
Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles

 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Oligomer Synthesis
2.3. Polyplex Formation
2.4. Polyplex Stability in the Presence of Salt
2.5. Erythrocyte Adhesion in the Presence or Absence of Serum
2.6. Particle Size and Zeta Potential
2.7. Ethidium Bromide Compaction Assay and Heparin Stress
2.8. Serum Stability of Polyplexes
2.9. Cell Culture
2.10. In Vitro Gene Transfer
2.11. Cellular Association
2.12. Cellular Internalization
2.13. In Vivo Gene Transfer
2.14. Statistical Analysis
3. Results and Discussion
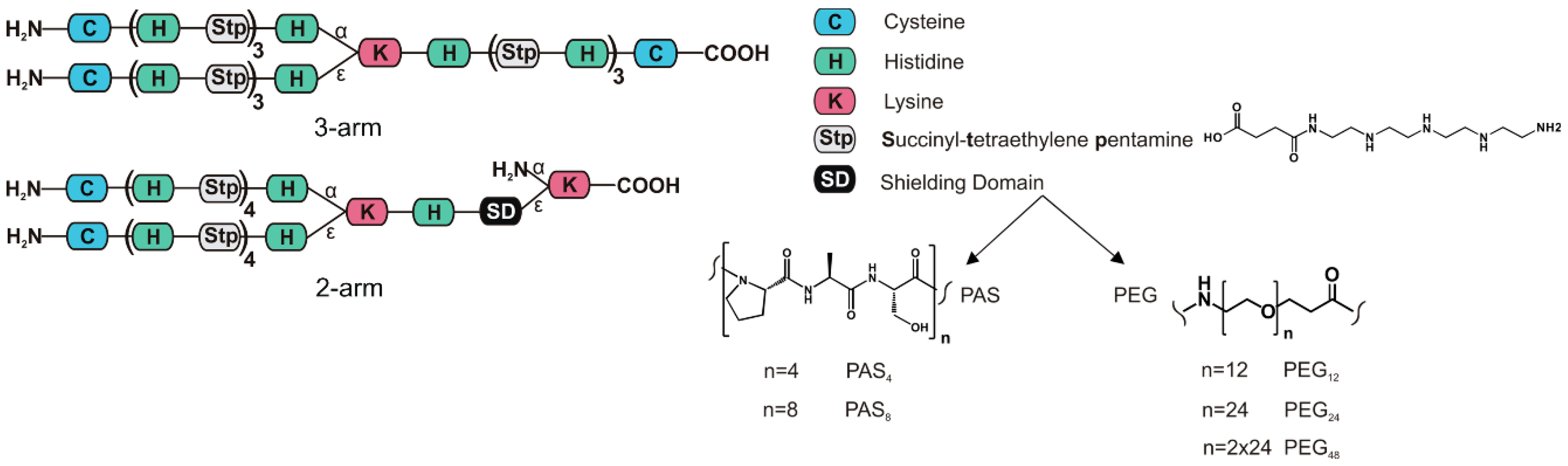
3.1. Peptide and Oligomer Synthesis
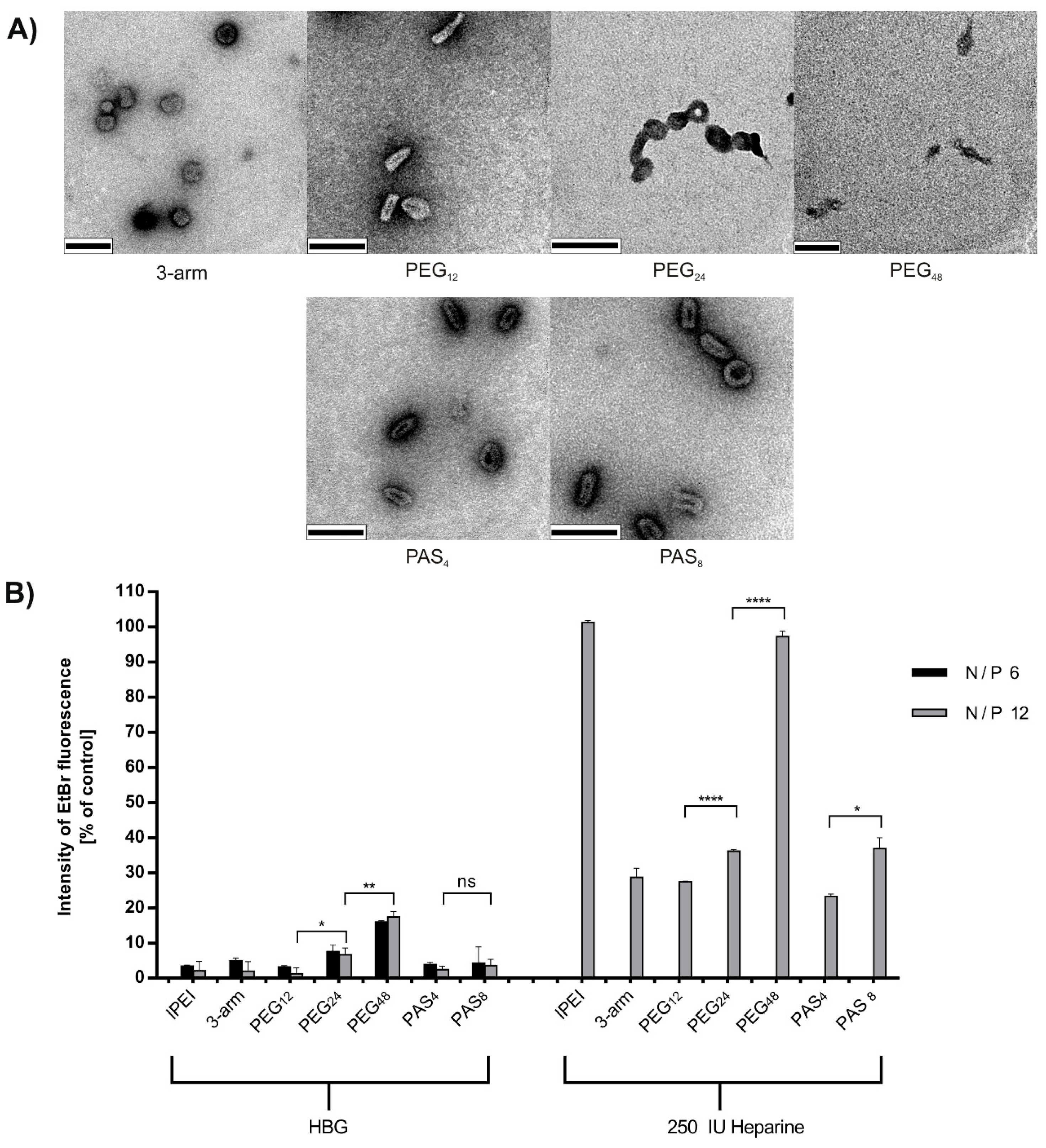
3.2. Physicochemical Polyplex Characterization
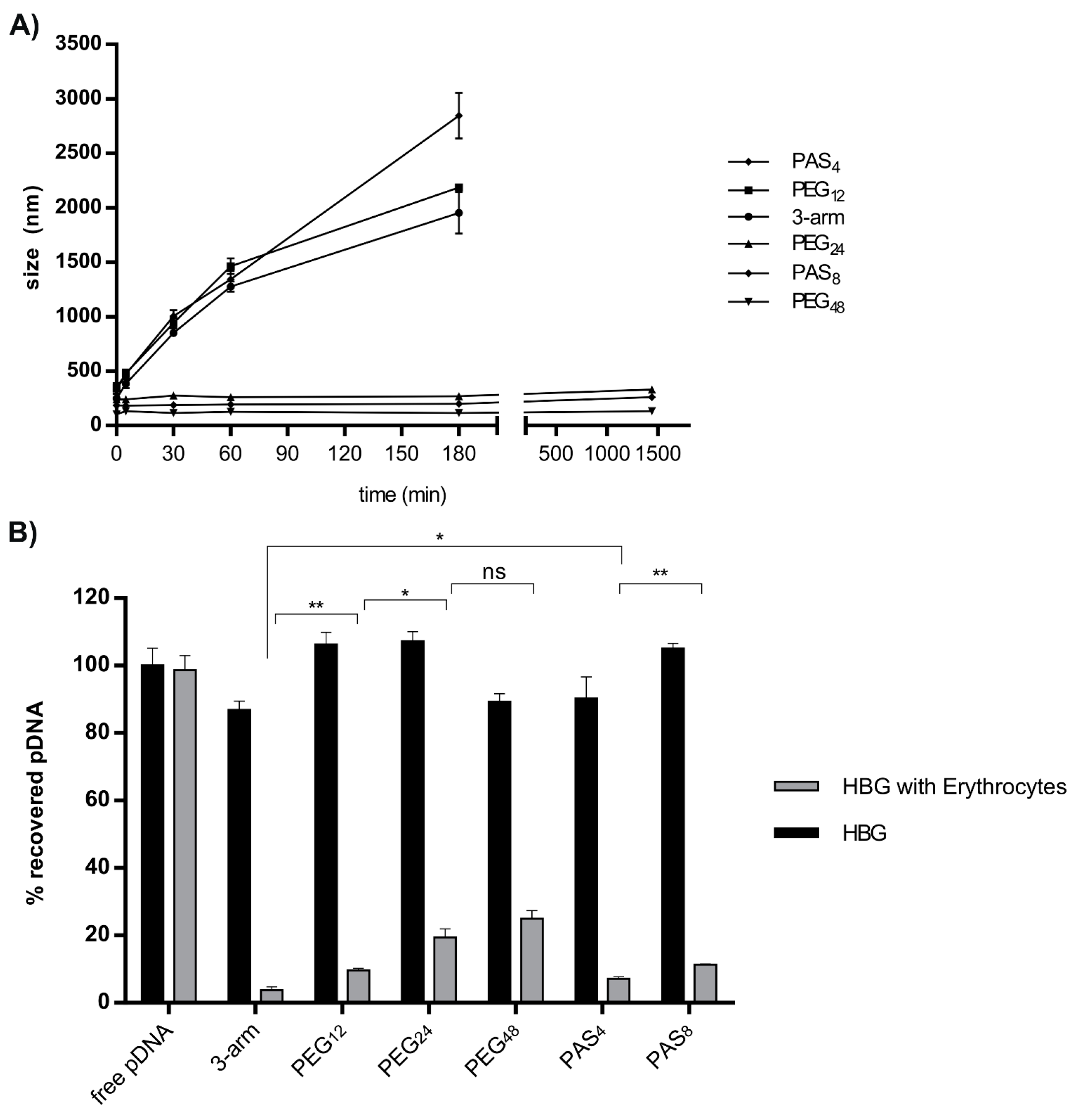
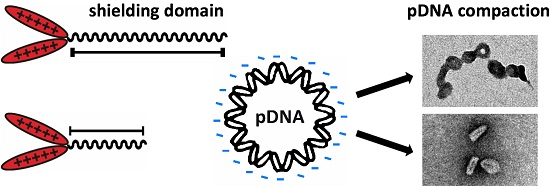
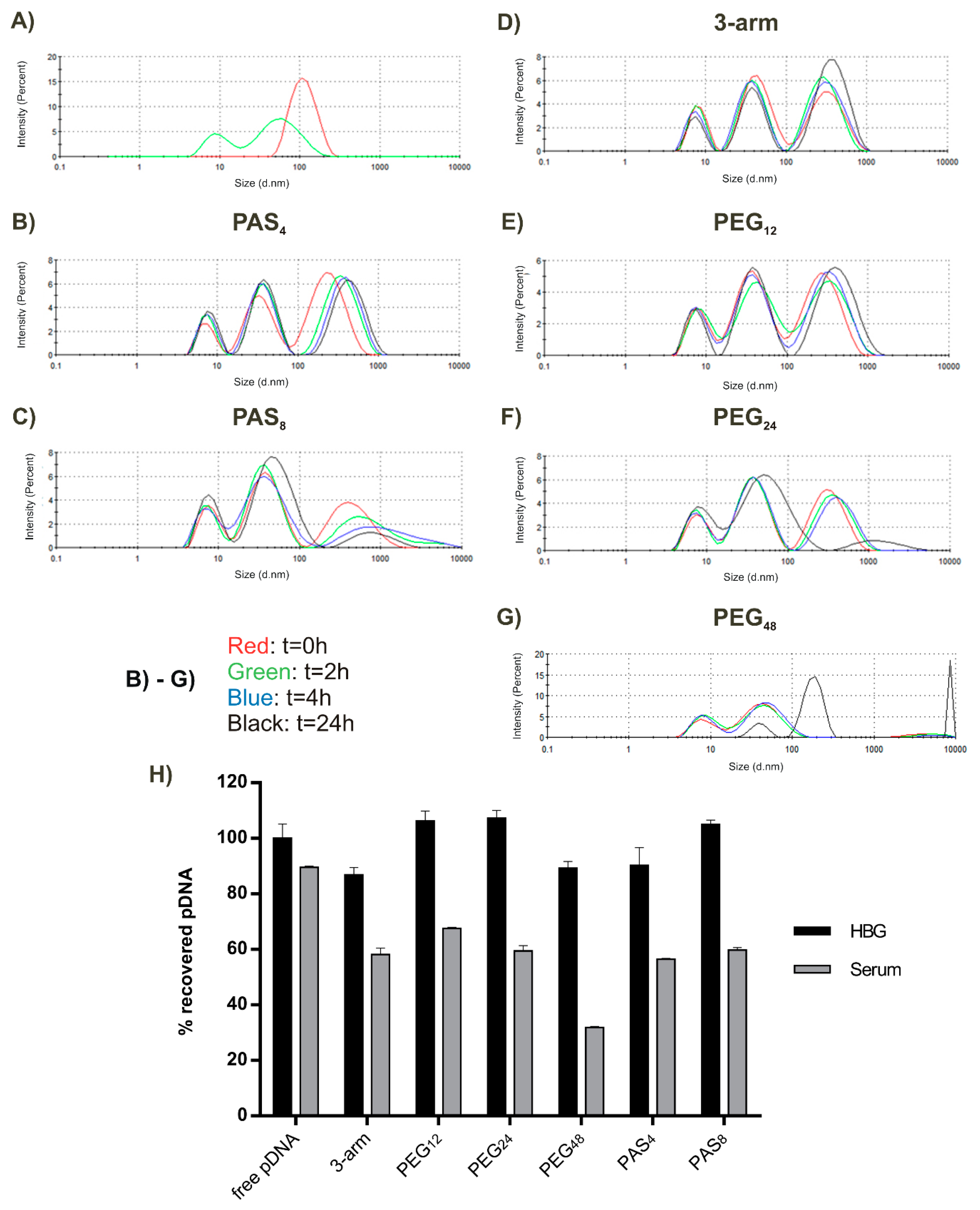
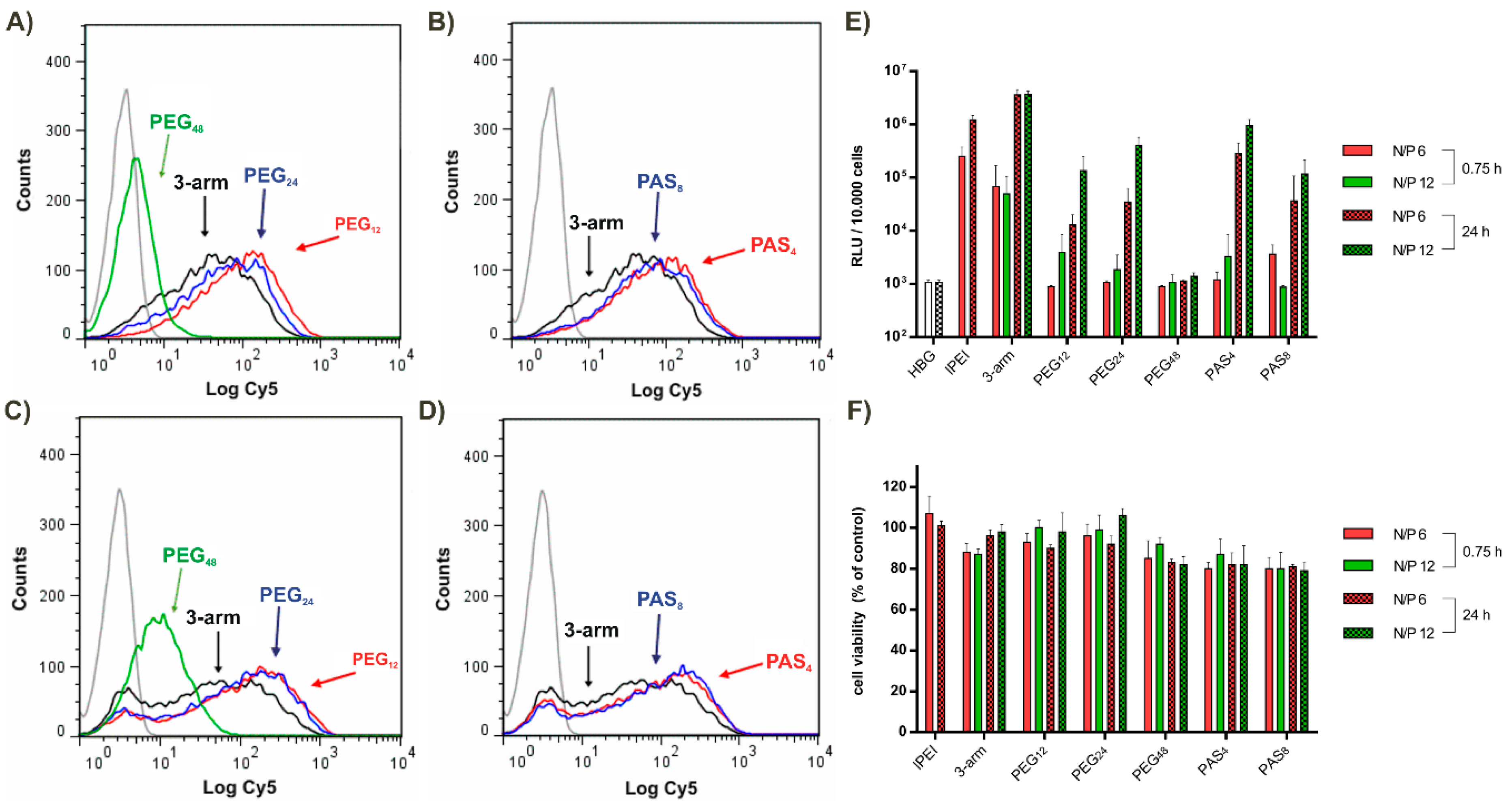
3.3. Steric Shielding
3.4. DNA Compaction
3.5. Serum Stability
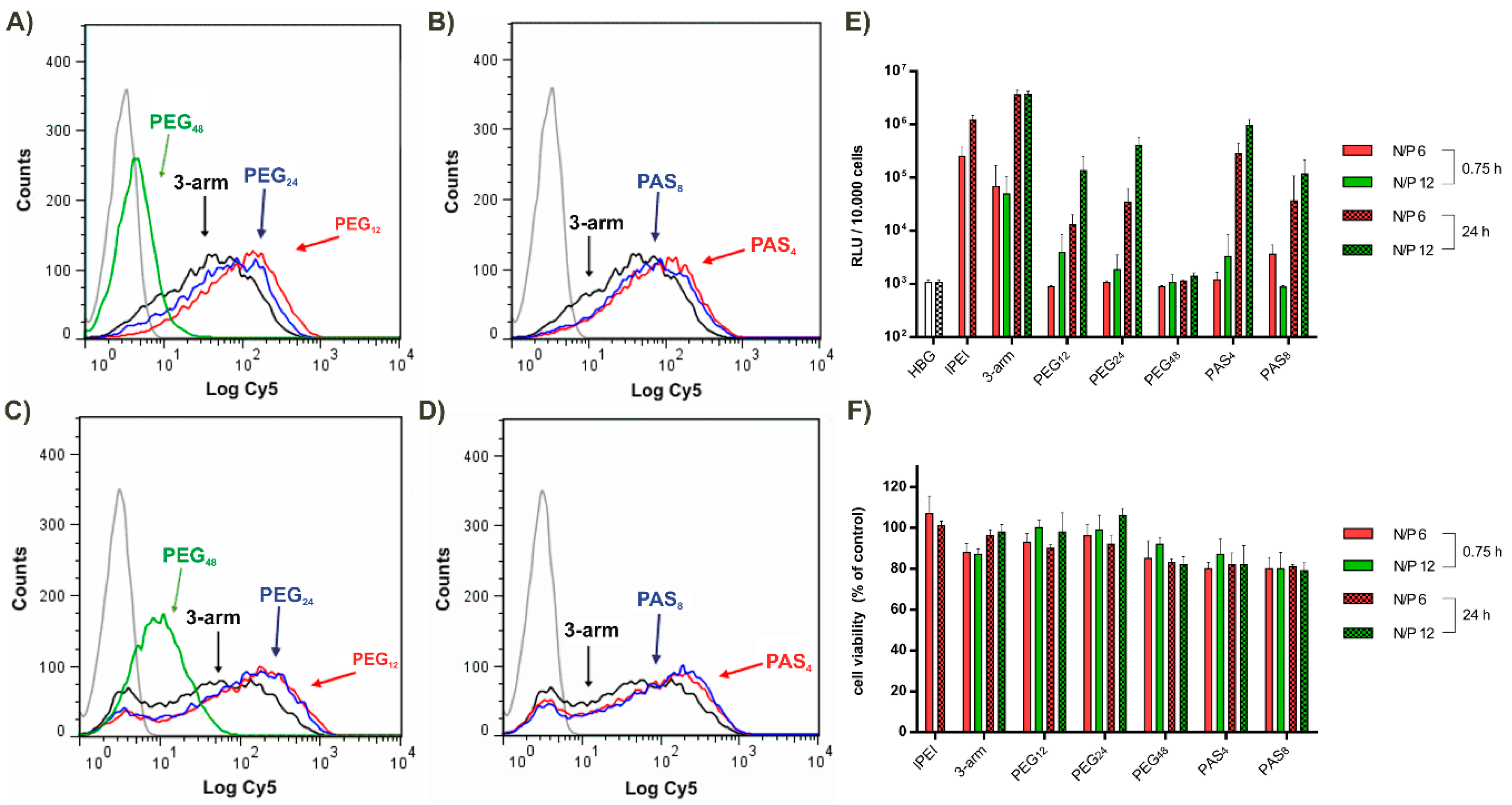
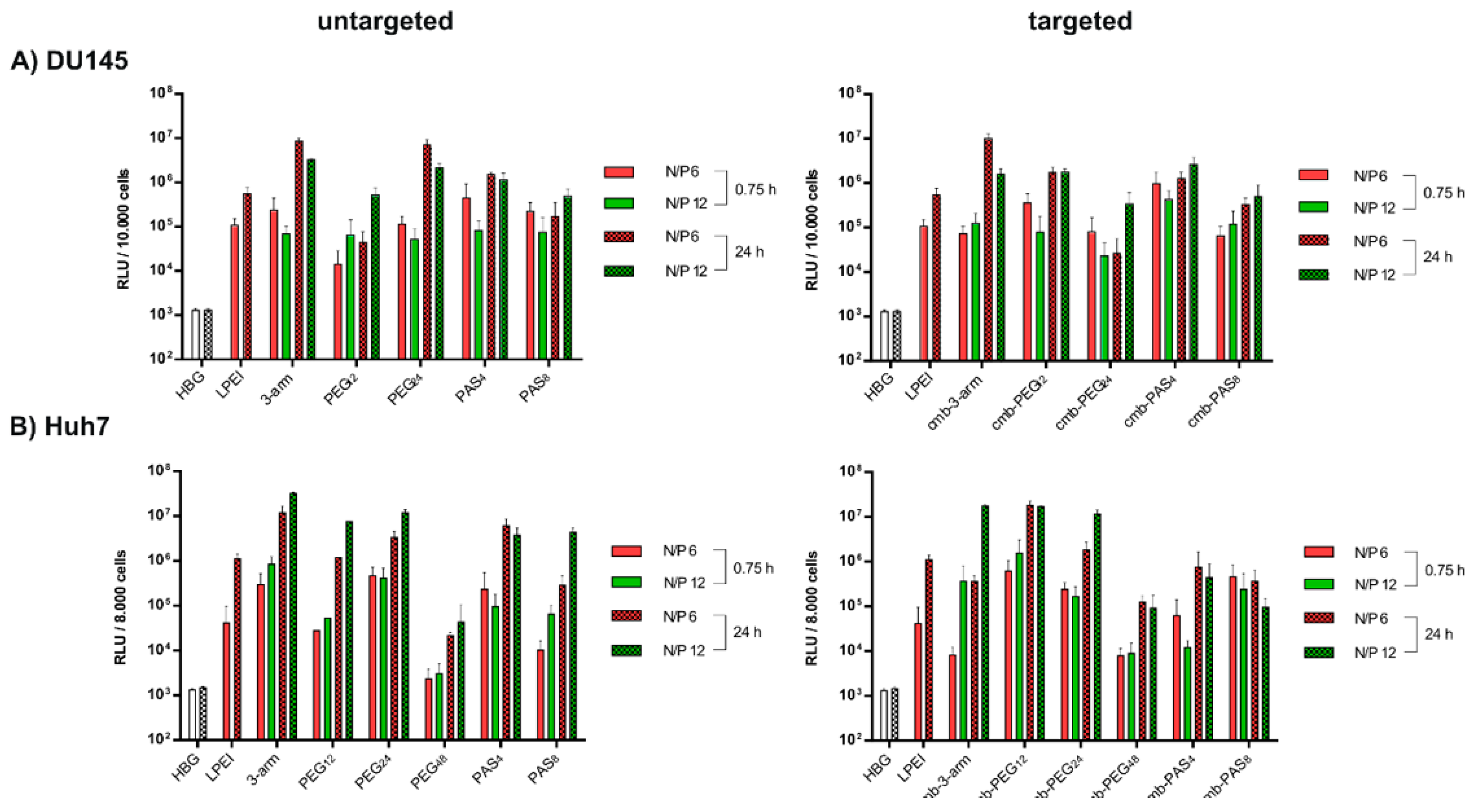
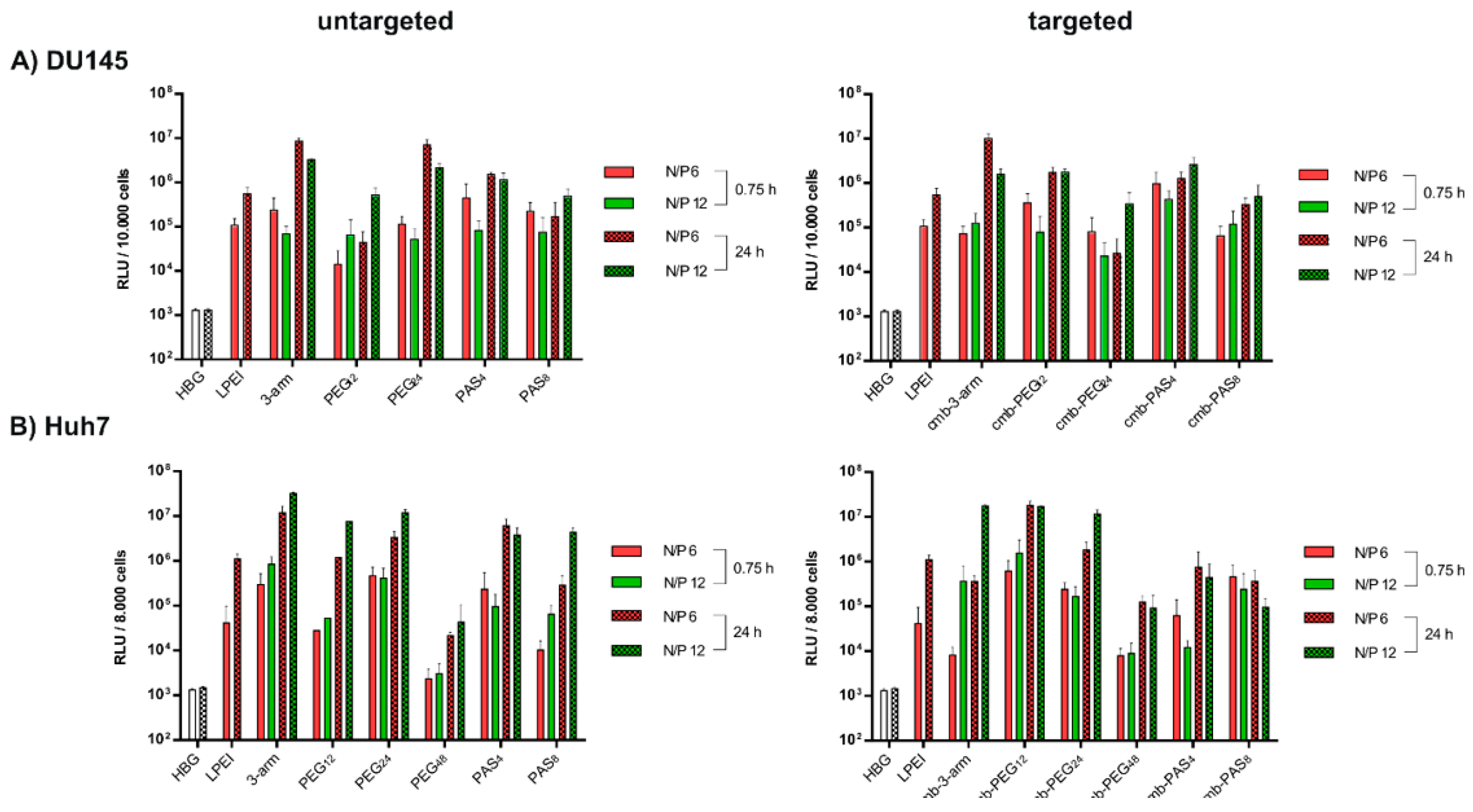
3.6. Tumor Cell Interactions In Vitro
3.7. Tumor Cell Interactions In Vitro without and with Targeting
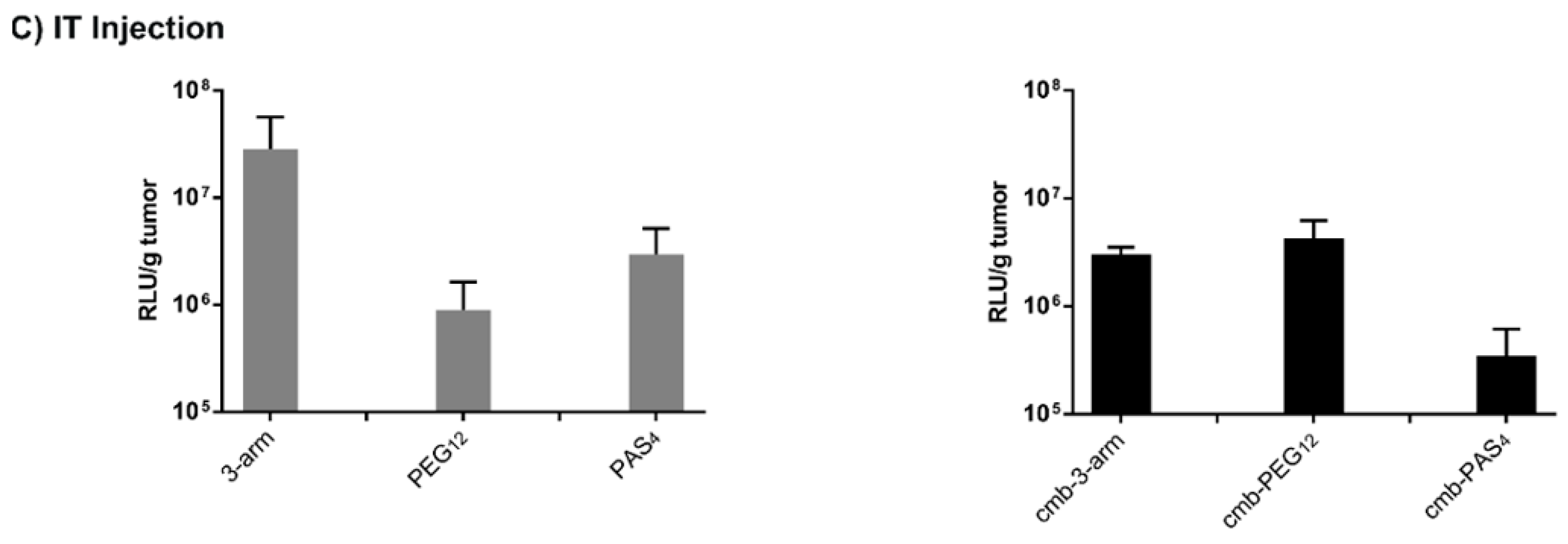
3.8. Tumor Cell Interactions In Vivo without and with Targeting
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Yla-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Methot, J.; Kastelein, J. Gene therapy for lipoprotein lipase deficiency. Curr. Opin. Lipidol. 2012, 23, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Al-Dosari, M.S.; Gao, X. Nonviral gene delivery: Principle, limitations, and recent progress. AAPS J. 2009, 11, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Lächelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, D.; Wagner, E. Gene therapy progress and prospects: Synthetic polymer-based systems. Gene Ther. 2008, 15, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Barenholz, Y.; Behr, J.P.; Cheng, S.H.; Cullis, P.; Huang, L.; Jessee, J.A.; Seymour, L.; Szoka, F.; Thierry, A.R.; et al. Nomenclature for synthetic gene delivery systems. Hum. Gene Ther. 1997, 8, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Cotten, M.; Foisner, R.; Birnstiel, M.L. Transferrin-polycation-DNA complexes: The effect of polycations on the structure of the complex and DNA delivery to cells. Proc. Natl. Acad. Sci. USA 1991, 88, 4255–4259. