3.1. Characterization of Materials
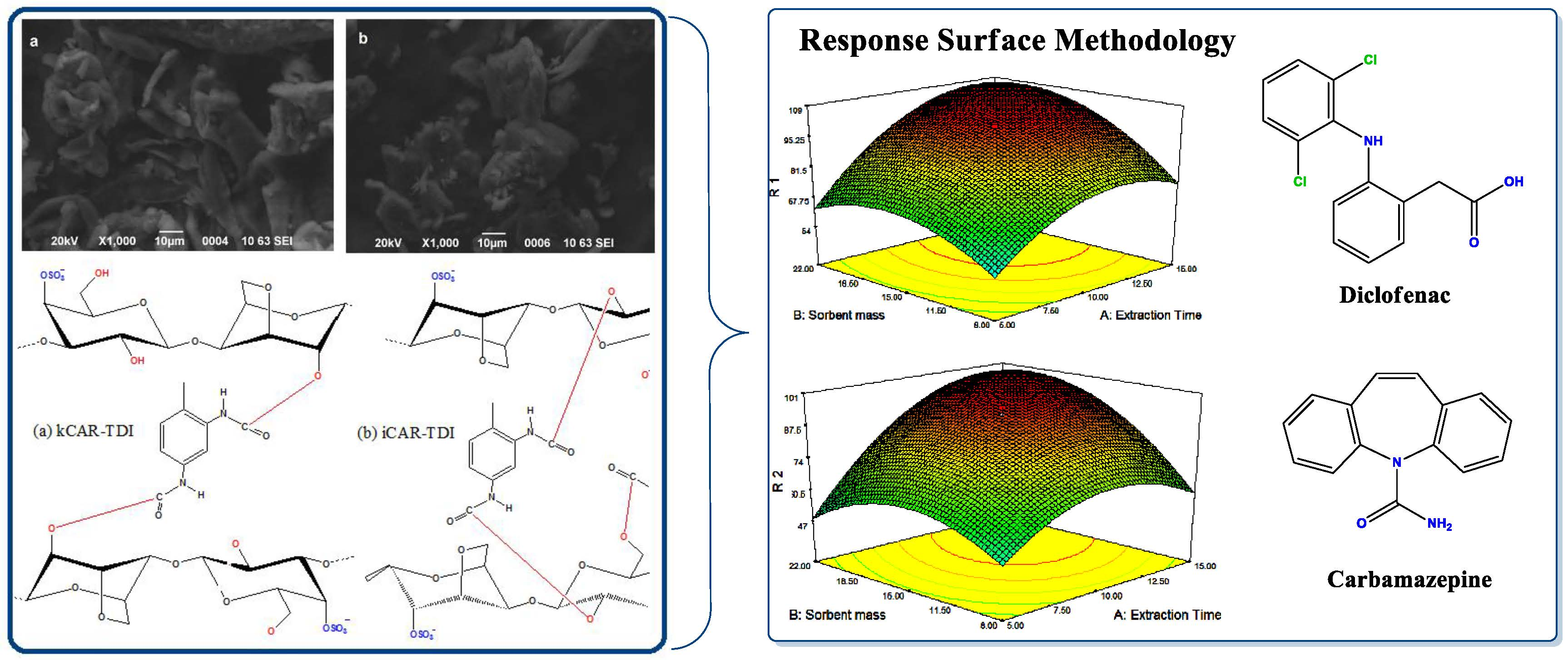
The morphology of the prepared materials was examined by SEM images. It was found that kCAR and iCAR have smooth surfaces without any specific shape (
Figure 1a,b). The modification with TDI caused more rigid and hard surface (
Figure 1c,d), while after chopping in cutting mill, the particles retained an irregular shape with sharp edges. This is an indication that the reaction had taken place.
