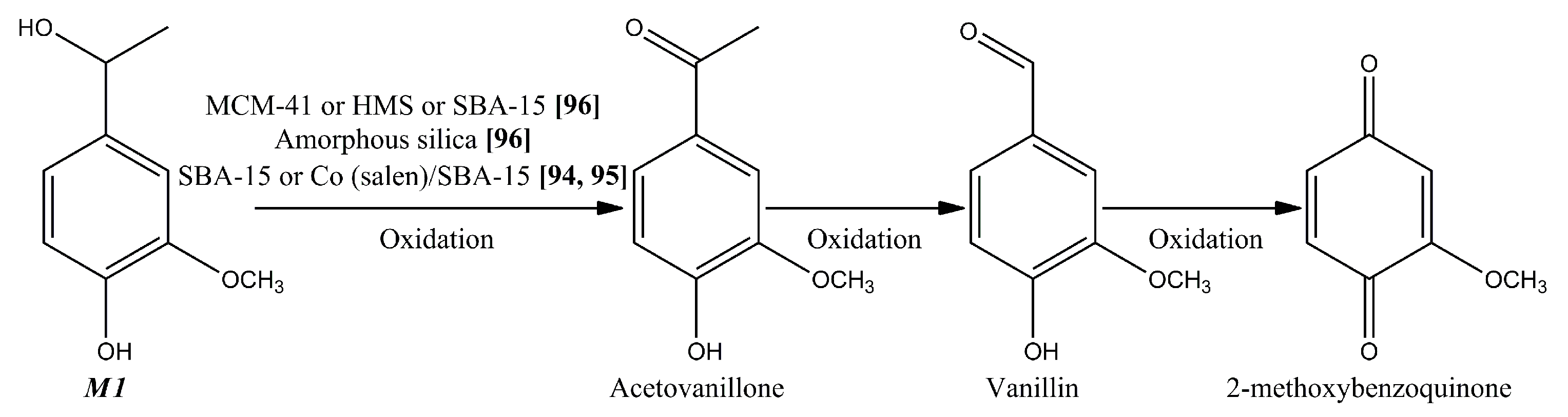
4.1.1. Apocynol
4.1.2. Veratryl alcohol/3-methoxy-4-hydroxybenzyl alcohol/4-hydroxybenzyl alcohol
4.2. Oxidative Cracking of Typical Inter-Unit Linkages in Lignin-Derived Oligomers
4.2.1. C–C Linkage
4.2.2. C–O Linkage
4.2.3. α-O-4 Containing Model Compound
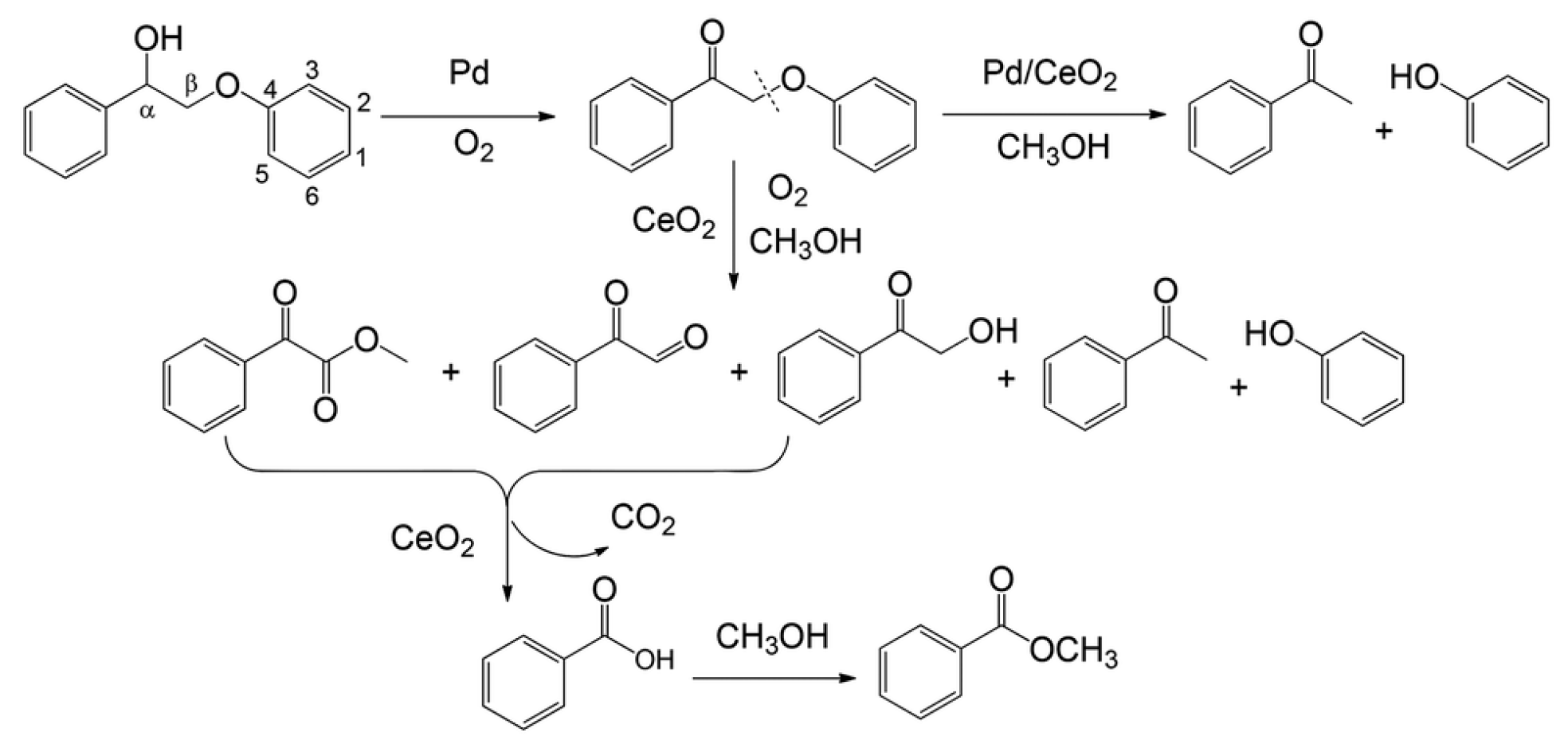
4.2.4. β-O-4 Containing Model Compound
5. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Sawin, J.L.; Sverrisson, F. Renewables 2014: Global Status Report; REN21 Secretariat: Paris, France, 2014; p. 214. [Google Scholar]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Melero, J.A.; Iglesias, J.; Garcia, A. Biomass as renewable feedstock in standard refinery units. Feasibility, opportunities and challenges. Energy Environ. Sci. 2012, 5, 7393–7420. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.O.; Perez, E.; Horvath, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Mamman, A.S.; Lee, J.-M.; Kim, Y.-C.; Hwang, I.T.; Park, N.-J.; Hwang, Y.K.; Chang, J.-S.; Hwang, J.-S. Furfural: Hemicellulose/xylose derived biochemical. Biofuels Bioprod. Biorefin. 2008, 2, 438–454. [Google Scholar] [CrossRef]
- Doherty, W.O.S.; Mousavioun, P.; Fellows, C.M. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crops Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Luque, R.; Sepúlveda-Escribanoa, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuelsvia aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R.; Palkovits, R.; Schüth, F. Depolymerization of cellulose using solid catalysts in ionic liquids. Angew. Chem. Int. Ed. 2008, 47, 8047–8050. [Google Scholar] [CrossRef] [PubMed]
- Gosselink, R.J.A.; de Jong, E.; Guran, B.; Abächerli, A. Co-ordination network for lignin-standardisation, production and applications adapted to market requirements. Ind. Crops Prod. 2004, 20, 121–129. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.N.; Goud, V.V.; Rout, P.K.; Dalai, A.K. Production of first and second generation biofuels: A comprehensive review. Renew. Sustain. Energy Rev. 2010, 14, 578–597. [Google Scholar] [CrossRef]
- Hu, L.H.; Pan, H.; Zhou, Y.H.; Zhang, M. Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: A brief review. BioResources 2011, 6, 3515–3525. [Google Scholar]
- Deuss, P.J.; Barta, K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar] [CrossRef]
- Kang, S.; Li, X.; Fan, J.; Chang, J. Hydrothermal conversion of lignin: A review. Renew. Sustain. Energy Rev. 2013, 27, 546–558. [Google Scholar] [CrossRef]
- Carpenter, D.; Westover, T.L.; Czernik, S.; Jablonski, W. Biomass feedstocks for renewable fuel production: A review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors. Green Chem. 2014, 16, 384–406. [Google Scholar] [CrossRef]
- Shen, D.; Xiao, R.; Gu, S.; Luo, K. The pyrolytic behavior of cellulose in lignocellulosic biomass: A review. RSC Adv. 2011, 1, 1641–1660. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Shen, D.; Jin, W.; Hu, J.; Xiao, R.; Luo, K. An overview on fast pyrolysis of the main constituents in lignocellulosic biomass to valued-added chemicals: Structures, pathways and interactions. Renew. Sustain. Energy Rev. 2015, 51, 761–774. [Google Scholar] [CrossRef]
- Chatel, G.; Rogers, R.D. Review: Oxidation of lignin using ionic liquids—An innovative strategy to produce renewable chemicals. ACS Sustain. Chem. Eng. 2014, 2, 322–339. [Google Scholar] [CrossRef]
- Behling, R.; Valange, S.; Chatel, G. Heterogeneous catalytic oxidation for lignin valorization into valuable chemicals: What results? What limitations? What trends? Green Chem. 2016, 18, 1839–1854. [Google Scholar] [CrossRef]