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Choi, J.L.; Chou, B.; Johnson, R.N.; Schellinger, J.G.; Pun, S.H. Effect of polyplex morphology on cellular uptake, intracellular trafficking, and transgene expression. ACS Nano 2013, 7, 10612–10620. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Whittaker, M.R.; Mak, C.W.; Davis, T.P. The importance of nanoparticle shape in cancer drug delivery. Expert Opin. Drug Deliv. 2015, 12, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Richardson, S.C. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: Opportunities and challenges. Mol. Pharm. 2012, 9, 2380–2402. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc′h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.P. The proton sponge: A trick to enter cells the viruses did not exploit. Chimia 1997, 51, 34–36. [Google Scholar]
- Sonawane, N.D.; Szoka, F.C.; Verkman, A.S. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Miyata, K.; Oba, M.; Ishii, T.; Suma, T.; Itaka, K.; Nishiyama, N.; Kataoka, K. Odd-even effect of repeating aminoethylene units in the side chain of n-substituted polyaspartamides on gene transfection profiles. J. Am. Chem. Soc. 2011, 133, 15524–15532. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Nishiyama, N.; Kataoka, K. Rational design of smart supramolecular assemblies for gene delivery: Chemical challenges in the creation of artificial viruses. Chem. Soc. Rev. 2012, 41, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.L.; Tan, J.K.Y.; Sellers, D.L.; Wei, H.; Horner, P.J.; Pun, S.H. Guanidinylated block copolymers for gene transfer: A comparison with amine-based materials for in vitro and in vivo gene transfer efficiency. Biomaterials 2015, 54, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Breunig, M.; Lungwitz, U.; Liebl, R.; Goepferich, A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl. Acad. Sci. USA 2007, 104, 14454–14459. [Google Scholar] [CrossRef] [PubMed]
- Kadlecova, Z.; Nallet, S.; Hacker, D.L.; Baldi, L.; Klok, H.A.; Wurm, F.M. Poly(ethyleneimine)-mediated large-scale transient gene expression: Influence of molecular weight, polydispersity and N-propionyl groups. Macromol. Biosci. 2012, 12, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, D.; Badgujar, N.; Wagner, E. Novel Fmoc-polyamino acids for solid-phase synthesis of defined polyamidoamines. Org. Lett. 2011, 13, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, D.; Troiber, C.; Wagner, E. New sequence-defined polyaminoamides with tailored endosomolytic properties for plasmid DNA delivery. Bioconjug. Chem. 2012, 23, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Kos, P.; Lächelt, U.; Herrmann, A.; Mickler, F.M.; Doblinger, M.; He, D.; Krhac Levacic, A.; Morys, S.; Bräuchle, C.; Wagner, E. Histidine-rich stabilized polyplexes for cMet-directed tumor-targeted gene transfer. Nanoscale 2015, 7, 5350–5362. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Müller, K.; Krhac Levacic, A.; Kos, P.; Lächelt, U.; Wagner, E. Combinatorial optimization of sequence-defined oligo(ethanamino)amides for folate receptor-targeted pDNA and siRNA delivery. Bioconjug. Chem. 2016, 27, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Dohmen, C.; Mas-Moruno, C.; Troiber, C.; Kos, P.; Schaffert, D.; Lächelt, U.; Teixido, M.; Günther, M.; Kessler, H.; et al. Solid-phase-assisted synthesis of targeting peptide-PEG-oligo(ethane amino)amides for receptor-mediated gene delivery. Org. Biomol. Chem. 2012, 10, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Edinger, D.; Fröhlich, T.; Schreiner, L.; Lächelt, U.; Troiber, C.; Rädler, J.; Hadwiger, P.; Vornlocher, H.P.; Wagner, E. Nanosized multifunctional polyplexes for receptor-mediated siRNA delivery. ACS Nano 2012, 6, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Lächelt, U.; Kos, P.; Mickler, F.M.; Herrmann, A.; Salcher, E.E.; Rödl, W.; Badgujar, N.; Bräuchle, C.; Wagner, E. Fine-tuning of proton sponges by precise diaminoethanes and histidines in pDNA polyplexes. Nanomedicine 2013, 10, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Rödl, W.; He, D.; Döblinger, M.; Lächelt, U.; Wagner, E. Combination of sequence-defined oligoaminoamides with transferrin-polycation conjugates for receptor-targeted gene delivery. J. Gene Med. 2015, 17, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.; Delgado, C.; Fisher, D.; Tilcock, C.; Gregoriadis, G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: Studies with poly(ethylene glycol)-coated vesicles. Biochim. Biophys. Acta 1991, 1062, 77–82. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.P.; Huang, L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef]
- Plank, C.; Mechtler, K.; Szoka, F.C., Jr.; Wagner, E. Activation of the complement system by synthetic DNA complexes: A potential barrier for intravenous gene delivery. Hum. Gene Ther. 1996, 7, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Kursa, M.; Walker, G.F.; Rössler, V.; Ogris, M.; Rödl, W.; Kircheis, R.; Wagner, E. Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug. Chem. 2003, 14, 222–231. [Google Scholar] [CrossRef] [PubMed]