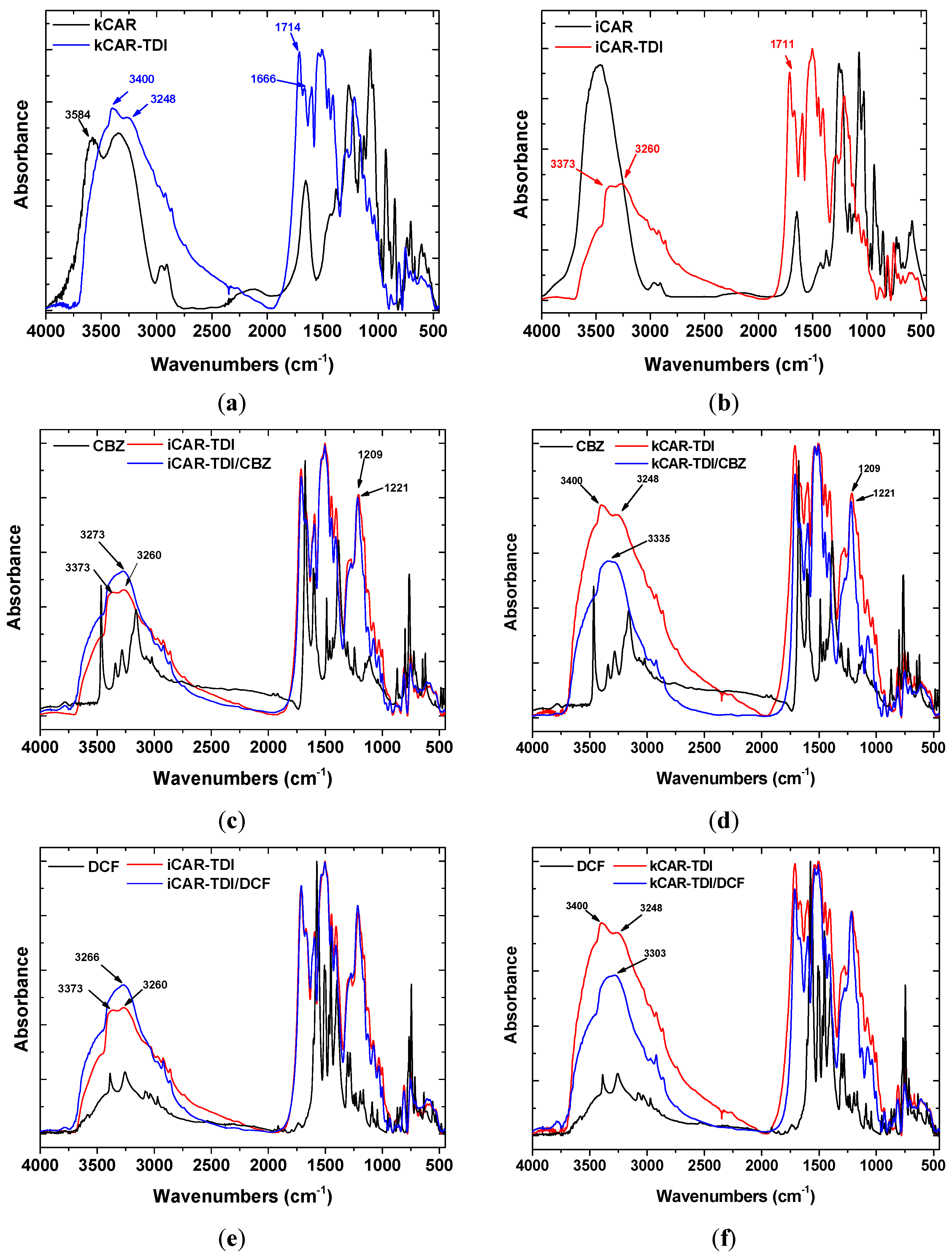
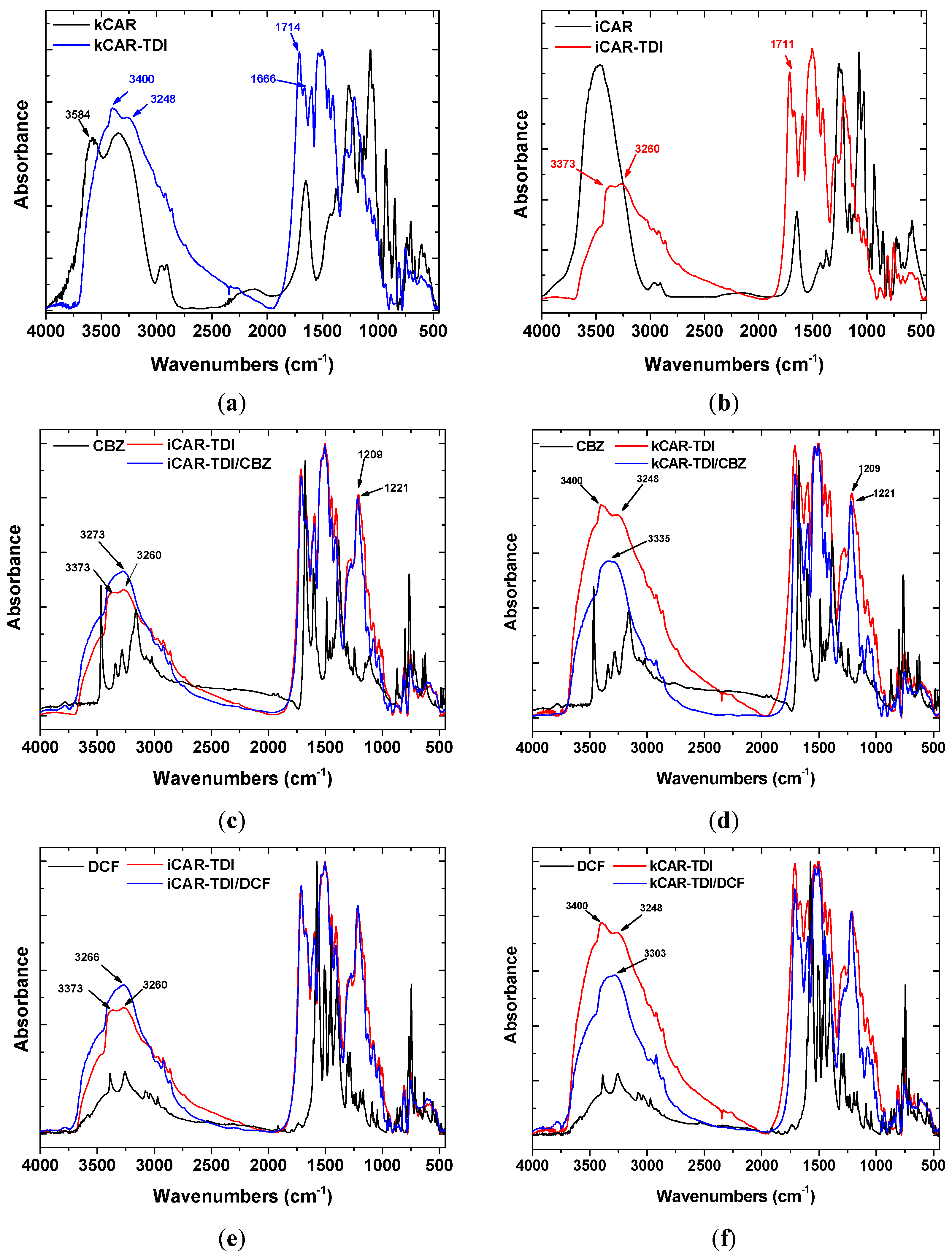
Figure 2a,b shows the FTIR spectra of neat and modified carrageenans before adsorption of CBZ, DCF. All the characteristic peaks of net carrageenans are present in the spectra: 3600–3200 cm
−1 (broad peak owing to O–H interactions), 1645–1755 cm
−1 (carbonyl group stretching), 1267 cm
−1 (O=S=O asymmetric stretching), 1156 cm
−1 (C–O–C asymmetric stretching), 1067–1074 cm
−1 (S–O symmetric stretching), 1027–1038 cm
−1 (C–O stretching), 928–935 cm
−1 (C–O–C stretching in 3,6-anhydrogalactose) and 852 cm
−1 (C–O–S) stretching in a (1-3)-
d-galactose [
32]. FTIR spectra of modified carrageenans are also present in
Figure 2. It was observed that kCAR-TDI (
Figure 2a) formed a new peak at 3248 cm
−1 owing to >N–H stretching vibrations. In advance, two new peaks were also recorded at 1666 and 1714 cm
−1 owing to >C=O stretching vibration. In addition, the absence of peak at 2275 cm
−1 (–N=C=O group) confirmed the absence of unreacted TDI. Similar findings were also presented for iCAR-TDI with >Ν–H group to be recorded at 3260 cm
−1 (
Figure 2b).