- Yokoyama, T. Revisiting the mechanism of β-O-4 bond cleavage during acidolysis of lignin. Part 6: A review. J. Wood Chem. Technol. 2014, 35, 27–42. [Google Scholar] [CrossRef]
- Mota, M.I.F.; Rodrigues Pinto, P.C.; Loureiro, J.M.; Rodrigues, A.E. Recovery of vanillin and syringaldehyde from lignin oxidation: A review of separation and purification processes. Sep. Purif. Technol. 2015, 45, 227–259. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidid, J.; Luque, R. Lignin depolymerisation strategies—Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
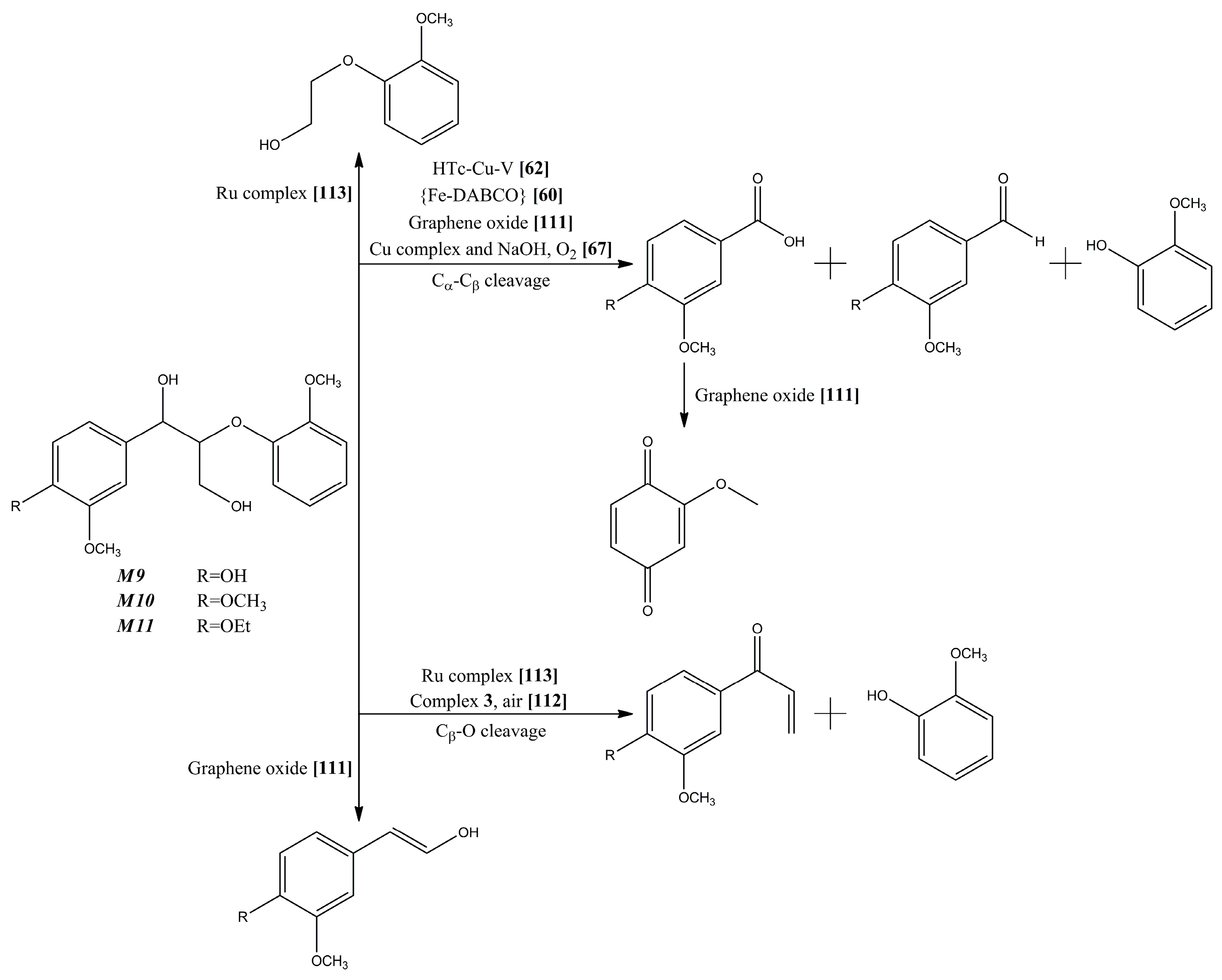
- Deuss, P.J.; Barta, K.; de Vries, J.G. Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals. Catal. Sci. Technol. 2014, 4, 1174–1196. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Collinson, S.R.; Thielemans, W. The catalytic oxidation of biomass to new materials focusing on starch, cellulose and lignin. Coord. Chem. Rev. 2010, 254, 1854–1870. [Google Scholar] [CrossRef]
- Dai, J.; Patti, A.F.; Saito, K. Recent developments in chemical degradation of lignin: Catalytic oxidation and ionic liquids. Tetrahedron Lett. 2016, 57, 4945–4951. [Google Scholar] [CrossRef]
- Ma, R.; Xu, Y.; Zhang, X. Catalytic oxidation of biorefinery lignin to value-added chemicals to support sustainable biofuel production. ChemSusChem 2015, 8, 24–51. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.C.; Kumar Sutar, A.; Lin, C.-C. Polymer-supported schiff base complexes in oxidation reactions. Coord. Chem. Rev. 2009, 253, 1926–1946. [Google Scholar] [CrossRef]
- Amadio, E.; Di Lorenzo, R.; Zonta, C.; Licini, G. Vanadium catalyzed aerobic carbon–carbon cleavage. Coord. Chem. Rev. 2015, 301–302, 147–162. [Google Scholar] [CrossRef]
- Stewart, D. Lignin as a base material for materials applications: Chemistry, application and economics. Ind. Crops Prod. 2008, 27, 202–207. [Google Scholar] [CrossRef]
- Crestini, C.; Crucianelli, M.; Orlandi, M.; Saladino, R. Oxidative strategies in lignin chemistry: A new environmental friendly approach for the functionalisation of lignin and lignocellulosic fibers. Catal. Today 2010, 156, 8–22. [Google Scholar] [CrossRef]
- Dong, C.; Feng, C.; Liu, Q.; Shen, D.; Xiao, R. Mechanism on microwave-assisted acidic solvolysis of black-liquor lignin. Bioresour. Technol. 2014, 162, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Liu, N.; Dong, C.; Xiao, R.; Gu, S. Catalytic solvolysis of lignin with the modified husys in formic acid assisted by microwave heating. Chem. Eng. J. 2015, 270, 641–647. [Google Scholar] [CrossRef]
- Nimz, H. Beech lignin—Proposal of a constitutional scheme. Angew. Chem. Int. Ed. 1974, 13, 313–321. [Google Scholar] [CrossRef]
- Erickson, M.; Larsson, S.; Miksche, G.E. Analysis using gas-chromatography of lignin oxidation-products. 8. Structure of spruce lignin. Acta Chem. Scand. 1973, 27, 903–914. [Google Scholar] [CrossRef]
- Hage, R.; Lienke, A. Applications of transition-metal catalysts to textile and wood-pulp bleaching. Angew. Chem. Int. Ed. 2006, 45, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xiao, R.; Shen, D.; Zhang, H. Structural analysis of lignin residue from black liquor and its thermal performance in thermogravimetric-fourier transform infrared spectroscopy. Bioresour. Technol. 2013, 128, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Cheng, C.; Liu, N.; Xiao, R. Lignin depolymerization (LDP) with solvolysis for selective production of renewable aromatic chemicals. In Production of Biofuels and Chemicals from Lignin; Fang, Z., Smith, R.L., Jr., Eds.; Springer: Berlin, Germany, 2016; p. 290. [Google Scholar]