- DeRouchey, J.; Walker, G.F.; Wagner, E.; Rädler, J.O. Decorated rods: A “bottom-up” self-assembly of monomolecular DNA complexes. J. Phys. Chem. B 2006, 110, 4548–4554. [Google Scholar] [CrossRef] [PubMed]
- Fella, C.; Walker, G.F.; Ogris, M.; Wagner, E. Amine-reactive pyridylhydrazone-based PEG reagents for pH-reversible PEI polyplex shielding. Eur. J. Pharm. Sci. 2008, 34, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Librizzi, D.; Pfestroff, A.; Schurrat, T.; Buyens, K.; Sanders, N.N.; De Smedt, S.C.; Behe, M.; Kissel, T. Stability of siRNA polyplexes from poly(ethylenimine) and poly(ethylenimine)-g-poly(ethylene glycol) under in vivo conditions: Effects on pharmacokinetics and biodistribution measured by fluorescence fluctuation spectroscopy and single photon emission computed tomography (spect) imaging. J. Control. Release 2009, 138, 148–159. [Google Scholar] [PubMed]
- Tockary, T.A.; Osada, K.; Motoda, Y.; Hiki, S.; Chen, Q.; Takeda, K.M.; Dirisala, A.; Osawa, S.; Kataoka, K. Rod-to-globule transition of pRNA/PEG-poly(l-lysine) polyplex micelles induced by a collapsed balance between DNA rigidity and PEG crowdedness. Small 2016, 12, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, P.; Mouton-Faivre, C. Anaphylaxis to macrogol 4000 after a parenteral corticoid injection. Allergy 2005, 60, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Lai, S.K. Anti-peg immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Wenande, E.; Garvey, L.H. Immediate-type hypersensitivity to polyethylene glycols: A review. Clin. Exp. Allergy 2016, 46, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Hunter, A.C.; Szebeni, J.; Moghimi, S.M. Poly(ethylene glycol)s generate complement activation products in human serum through increased alternative pathway turnover and a MASP-2-dependent process. Mol. Immunol. 2008, 46, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Dadswell, C.M.; Savay, S.; Alving, C.R.; Szebeni, J. Causative factors behind poloxamer 188 (pluronic f68, flocor)-induced complement activation in human sera. A protective role against poloxamer-mediated complement activation by elevated serum lipoprotein levels. Biochim. Biophys. Acta 2004, 1689, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Ulbrich, K. HPMA copolymers: 30 years of advances. Adv. Drug Deliv. Rev. 2010, 62, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, V.; Wolfert, M.A.; Dash, P.R.; Oupicky, D.; Ulbrich, K.; Seymour, L.W.; Schacht, E.H. Novel vectors for gene delivery formed by self-assembly of DNA with poly(l-lysine) grafted with hydrophilic polymers. Biochim. Biophys. Acta 1998, 1380, 354–368. [Google Scholar] [CrossRef]