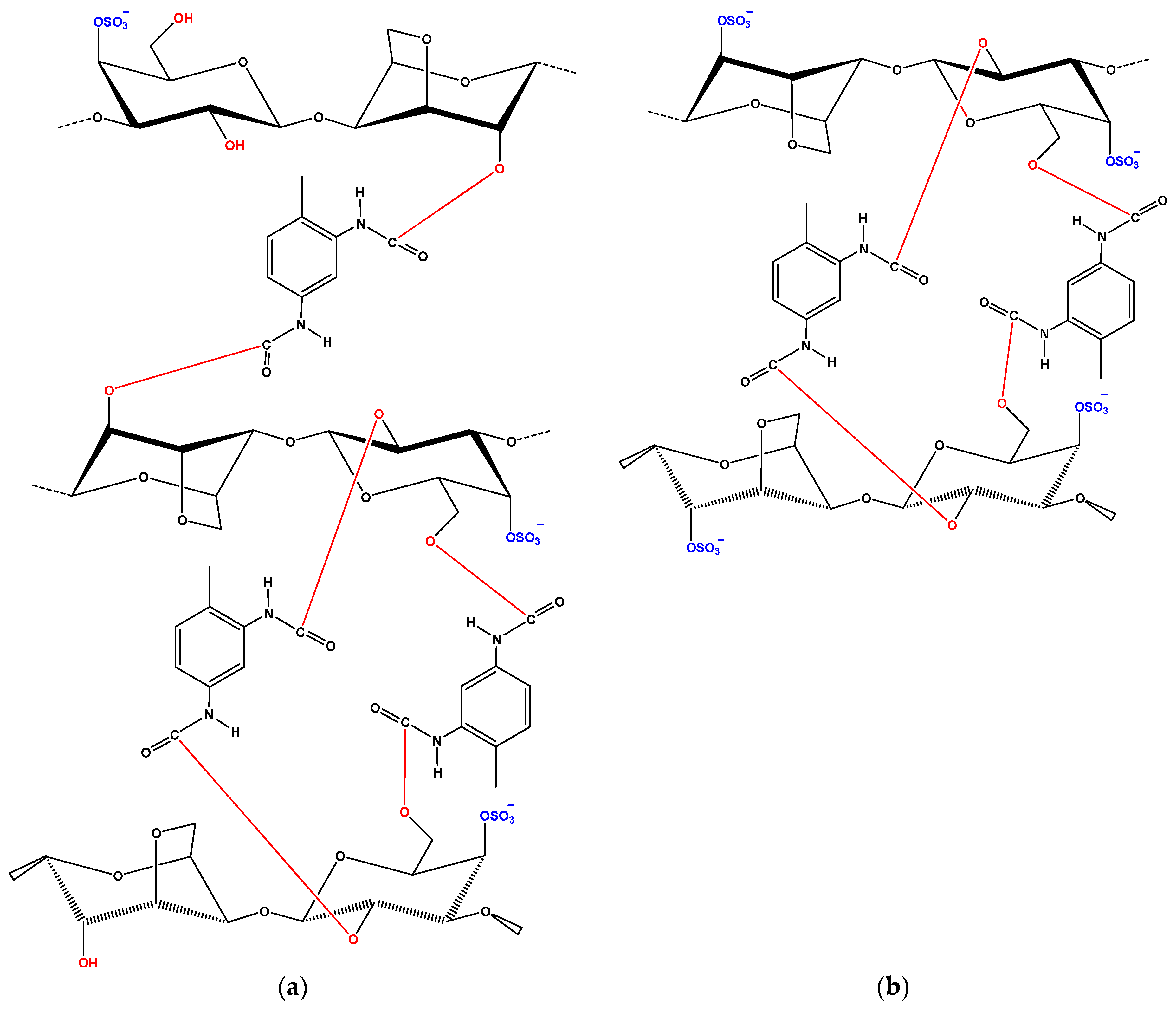
Before analyzing the loaded-adsorbents (after adsorption of CBZ or DCF), it is necessary to present the FTIR of each drug separately. CBZ spectrum corresponds to those previously reported for the polymorph form III. Characteristic peaks were observed at 3461 (−NH valence vibration), 1676 (−CO−R vibration), 1594 and 1602 cm−1 (in the range of −C=C− and −C=O vibrations; −NH deformation). On the other hand, DCF spectrum exhibited distinctive peaks at 3381 cm−1 due to N−H stretching of the secondary amine, 1571 cm−1 owing to –C=O stretching of the carboxyl ion and at 748 cm−1 owing to C−Cl stretching.