- Choi, Y.S.; Singh, R.; Zhang, J.; Balasubramanian, G.; Sturgeon, M.R.; Katahira, R.; Chupka, G.; Beckham, G.T.; Shanks, B.H. Pyrolysis reaction networks for lignin model compounds: Unraveling thermal deconstruction of β-O-4 and α-O-4 compounds. Green Chem. 2016, 18, 1762–1773. [Google Scholar] [CrossRef]
- Forsythe, W.G.; Garrett, M.D.; Hardacre, C.; Nieuwenhuyzen, M.; Sheldrake, G.N. An efficient and flexible synthesis of model lignin oligomers. Green Chem. 2013, 15, 3031–3038. [Google Scholar] [CrossRef]
- Sedai, B.; Díaz-Urrutia, C.; Baker, R.T.; Wu, R.; Silks, L.A.P.; Hanson, S.K. Aerobic oxidation of β-1 lignin model compounds with copper and oxovanadium catalysts. ACS Catal. 2013, 3, 3111–3122. [Google Scholar] [CrossRef]
- Reddy, G.V.B.; Sridhar, M.; Gold, M.H. Cleavage of nonphenolic β-1 diarylpropane lignin model dimers by manganese peroxidase from phanerochaete chrysosporium. Evidence for a hydrogen abstraction mechanism. Eur. J. Biochem. 2003, 270, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Tran, F.; Lancefield, C.S.; Kamer, P.C.J.; Lebl, T.; Westwood, N.J. Selective modification of the β-β linkage in DDQ-treated kraft lignin analysed by 2D NMR spectroscopy. Green Chem. 2015, 17, 244–249. [Google Scholar] [CrossRef]
- Ouyang, X.-P.; Liu, C.-L.; Pang, Y.-X.; Qiu, X.-Q. Synthesis of a trimeric lignin model compound composed of α-O-4 and β-O-4 linkages under microwave irradiation. Chin. Chem. Lett. 2013, 24, 1091–1094. [Google Scholar] [CrossRef]
- Mimms, A.; Kocurek, M.; Pyatte, J.; Wright, E. (Eds.) Kraft Pulping Chemistry and Process; Tappi Press: Peachtree Corners, GA, USA, 1989. [Google Scholar]
- Glasser, W.; Dave, V.; Frazier, C. Molecular weight distribution of (semi-)commercial lignin derivatives. J. Wood Chem. Technol. 1993, 13, 545–559. [Google Scholar] [CrossRef]
- Pan, X.J.; Arato, C.; Gilkes, N.; Gregg, D.; Mabee, W.; Pye, K.; Xiao, Z.Z.; Zhang, X.; Saddler, J. Biorefining of softwoods using ethanol organosolv pulping: Preliminary evaluation of process streams for manufacture of fuel-grade ethanol and co-products. Biotechnol. Bioeng. 2005, 90, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Scholze, B.; Meier, D. Characterization of the water-insoluble fraction from pyrolysis oil (pyrolytic lignin). Part I. PY-GC/MS, FTIR, and functional groups. J. Anal. Appl. Pyrolysis 2001, 60, 41–54. [Google Scholar] [CrossRef]
- Scholze, B.; Hanser, C.; Meier, D. Characterization of the water-insoluble fraction from fast pyrolysis liquids (pyrolytic lignin): Part II. GPC, carbonyl goups, and 13C NMR. J. Anal. Appl. Pyrolysis 2001, 58, 387–400. [Google Scholar] [CrossRef]
- Samuel, R.; Pu, Y.; Raman, B.; Ragauskas, A. Structural characterization and comparison of switchgrass ball-milled lignin before and after dilute acid pretreatment. Appl. Biochem. Biotechnol. 2010, 162, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Iten, L.; Cotta, M.; Wu, Y. Dilute acid pretreatment, enzymatic saccharification and fermentation of wheat straw to ethanol. Process Biochem. 2005, 40, 3693–3700. [Google Scholar] [CrossRef]
- Chan, J.M.W.; Bauer, S.; Sorek, H.; Sreekumar, S.; Wang, K.; Toste, F.D. Studies on the vanadium-catalyzed nonoxidative depolymerization of miscanthus giganteus-derived lignin. ACS Catal. 2013, 3, 1369–1377. [Google Scholar] [CrossRef]
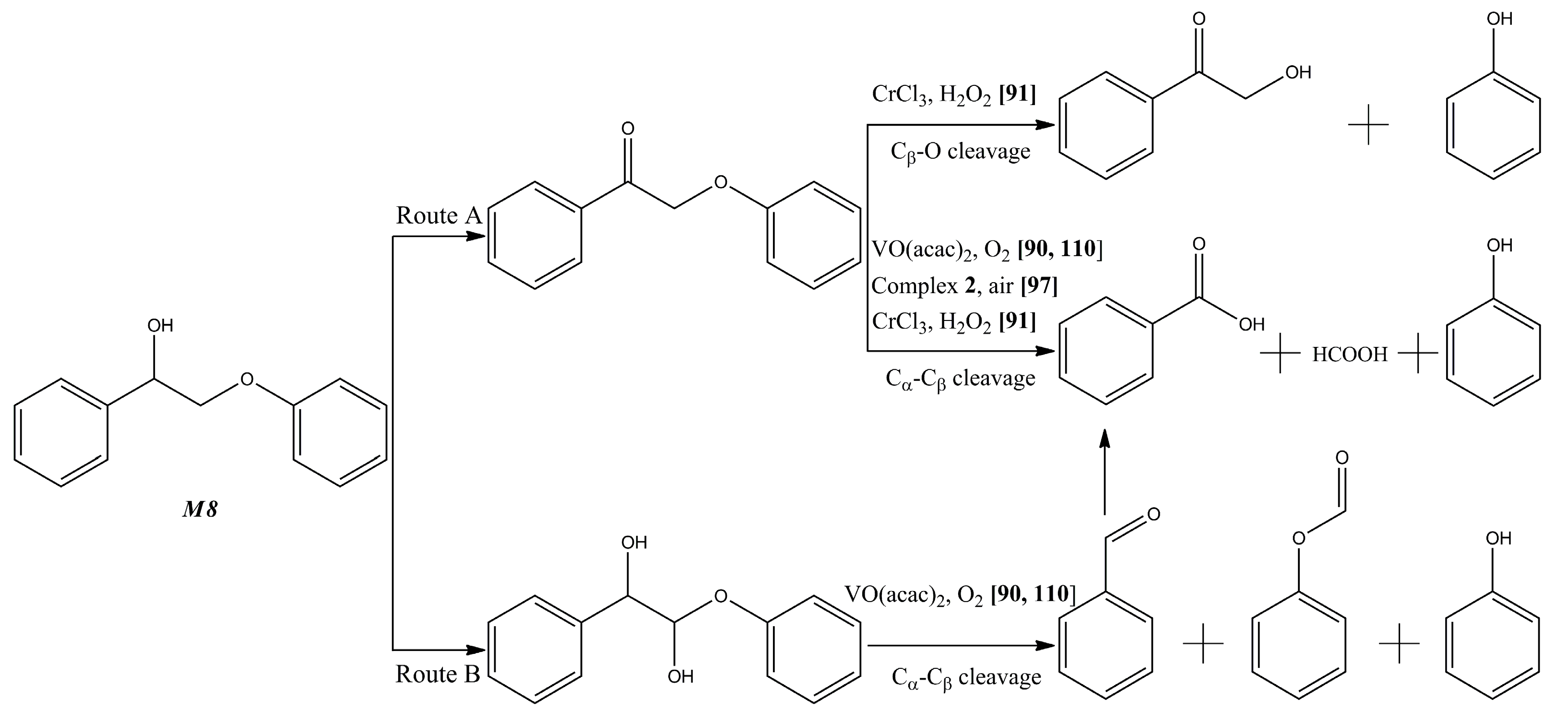
- Díaz-Urrutia, C.; Chen, W.-C.; Crites, C.-O.; Daccache, J.; Korobkov, I.; Baker, R.T. Towards lignin valorisation: Comparing homogeneous catalysts for the aerobic oxidation and depolymerisation of organosolv lignin. RSC Adv. 2015, 5, 70502–70511. [Google Scholar] [CrossRef]