- Oupicky, D.; Howard, K.A.; Konak, C.; Dash, P.R.; Ulbrich, K.; Seymour, L.W. Steric stabilization of poly-l-lysine/DNA complexes by the covalent attachment of semitelechelic poly[N-(2-hydroxypropyl)methacrylamide]. Bioconjug. Chem. 2000, 11, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Noga, M.; Edinger, D.; Kläger, R.; Wegner, S.V.; Spatz, J.P.; Wagner, E.; Winter, G.; Besheer, A. The effect of molar mass and degree of hydroxyethylation on the controlled shielding and deshielding of hydroxyethyl starch-coated polyplexes. Biomaterials 2013, 34, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Tian, C.; Ling, J.; Wang, Y. R8-modified polysarcosine-b-polylysine polypeptide to enhance circulation stability and gene delivery efficiency. J. Control. Release 2015, 213, e50–e51. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Birke, A.; Huesmann, D.; Weber, B.; Fischer, K.; Reske-Kunz, A.; Bros, M.; Barz, M. Introducing peptoplexes: Polylysine-block-polysarcosine based polyplexes for transfection of HEK 293t cells. Macromol. Biosci. 2014, 14, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tetley, L.; Uchegbu, I.F. The level of hydrophobic substitution and the molecular weight of amphiphilic poly-l-lysine-based polymers strongly affects their assembly into polymeric bilayer vesicles. J. Colloid Interface Sci. 2001, 237, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Wightman, L.; Schreiber, A.; Robitza, B.; Rössler, V.; Kursa, M.; Wagner, E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther. 2001, 8, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Binder, U.; Borger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. Pasylation: A biological alternative to pegylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mendler, C.T.; Friedrich, L.; Laitinen, I.; Schlapschy, M.; Schwaiger, M.; Wester, H.J.; Skerra, A. High contrast tumor imaging with radio-labeled antibody fab fragments tailored for optimized pharmacokinetics via pasylation. MAbs 2015, 7, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.F.; Fella, C.; Pelisek, J.; Fahrmeir, J.; Boeckle, S.; Ogris, M.; Wagner, E. Toward synthetic viruses: Endosomal pH-triggered deshielding of