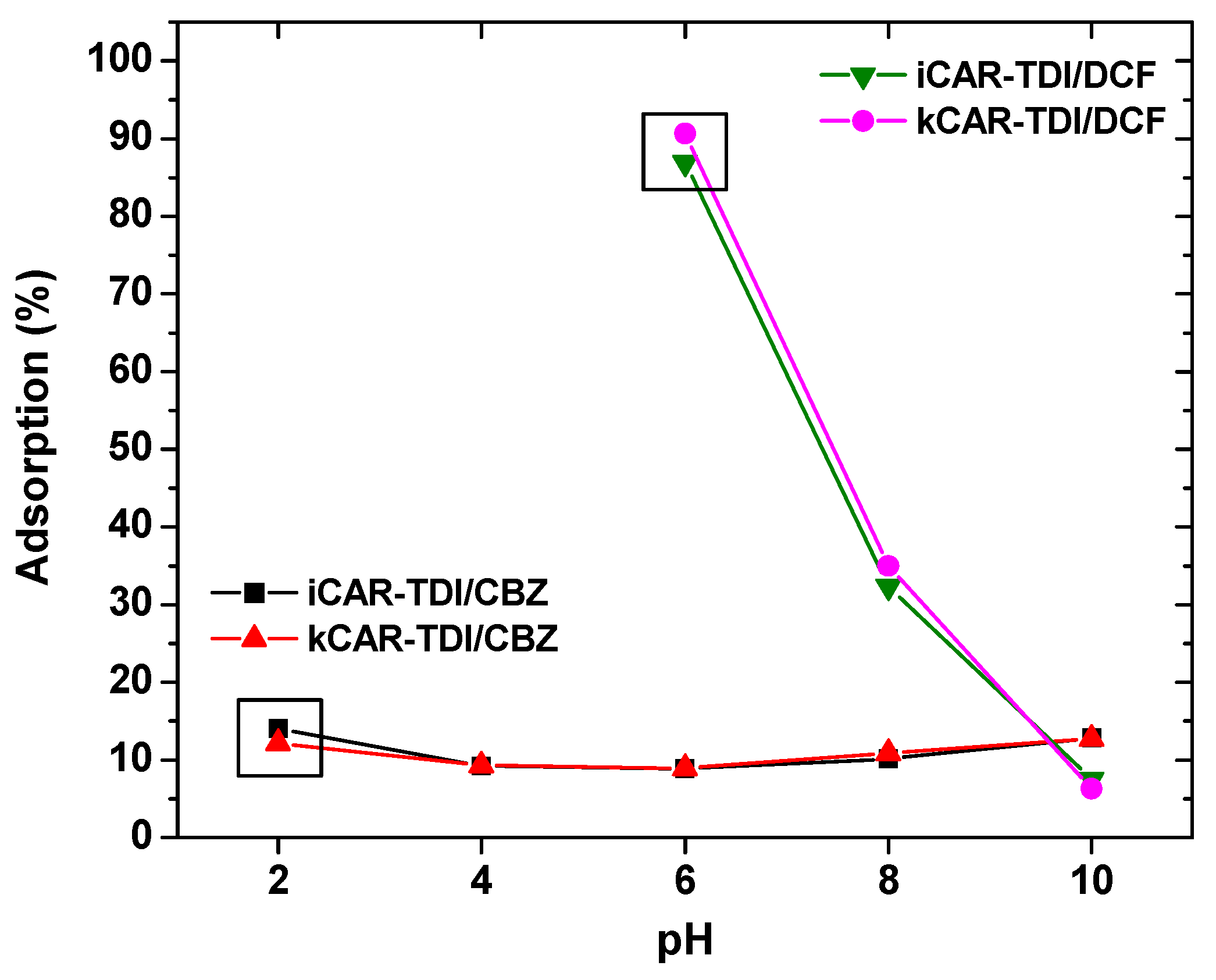
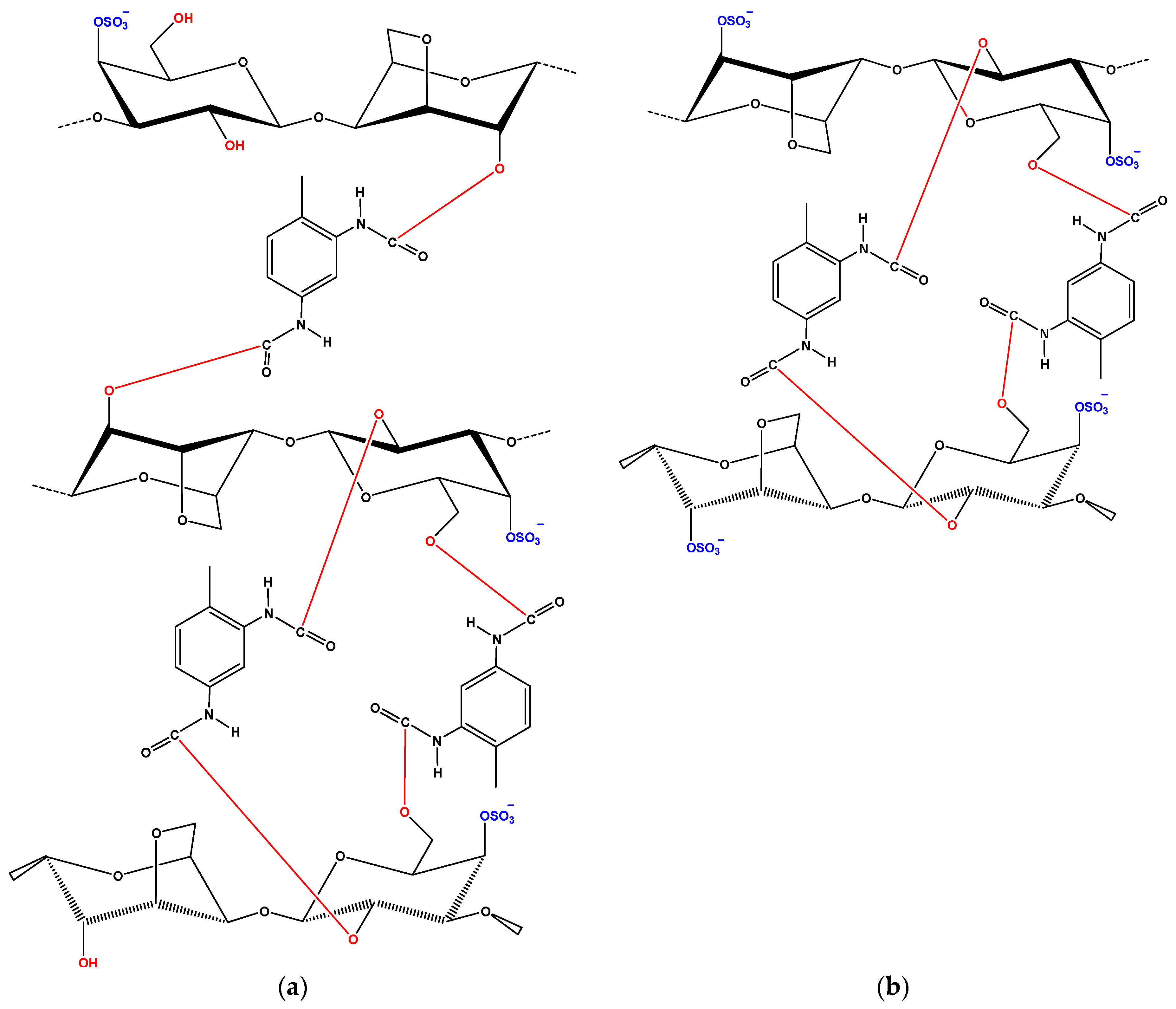
After adsorption, there were shifts of wavenumbers. For CBZ adsorption, in the case of iCAR-TDI (
Figure 2c), a shift from 3260 to 3273 cm
−1 was due to the hydrogen bond formation between CBZ molecule and material. Another shift was observed from 1209 to 1221 cm
−1 owing to interactions between sulfur and amino groups or oxygen atom of CBZ molecule. Similar findings were observed for kCAR-TDI (
Figure 2d).