- Mottweiler, J.; Rinesch, T.; Besson, C.; Buendia, J.; Bolm, C. Iron-catalysed oxidative cleavage of lignin and β-O-4 lignin model compounds with peroxides in DMSO. Green Chem. 2015, 17, 5001–5008. [Google Scholar] [CrossRef]
- Zhou, X.-F.; Lu, X.-J. Co(salen) supported on graphene oxide for oxidation of lignin. J. Appl. Polym. Sci. 2016, 44133, 1–9. [Google Scholar] [CrossRef]
- Mottweiler, J.; Puche, M.; Rauber, C.; Schmidt, T.; Concepcion, P.; Corma, A.; Bolm, C. Copper-and vanadium-Catalyzed oxidative cleavage of lignin using dioxygen. ChemSusChem 2015, 8, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Crestini, C.; Caponi, M.C.; Argyropoulos, D.S.; Saladino, R. Immobilized methyltrioxo rhenium (MTO)/H2O2 systems for the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2006, 14, 5292–5302. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Chen, X.; Ma, D.; Yan, N. A metal-free, carbon-based catalytic system for the oxidation of lignin model compounds and lignin. ChemPlusChem 2014, 79, 825–834. [Google Scholar] [CrossRef]
- Demesa, A.G.; Laari, A.; Turunen, I.; Sillanpää, M. Alkaline partial wet oxidation of lignin for the production of carboxylic acids. Chem. Eng. Technol. 2015, 38, 2270–2278. [Google Scholar] [CrossRef]
- Ouyang, X.; Ruan, T.; Qiu, X. Effect of solvent on hydrothermal oxidation depolymerization of lignin for the production of monophenolic compounds. Fuel Process. Technol. 2016, 144, 181–185. [Google Scholar] [CrossRef]
- Azarpira, A.; Ralph, J.; Lu, F. Catalytic alkaline oxidation of lignin and its model compounds: A pathway to aromatic biochemicals. BioEnergy Res. 2013, 7, 78–86. [Google Scholar] [CrossRef]
- Voitl, T.; von Rohr, P.R. Oxidation of lignin using aqueous polyoxometalates in the presence of alcohols. ChemSusChem 2008, 1, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Voitl, T.; von Rohr, P.R. Demonstration of a process for the conversion of kraft lignin into vanillin and methyl vanillate by acidic oxidation in aqueous methanol. Ind. Eng. Chem. Res. 2010, 49, 520–525. [Google Scholar] [CrossRef]
- Werhan, H.; Mir, J.M.; Voitl, T.; von Rohr, P.R. Acidic oxidation of kraft lignin into aromatic monomers catalyzed by transition metal salts. Holzforschung 2011, 65. [Google Scholar] [CrossRef]
- Napoly, F.; Kardos, N.; Jean-Gérard, L.; Goux-Henry, C.; Andrioletti, B.; Draye, M. H2O2-mediated kraft lignin oxidation with readily available metal salts: What about the effect of ultrasound? Ind. Eng. Chem. Res. 2015, 54, 6046–6051. [Google Scholar] [CrossRef]
- Partenheimer, W. The aerobic oxidative cleavage of lignin to produce hydroxyaromatic benzaldehydes and carboxylic acids via metal/bromide catalysts in acetic acid/water mixtures. Adv. Synth. Catal. 2009, 351, 456–466. [Google Scholar] [CrossRef]
- Gu, X.; He, M.; Shi, Y.; Li, Z. La-modified SBA-15/H2O2 systems for the microwave assisted oxidation of organosolv beech wood lignin. Maderas Cienc. Tecnol. 2012, 14, 31–41. [Google Scholar] [CrossRef]
- Deng, W.; Zhang, H.; Wu, X.; Li, R.; Zhang, Q.; Wang, Y. Oxidative conversion of lignin and lignin model compounds catalyzed by CeO2-supported pd nanoparticles. Green Chem. 2015, 17, 5009–5018. [Google Scholar] [CrossRef]
- Deng, H.; Lin, L.; Sun, Y.; Pang, C.; Zhuang, J.; Ouyang, P.; Li, Z.; Liu, S. Perovskite-type oxide LaMnO3: An efficient and recyclable heterogeneous catalyst for the wet aerobic oxidation of lignin to aromatic aldehydes. Catal. Lett. 2008, 126, 106–111. [Google Scholar] [CrossRef]