targeted polyplexes greatly enhances gene transfer in vitro and in vivo. Mol. Ther. 2005, 11, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. A multifunctional envelope type nano device (mend) for gene delivery to tumours based on the epr effect: A strategy for overcoming the PEG dilemma. Adv. Drug Deliv. Rev. 2011, 63, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Morys, S.; Wagner, E.; Lächelt, U. From artificial amino acids to sequence-defined targeted oligoaminoamides. Methods Mol. Biol. 2016, 1445, 235–258. [Google Scholar] [PubMed]
- Schaffert, D.; Troiber, C.; Salcher, E.E.; Fröhlich, T.; Martin, I.; Badgujar, N.; Dohmen, C.; Edinger, D.; Kläger, R.; Maiwald, G.; et al. Solid-phase synthesis of sequence-defined t-, i-, and u-shape polymers for pDNA and siRNA delivery. Angew. Chem. Int. Ed. Engl. 2011, 50, 8986–8989. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.X.; Goldgeier, L.; Zhu, J.S.; Cambell, P.; Ambulos, N.; Mixson, A.J. Histidine-lysine peptides as carriers of nucleic acids. Drug News Perspect. 2007, 20, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Salcher, E.E.; Kos, P.; Fröhlich, T.; Badgujar, N.; Scheible, M.; Wagner, E. Sequence-defined four-arm oligo(ethanamino)amides for pDNA and siRNA delivery: Impact of building blocks on efficacy. J. Control. Release 2012, 164, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Troiber, C.; Edinger, D.; Kos, P.; Schreiner, L.; Kläger, R.; Herrmann, A.; Wagner, E. Stabilizing effect of tyrosine trimers on pDNA and siRNA polyplexes. Biomaterials 2013, 34, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Ogris, M.; Brunner, S.; Schuller, S.; Kircheis, R.; Wagner, E. Pegylated DNA/transferrin-pei complexes: Reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999, 6, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Beck, R.W.; Prevette, L.E. Impact of molecular weight and degree of conjugation on the thermodynamics of DNA complexation and stability of polyethylenimine-graft-poly(ethylene glycol) copolymers. Biophys. Chem. 2015, 203–204, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Williford, J.M.; Archang, M.M.; Minn, I.; Ren, Y.; Wo, M.; Vandermark, J.; Fisher, P.B.; Pomper, M.G.; Mao, H.Q. Critical length of PEG grafts on lPEI/DNA nanoparticles for efficient in vivo delivery. ACS Biomater. Sci. Eng. 2016, 2, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Ogris, M.; Steinlein, P.; Kursa, M.; Mechtler, K.; Kircheis, R.; Wagner, E. The size