- Deng, H.; Lin, L.; Sun, Y.; Pang, C.; Zhuang, J.; Ouyang, P.; Li, J.; Liu, S. Activity and stability of perovskite-type oxide LaCoO3 catalyst in lignin catalytic wet oxidation to aromatic aldehydes process. Energy Fuels 2009, 23, 19–24. [Google Scholar] [CrossRef]
- Dawange, M.; Galkin, M.V.; Samec, J.S.M. Selective aerobic benzylic alcohol oxidation of lignin model compounds: Route to aryl ketones. ChemCatChem 2015, 7, 401–404. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, B.; Cui, M.; Deng, J.; Li, X.; Ma, Y.; Fu, Y. Chemoselective oxidant-free dehydrogenation of alcohols in lignin using Cp*Ir catalysts. Green Chem. 2016, 18, 2029–2036. [Google Scholar] [CrossRef]
- Cedeno, D.; Bozell, J. Catalytic oxidation of para-substituted phenols with cobalt-schiff base complexes/O2-selective conversion of syringyl and guaiacyl lignin models to benzoquinones. Tetrahedron Lett. 2012, 53, 2380–2383. [Google Scholar] [CrossRef]
- Badamali, S.; Luque, R.; Clark, J.; Breeden, S. Co(salen)/SBA-15 catalysed oxidation of a β-O-4 phenolic dimer under microwave irradiation. Catal. Commun. 2011, 12, 993–995. [Google Scholar] [CrossRef]
- Crestini, C.; Pro, P.; Neri, V.; Saladino, R. Methyltrioxorhenium: A new catalyst for the activation of hydrogen peroxide to the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2005, 13, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Lin, Z.; Deng, Y.; Yang, D.; Qiu, X. Oxidative degradation of soda lignin assisted by microwave irradiation. Chin. J. Chem. Eng. 2010, 18, 695–702. [Google Scholar] [CrossRef]
- Wu, A.; Lauzon, J.M.; Andriani, I.; James, B.R. Breakdown of lignins, lignin model compounds, and hydroxy-aromatics, to C1 and C2 chemicals via metal-free oxidation with peroxide or persulfate under mild conditions. RSC Adv. 2014, 4, 17931–17934. [Google Scholar] [CrossRef]
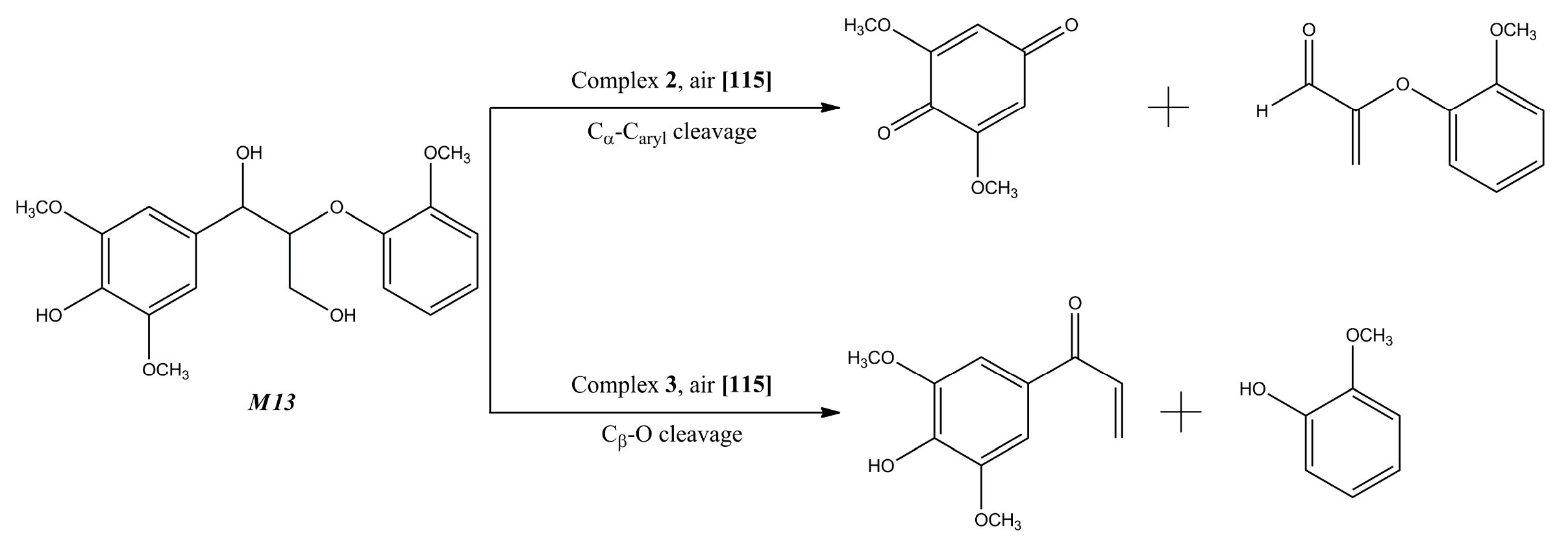
- Maziero, P.; de Oliveira Neto, M.; Machado, D.; Batista, T.; Schmitt Cavalheiro, C.C.; Neumann, M.G.; Craievich, A.F.; de Moraes Rocha, G.J.; Polikarpov, I.; Gonçalves, A.R. Structural features of lignin obtained at different alkaline oxidation conditions from sugarcane bagasse. Ind. Crops Prod. 2012, 35, 61–69. [Google Scholar] [CrossRef]
- Chen, C.; Pakdel, H.; Roy, C. Production of monomeric phenols by thermochemical conversion of biomass: A review. Bioresour. Technol. 2001, 79, 277–299. [Google Scholar] [CrossRef]
- Bujanovic, B.; Reiner, R.S.; Ralph, S.A.; Atalla, R.H. Polyoxometalate delignification of birch kraft pulp and effect on residual lignin. J. Wood Chem. Technol. 2011, 31, 121–141. [Google Scholar] [CrossRef]
- Sik Kim, Y.; Chang, H.-m.; Kadla, J.F. Polyoxometalate (POM) oxidation of milled wood lignin (MWL). J. Wood Chem. Technol. 2007, 27, 225–241. [Google Scholar] [CrossRef]
- Partenheimer, W. Methodology and scope of metal bromide autoxidation of hydrocarbons. Catal. Today 1995, 23, 69–158. [Google Scholar] [CrossRef]
- Rothenberg, G. Catalysis: Concepts and Green Applications; Wiely-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Ma, Y.; Du, Z.; Liu, J.; Xia, F.; Xu, J. Selective oxidative C–C bond cleavage of a lignin model compound in the presence of acetic acid with a vanadium catalyst. Green Chem. 2015, 17, 4968–4973. [Google Scholar] [CrossRef]
- Pan, J.; Fu, J.; Lu, X. Microwave-assisted oxidative degradation of lignin model compounds with metal salts. Energy Fuels 2015, 29, 4503–4509. [Google Scholar] [CrossRef]
- Shilpy, M.; Ehsan, M.A.; Ali, T.H.; Abd Hamid, S.B.; Ali, M.E. Performance of cobalt titanate towards H2O2 based catalytic oxidation of lignin model compound. RSC Adv. 2015, 5, 79644–79653. [Google Scholar] [CrossRef]
- Tumula, V.; Bondwal, S.; Bisht, P.; Pendem, C.; Kumar, J. Oxidation of sulfides to sulfones with hydrogen peroxide in the presence of acetic acid and amberlyst 15. React. Kinet. Mech. Catal. 2012, 107, 449–466. [Google Scholar] [CrossRef]
- Badamali, S.; Clark, J.; Breeden, S. Microwave assisted selective oxidation of lignin model phenolic monomer over SBA-15. Catal. Commun. 2008, 9, 2168–2170. [Google Scholar] [CrossRef]
- Badamali, S.; Luque, R.; Clark, J.; Breeden, S. Microwave assisted oxidation of a lignin model phenolic monomer using Co(salen)/SBA-15. Catal. Commun. 2009, 10, 1010–1013. [Google Scholar] [CrossRef]
- Badamali, S.K.; Luque, R.; Clark, J.H.; Breeden, S.W. Unprecedented oxidative properties of mesoporous silica materials: Towards microwave-assisted oxidation of lignin model compounds. Catal. Commun. 2013, 31, 1–4. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Thorn, D.L. Aerobic oxidation of lignin models using a base metal vanadium catalyst. Inorg. Chem. 2010, 49, 5611–5618. [Google Scholar] [CrossRef] [PubMed]
- Mate, V.R.; Shirai, M.; Rode, C.V. Heterogeneous Co3O4 catalyst for selective oxidation of aqueous veratryl alcohol using molecular oxygen. Catal. Commun. 2013, 33, 66–69. [Google Scholar] [CrossRef]
- Mate, V.R.; Jha, A.; Joshi, U.D.; Patil, K.R.; Shirai, M.; Rode, C.V. Effect of preparation parameters on characterization and activity of Co3O4 catalyst in liquid phase oxidation of lignin model substrates. Appl. Catal. A 2014, 487, 130–138. [Google Scholar] [CrossRef]
- Gu, X.; He, M.; Shi, Y.; Li, Z. La-containing SBA-15/H2O2 systems for the microwave assisted oxidation of a lignin model phenolic monomer. Maderas Cienc. Tecnol. 2010, 12, 181–188. [Google Scholar] [CrossRef]
- Gu, X.; He, M.; Shi, Y.; Li, Z. Production of armotic aldehyde by microwave catalytic oxidation of a lignin model compound with La-containing SBA-15/H2O2 systems. BioResources 2010, 5, 2029–2039. [Google Scholar]
- Lanfranchi, M.; Prati, L.; Rossi, M.; Tiripicchio, A. The oxidation of ethane-1,2-diol resulting from molecular oxygen activation by copper: Formation of an oxoethanoate complex precedes carbon-carbon bond cleavage. J. Chem. Soc. Chem. Commun. 1993, 22, 1698–1699. [Google Scholar] [CrossRef]
- Prati, L.; Rossi, M. Stepwise oxidation of 1,2-diols resulting from molecular oxygen activation by copper. J. Mol. Catal. A 1996, 110, 221–226. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T. Knocking on wood: Base metal complexes as catalysts for selective oxidation of lignin models and extracts. Acc. Chem. Res. 2015, 48, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Elkurtehi, A.I.; Walsh, A.G.; Dawe, L.N.; Kerton, F.M. Vanadium aminophenolate complexes and their catalytic activity in aerobic and H2O2-mediated oxidation reactions. Eur. J. Inorg. Chem. 2016, 2016, 3123–3130. [Google Scholar] [CrossRef]
- Ede, R.M.; Ralph, J.; Torr, K.M.; Dawson, B.S.W. A 2D NMR investigation of the heterogeneity of distribution of diarylpropane structures in extracted pinus radiata lignins. Holzforschung 1996, 50, 161–164. [Google Scholar]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Silks, L.A.P.; Thorn, D.L. Mechanism of alcohol oxidation by dipicolinate vanadium(V): Unexpected role of pyridine. J. Am. Chem. Soc. 2010, 132, 17804–17816. [Google Scholar] [CrossRef] [PubMed]
- Haibach, M.C.; Lease, N.; Goldman, A.S. Catalytic cleavage of ether C–O bonds by pincer iridium complexes. Angew. Chem. Int. Ed. 2014, 53, 10160–10163. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Urrutia, C.; Sedai, B.; Leckett, K.C.; Baker, R.T.; Hanson, S.K. Aerobic oxidation of 2-phenoxyethanol lignin model compounds using vanadium and copper catalysts. ACS Sustain. Chem. Eng. 2016, 4, 6244–6251. [Google Scholar] [CrossRef]