of DNA/transferrin-pei complexes is an important factor for gene expression in cultured cells. Gene Ther. 1998, 5, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Polymers for siRNA delivery: Inspired by viruses to be targeted, dynamic, and precise. Acc. Chem. Res. 2012, 45, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Lächelt, U.; Bartek, J.; Wagner, E.; Moghimi, S.M. Polyplex evolution: Understanding biology, optimizing performance. Mol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Knorr, V.; Allmendinger, L.; Walker, G.F.; Paintner, F.F.; Wagner, E. An acetal-based pegylation reagent for pH-sensitive shielding of DNA polyplexes. Bioconjug. Chem. 2007, 18, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Park, E.H.; Cheong, S.J.; Lee, C.M.; Jeong, H.J.; Kim, D.W.; Lim, S.T.; Sohn, M.H. In vivo imaging of mesenchymal-epithelial transition factor (c-Met) expression using an optical imaging system. Bioconjug. Chem. 2009, 20, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Broda, E.; Mickler, F.M.; Lächelt, U.; Morys, S.; Wagner, E.; Bräuchle, C. Assessing potential peptide targeting ligands by quantification of cellular adhesion of model nanoparticles under flow conditions. J. Control. Release 2015, 213, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Hepatocyte growth factor and its receptor signalling complex as targets in cancer therapy. Anticancer Agents Med. Chem. 2010, 10, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Urnauer, S.; Morys, S.; Krhac Levacic, A.; Müller, A.M.; Schug, C.; Schmohl, K.A.; Schwenk, N.; Zach, C.; Carlsen, J.; Bartenstein, P.; et al. Sequence-defined cMET/HGFR-targeted polymers as gene delivery vehicles for the theranostic sodium iodide symporter (NIS) gene. Mol. Ther. 2016, 24, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Park, E.H.; Cheong, S.J.; Lee, C.M.; Kim, D.W.; Jeong, H.J.; Lim, S.T.; Sohn, M.H.; Kim, K.; Chung, J. Characterization, biodistribution and small-animal spect of I-125-labeled c-Met binding peptide in mice bearing c-Met receptor tyrosine kinase-positive tumor xenografts. Nuclear Med. Biol. 2009, 36, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Polymeric filomicelles and nanoworms: Two decades of synthesis and application. Polym. Chem. 2016, 7, 4295–4312. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano 2013, 7, 2935–2947. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer # | Structure | Abbreviation |
|---|---|---|
| 689 | [C-(H-Stp)3-H]α,ε-K-H-(Stp-H)3-C | 3-arm |
| 1088 | {[C-(H-Stp)4-H]α,ε-K-H-dPEG12}ε-K | PEG12 |
| 1091 | {[C-(H-Stp)4-H]α,ε-K-H-dPEG24}ε-K | PEG24 |
| 1120 | {[C-(H-Stp)4-H]α,ε-K-H-dPEG24-dPEG24}ε-K | PEG48 |
| 1094 | {[C-(H-Stp)4-H]α,ε-K-H-(PAS)4}ε-K | PAS4 |
| 1097 | {[C-(H-Stp)4-H]α,ε-K-H-(PAS)8}ε-K | PAS8 |
| Polymer # | Abbreviation | Average size (nm) | Average PDI | Average zeta potential (mV) |
|---|---|---|---|---|
| 689 | 3-arm | 126.8 ± 2.7 | 0.15 ± 0.02 | 32.0 ± 3.5 |
| 1088 | PEG12 | 97.9 ± 1.1 | 0.14 ± 0.02 | 6.7 ± 2.3 |
| 1091 | PEG24 | 111.6 ± 0.9 | 0.16 ± 0.01 | 3.1 ± 0.6 |
| 1120 | PEG48 | 87.2 ± 1.4 | 0.33 ± 0.05 | 1.6 ± 0.5 |
| 1094 | PAS4 | 127.7 ± 0.8 | 0.12 ± 0.01 | 7.1 ± 1.5 |
| 1097 | PAS8 | 147.0 ± 1.8 | 0.15 ± 0.01 | 3.4 ± 0.8 |
| Experiment | Oligomers | |||||
|---|---|---|---|---|---|---|
| 3-arm | PEG12 | PEG24 | PEG48 | PAS4 | PAS8 | |
| PBS resistance | – | – | + | + | – | + |
| Erythrocyte adhesion | – | ++ | ++ | +++ | + | + |
| Particle compaction | +++ | ++ | + | – | ++ | + |
| Polyplex shape | globule | rod-like | doughnut aggregates | cord-like | rod-like | doughnut |
| Heparin resistance | + | + | – | – | + | + |
| Serum stability | + | + | – | – | + | – |
| Shielding | – | + | ++ | +++ | + | ++ |
| Transfection (short term) | + | – | – | – | – | – |
| Transfection (long term) | + | + | + | v | + | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morys, S.; Krhac Levacic, A.; Urnauer, S.; Kempter, S.; Kern, S.; Rädler, J.O.; Spitzweg, C.; Lächelt, U.; Wagner, E. Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles. Polymers 2017, 9, 142. https://doi.org/10.3390/polym9040142
Morys S, Krhac Levacic A, Urnauer S, Kempter S, Kern S, Rädler JO, Spitzweg C, Lächelt U, Wagner E. Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles. Polymers. 2017; 9(4):142. https://doi.org/10.3390/polym9040142
Chicago/Turabian StyleMorys, Stephan, Ana Krhac Levacic, Sarah Urnauer, Susanne Kempter, Sarah Kern, Joachim O. Rädler, Christine Spitzweg, Ulrich Lächelt, and Ernst Wagner. 2017. "Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles" Polymers 9, no. 4: 142. https://doi.org/10.3390/polym9040142
APA StyleMorys, S., Krhac Levacic, A., Urnauer, S., Kempter, S., Kern, S., Rädler, J. O., Spitzweg, C., Lächelt, U., & Wagner, E. (2017). Influence of Defined Hydrophilic Blocks within Oligoaminoamide Copolymers: Compaction versus Shielding of pDNA Nanoparticles. Polymers, 9(4), 142. https://doi.org/10.3390/polym9040142