- Ma, Y.; Du, Z.; Xia, F.; Ma, J.; Gao, J.; Xu, J. Mechanistic studies on the VO(acac)2-catalyzed oxidative cleavage of lignin model compounds in acetic acid. RSC Adv. 2016, 6, 110229–110234. [Google Scholar] [CrossRef]
- Blandez, J.F.; Navalón, S.; Alvaro, M.; Garcia, H. Graphenes as metal-free catalysts for the oxidative depolymerization of lignin models. ChemCatChem 2015, 7, 3020–3026. [Google Scholar] [CrossRef]
- Son, S.; Toste, F.D. Non-oxidative vanadium-catalyzed C–O bond cleavage: Application to degradation of lignin model compounds. Angew. Chem. Int. Ed. 2010, 49, 3791–3794. [Google Scholar] [CrossRef] [PubMed]
- vom Stein, T.; Hartog, T.; Buendia, J.; Stoychev, S.; Mottweiler, J.; Bolm, C.; Leitner, W. Ruthenium-catalyzed C–C bond cleavage in lignin model substrates. Angew. Chem. Int. Ed. 2015, 54, 5859–5863. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.; Huh, S.; Jun, M. Syntheses of ruthenium(II) complexes containing polyphosphine ligands and their applications in the homogeneous hydrogenation. Polyhedron 1999, 18, 469–479. [Google Scholar] [CrossRef]
- Hanson, S.K.; Wu, R.; Silks, L.A. C–C or C–O bond cleavage in a phenolic lignin model compound: Selectivity depends on vanadium catalyst. Angew. Chem. Int. Ed. 2012, 51, 3410–3413. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Y.; Yan, L.; Yu, H.-Z.; Zhang, Q.; Fu, Y. Mechanism of vanadium-catalyzed selective C–O and C–C cleavage of lignin model compound. ACS Catal. 2016, 6, 4399–4410. [Google Scholar] [CrossRef]
- Parker, H.J.; Chuck, C.J.; Woodman, T.; Jones, M.D. Degradation of β-O-4 model lignin species by vanadium Schiff-base catalysts: Influence of catalyst structure and reaction conditions on activity and selectivity. Catal. Today 2016, 269, 40–47. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Zhang, X.; Li, L.; Li, H.; Luo, N.; Wang, F. Two-step, catalytic C–C bond oxidative cleavage process converts lignin models and extracts to aromatic acids. ACS Catal. 2016, 6, 6086–6090. [Google Scholar] [CrossRef]
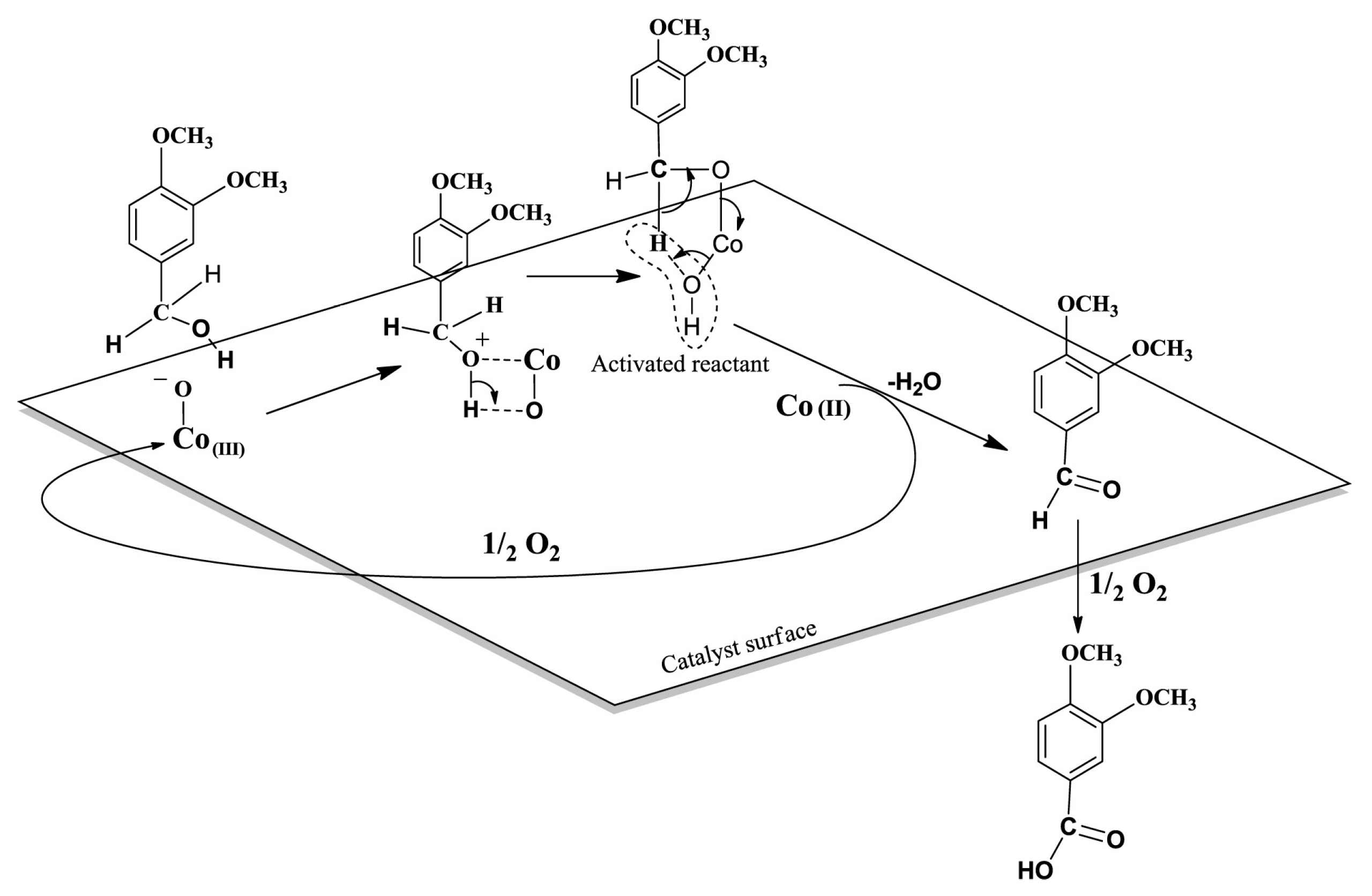

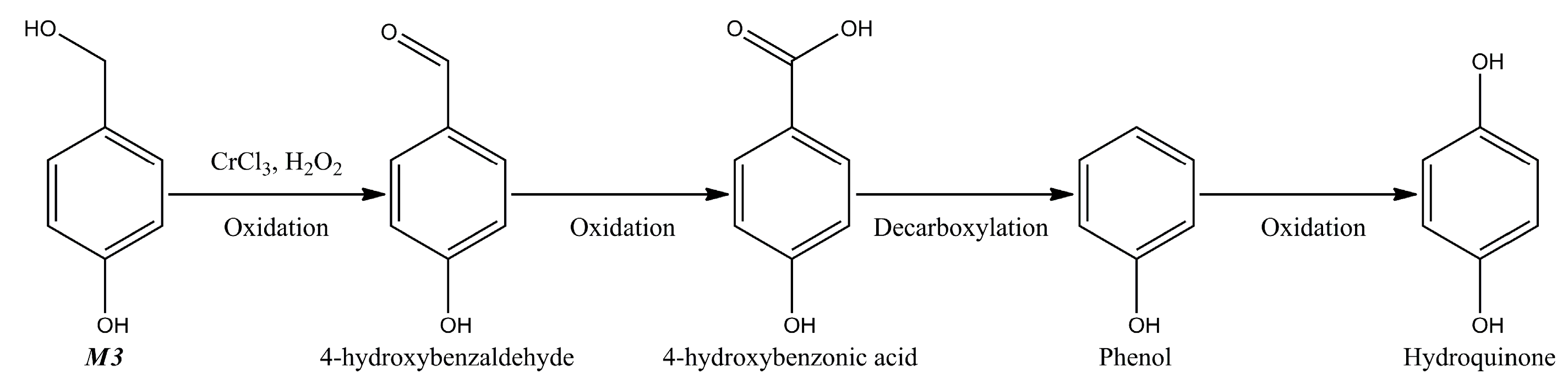

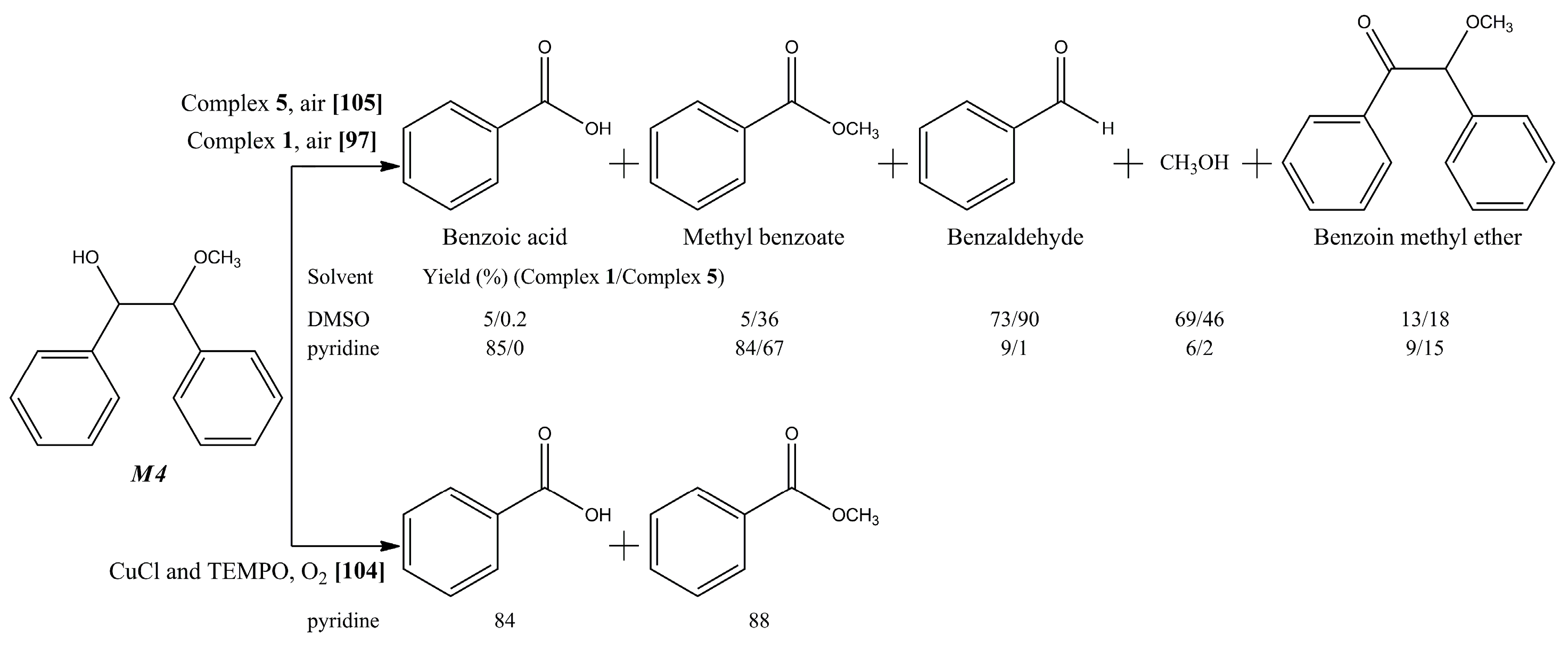
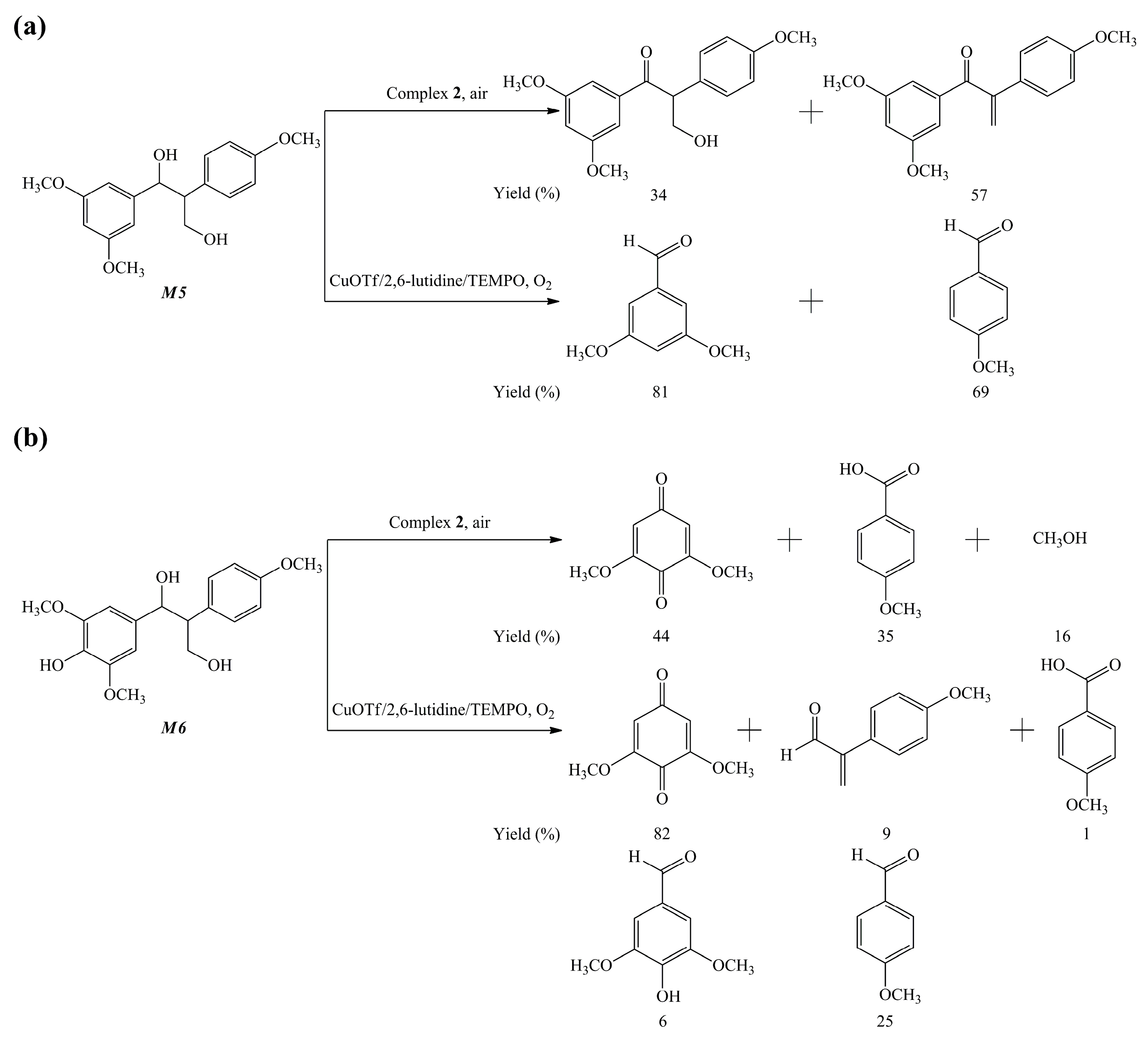
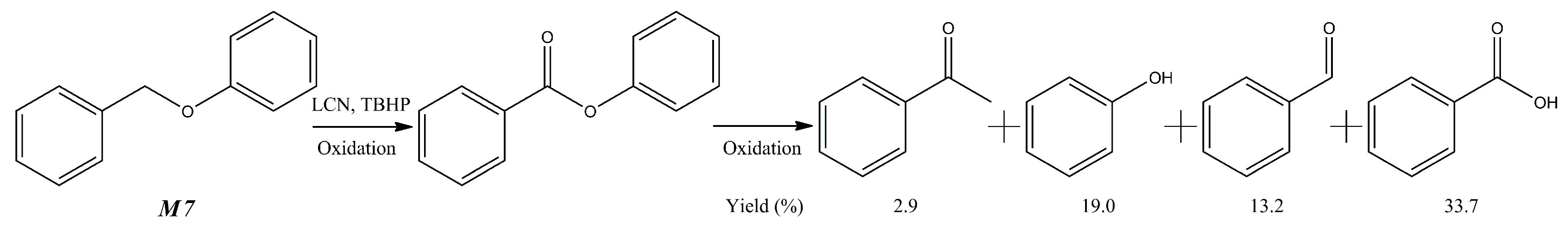
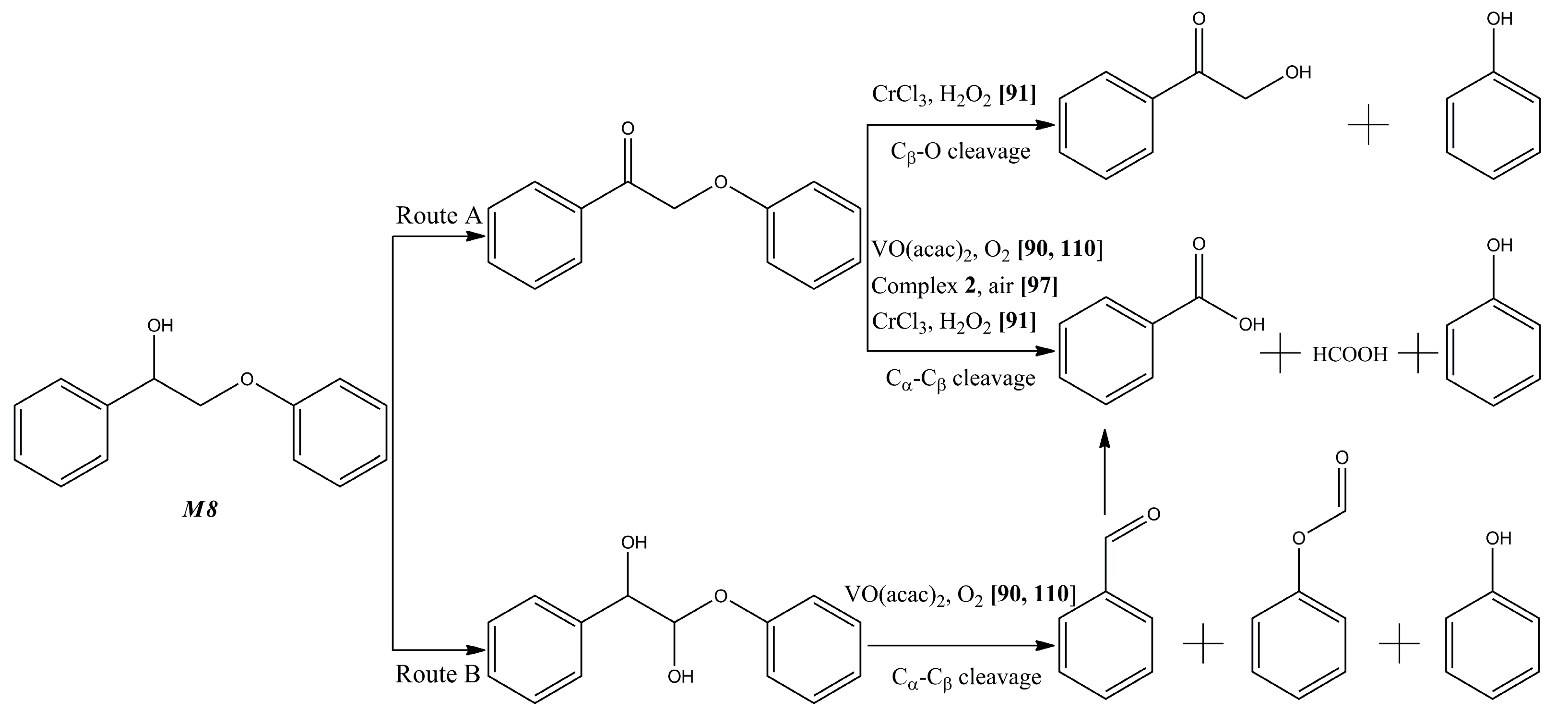
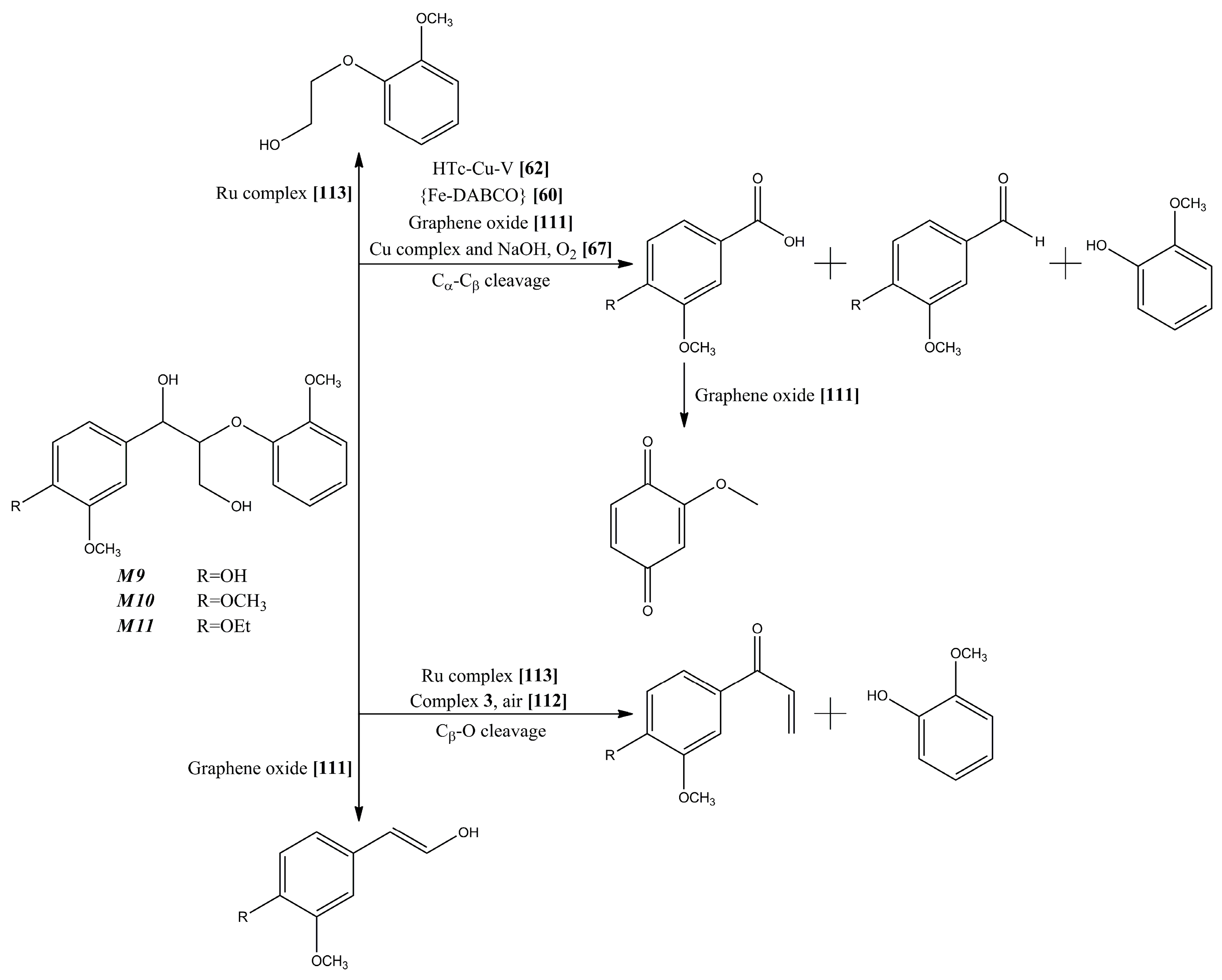
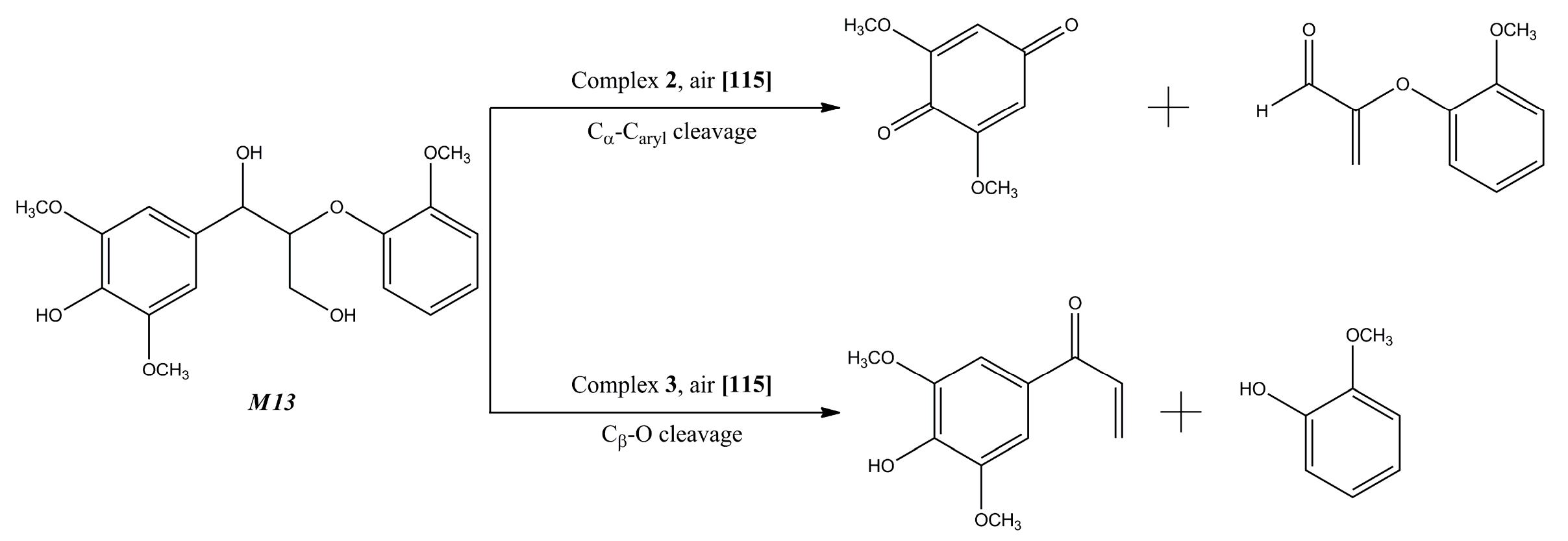
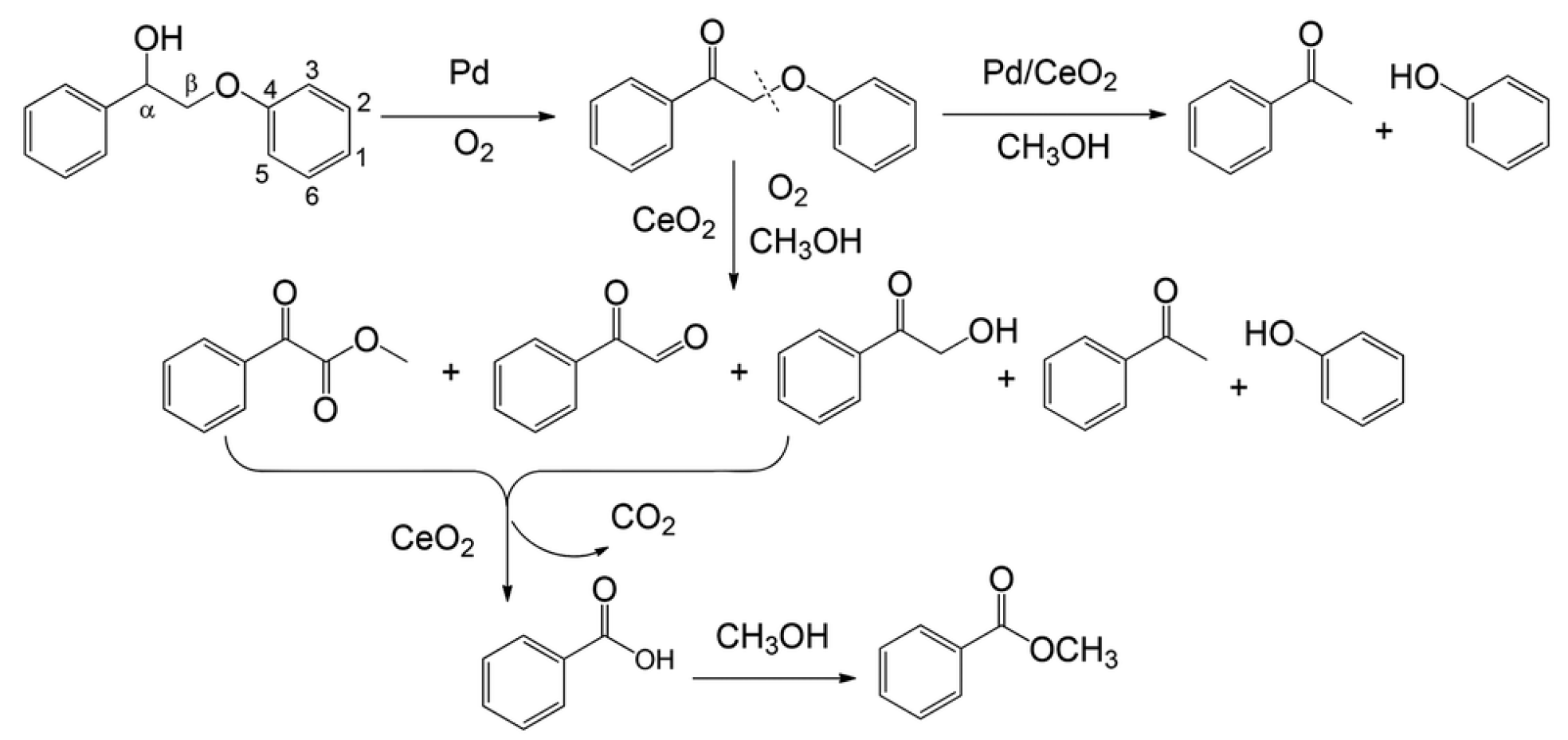


{kind=link}
{kind=link}
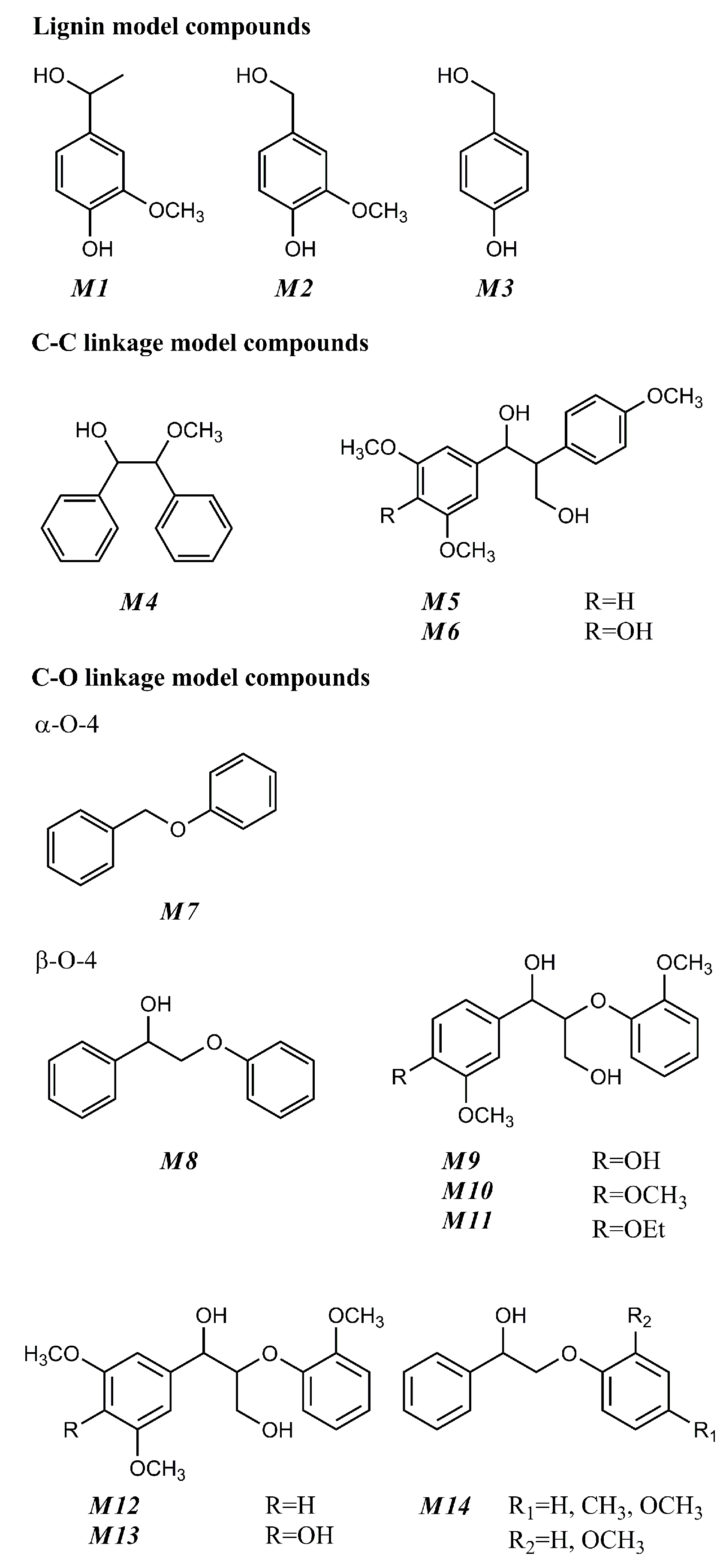{kind=link}
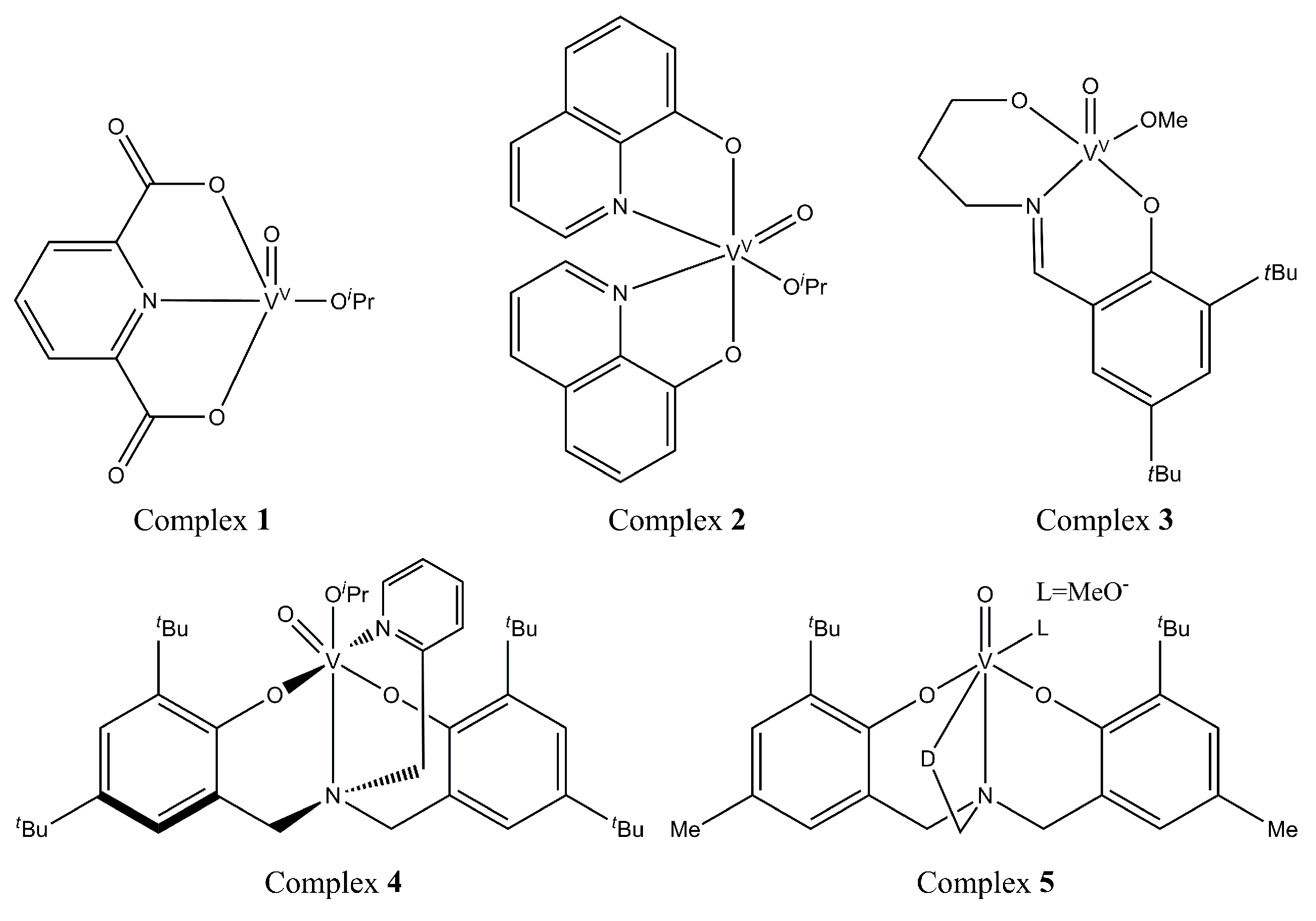{kind=link}
{kind=link}
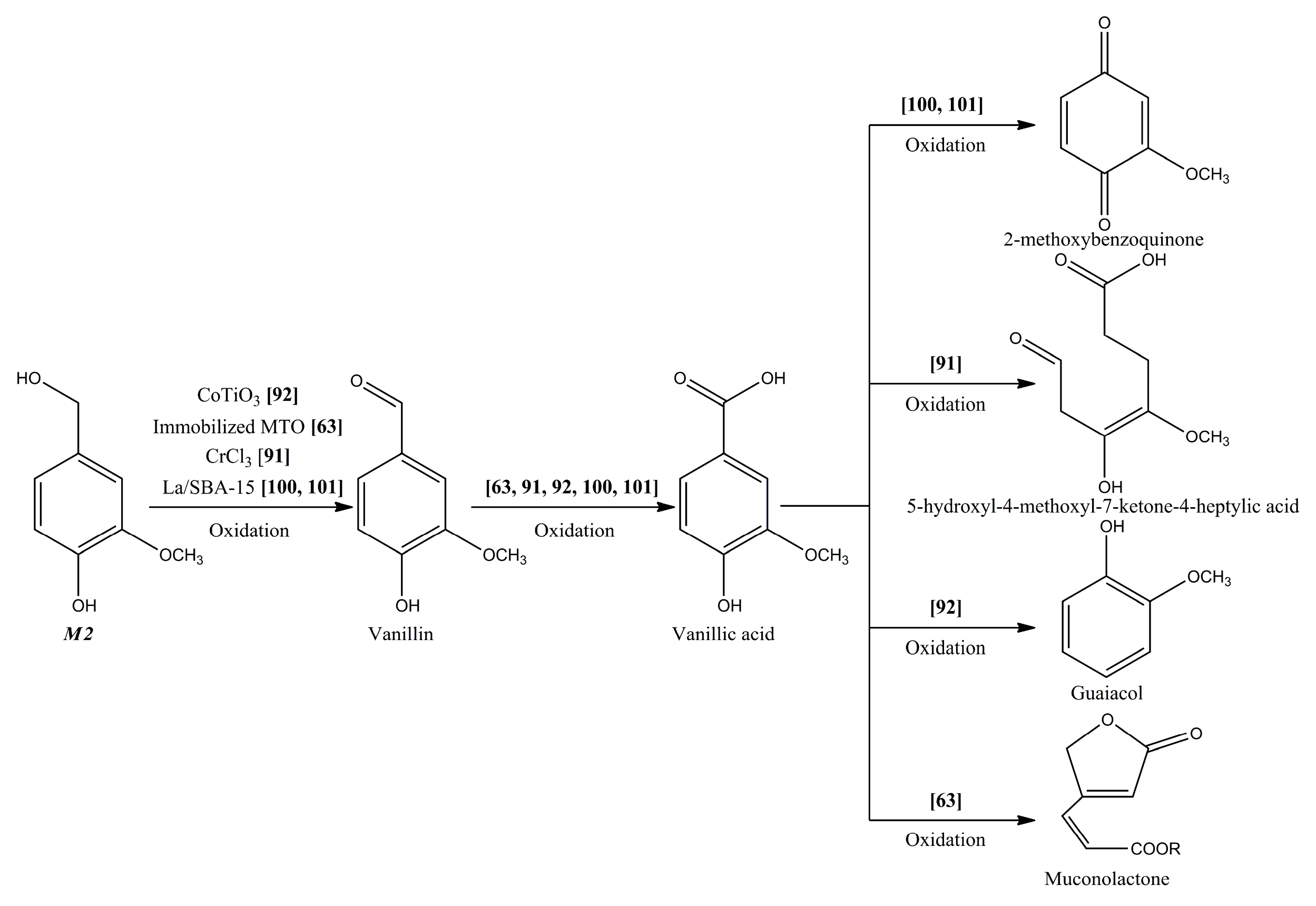{kind=link}
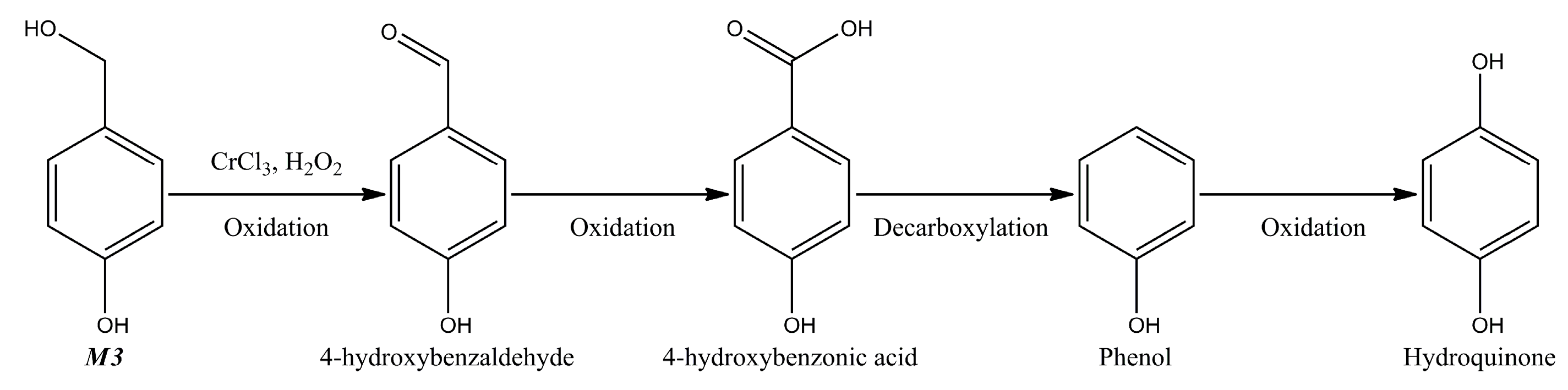{kind=link}
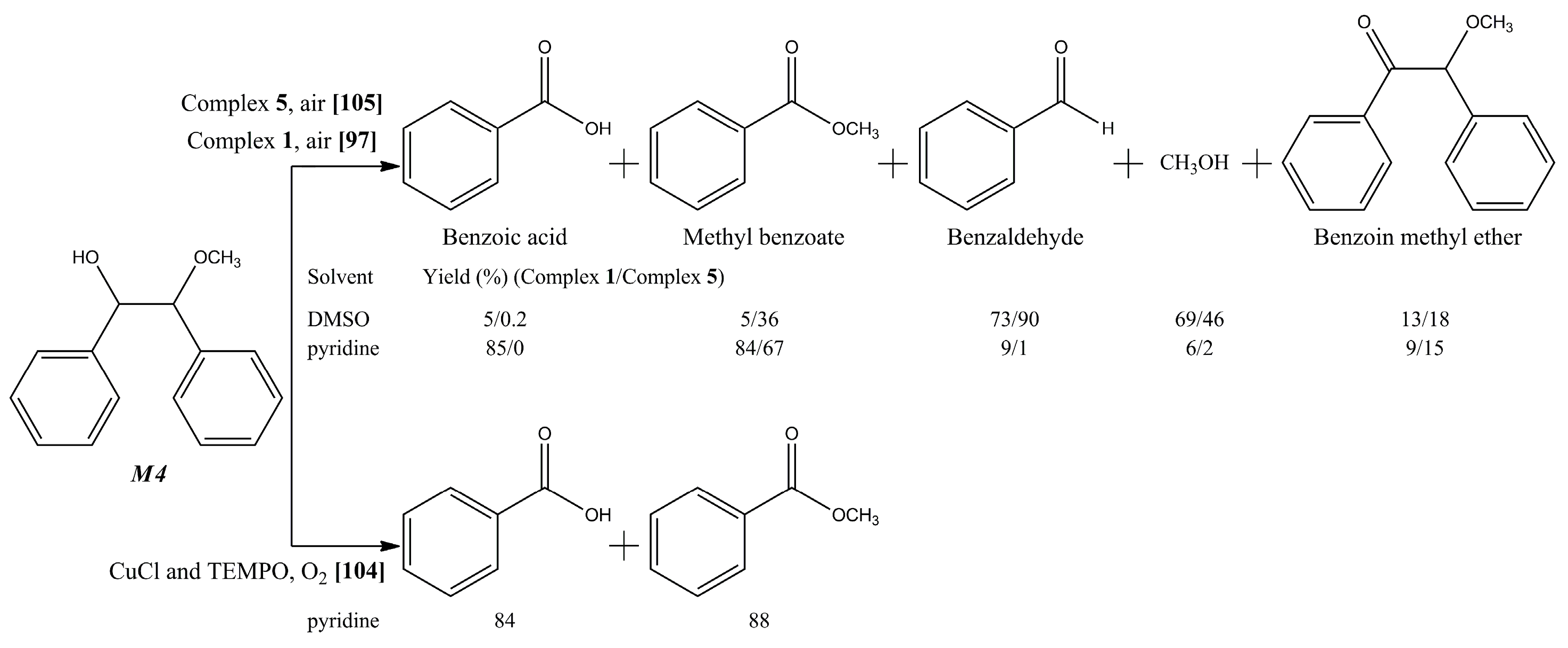{kind=link}
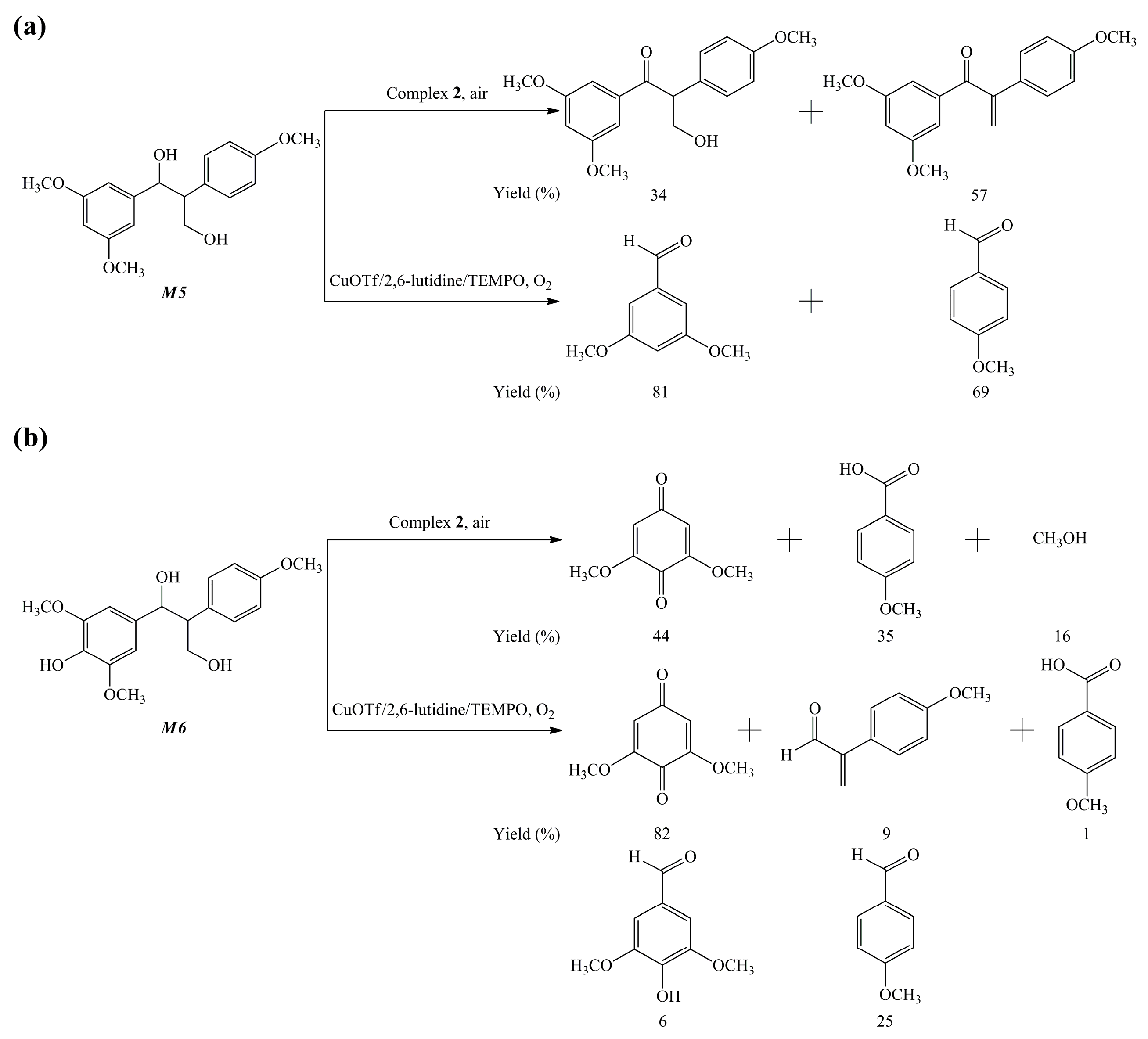{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Feedstock | Conditions | Solvent | Catalyst | Products | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Organosolv lignin (from M. giganteus) | 80 °C, 24 h, air | Acetonitrile-THF or ethyl acetate-THF | Vanadium complexes bearing Schiff base ligands | Monophenolic compounds (vanillin, syringic acid, syringaldehyde) | 0.78 wt %, 0.67 wt %, 0.59 wt % (Catalyst = Complex 3) | [58] |
| 2 | Organosolv lignin | 100 °C, 18 h, 0.8 MPa synthetic air | Ethyl acetate | Vanadium complexes and other organometallic catalysts | Bio-oil | Mw = 575 Da (Catalyst = Complex 2) | [59] |
| 3 | Organosolv lignin and Kraft lignin | 100 °C, 8 h, H2O2 | DMSO and acetic acid | {Fe-DABCO} | Bio-oil | / | [60] |
| 4 | Organosolv lignin | 80 °C, 24 h, air | Acetonitrile-THF | Co(salen) supported on graphene oxide | Vanillin (main) | 3067 g | [61] |
| 5 | Organosolv lignin and Kraft lignin | 135 °C, 40 h, 1.0 MPa O2 | Pyridine | V(acac)3 and Cu(NO3)2·3H2O or HTc-Cu-V | Bio-oil | Mw = 300 Da | [62] |
| 6 | Hydrolytic sugar cane lignin and red spruce kraft lignin | 25 °C, 24 h, H2O2 | Acetic acid | MTO or poly(4-vinylpyridine)/MTO or polystyrene/MTO | Bio-oil | / | [63] |
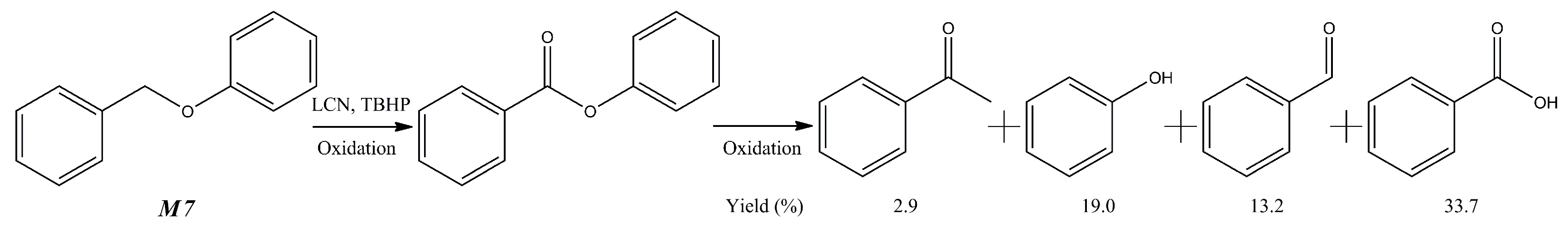
| 7 | Organosolv ligin (from birch wood) | 140 °C, 24 h, 0.1 MPa O2 | H2O, tert-butyl hydroperoxide (TBHP) | Nitrogen-containing graphene material (LCN) | Bio-oil | 45.8 wt % | [64] |
| 8 | Alkali lignin | 175–225 °C, 0–1 h, 0.5–1.5 MPa O2 | Water | NaOH | Formic acid, acetic acid, succinic acid, oxalic acid, glutaconic acid | 44.0 wt % | [65] |
| 9 | Wheat alkali lignin | 150 °C, 1 h, H2O2 | Water/methanol/1,4-dioxane/tetrahydrofuran/ethanol | CuO, Fe2(SO4)3 and NaOH | Monophenolic compounds | 17.92 wt % (in methanol-water) | [66] |
| 10 | Wood lignin from Loblolly pine | 80 °C, 24 h, 0.27/1.24 MPa O2 | Methanol | Copper-phenanthroline complex and NaOH | Vanillic acid, vanillin | 3.5 wt %, 12.6 wt % | [67] |
| 11 | Kraft lignin | 170 °C, 0.3 h, 0.5 MPa O2 | Methanol-water and H2SO4 | H3PMo12O40 | Vanillin, methyl vanillate | 5.2 wt % | [68] |
| 12 | Kraft lignin | 170 °C, 0.3 h, 1.0 MPa O2 | Methanol-water and H2SO4 | H3PMo12O40 | Vanillin, methyl vanillate | 4.6 wt %, 4.2 wt % | [69] |
| 13 | Kraft lignin | 170 °C, 1 h, 1.0 MPa O2 | Methanol-water and H2SO4 | CuSO4; FeCl3; | Vanillin, methyl vanillate | 6.3 wt % | [70] |
| CuCl2; CoCl2 | |||||||
| 14 | Kraft lignin | 45 °C, 1 h, H2O2 | Acetone-water | Metal salt catalysts | Vanillin-based monomers | 0.51 wt % | [71] |
| 15 | Organosolv lignin | 180 °C, 2 h, 13.8 MPa air | Acetic Acid-water | Co/Mn/Zr/Br mixture | Vanillin, vanillin acid, syringaldehyde, syringic acid | 0.99 wt %, 2.91 wt %, 2.52 wt %, 4.51 wt % | [72] |
| 16 | Organosolv beech wood lignin | 200W, 5–30 min, H2O2 | NaOH solution | La/SBA-15 | Vanillin, syringaldehyde | 9.94 wt %, 15.66 wt %. | [73] |
| 17 | Organosolv lignin | 185 °C, 24 h, 0.1 MPa O2 | Methanol | Pd/CeO2 | Vanillin, guaiacol, 4-hydroxybenzaldehyde | 5.2 wt %, 0.87 wt %, 2.4 wt % | [74] |
| 18 | Enzymatic hydrolysis lignin | 120 °C, 0–3 h, 0.5 MPa O2 | NaOH solution | LaMnO3 | Vanillin, p-hydroxybenzyl aldehyde, syringaldehyde | 4.32 wt %, 2.03 wt %, 9.33 wt % | [75] |
| 19 | Enzymatic hydrolysis lignin | 120 °C, 0–3 h, 0.5 MPa O2 | NaOH solution | LaCoO3 | Vanillin, p-hydroxybenzyl aldehyde, syringaldehyde | 4.55 wt %, 2.23 wt %, 9.99 wt % | [76] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.; Wang, J.; Shen, D.; Xue, J.; Guan, S.; Gu, S.; Luo, K.H. Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review. Polymers 2017, 9, 240. https://doi.org/10.3390/polym9060240
Cheng C, Wang J, Shen D, Xue J, Guan S, Gu S, Luo KH. Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review. Polymers. 2017; 9(6):240. https://doi.org/10.3390/polym9060240
Chicago/Turabian StyleCheng, Chongbo, Jinzhi Wang, Dekui Shen, Jiangtao Xue, Sipian Guan, Sai Gu, and Kai Hong Luo. 2017. "Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review" Polymers 9, no. 6: 240. https://doi.org/10.3390/polym9060240
APA StyleCheng, C., Wang, J., Shen, D., Xue, J., Guan, S., Gu, S., & Luo, K. H. (2017). Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review. Polymers, 9(6), 240. https://doi.org/10.3390/polym9060240