For DCF adsorption, in the case of iCAR-TDI (
Figure 2e), a shift from 3260 to 3266 cm
−1 was due to hydrogen bond formation between CBZ molecule and material. Analogous were the observations for kappa-modified carrageenan and recorded shifts are presenting in corresponding figures.
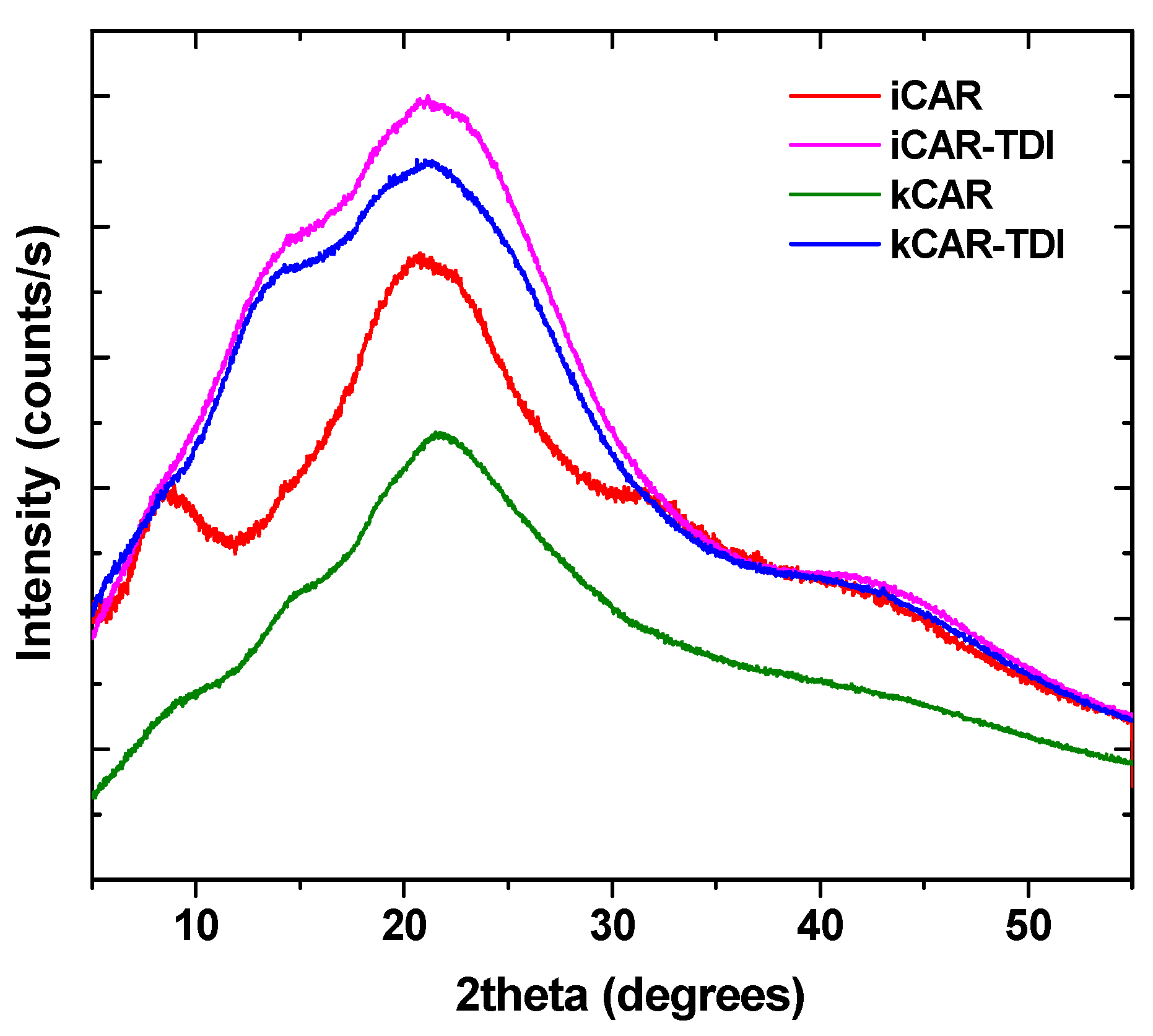
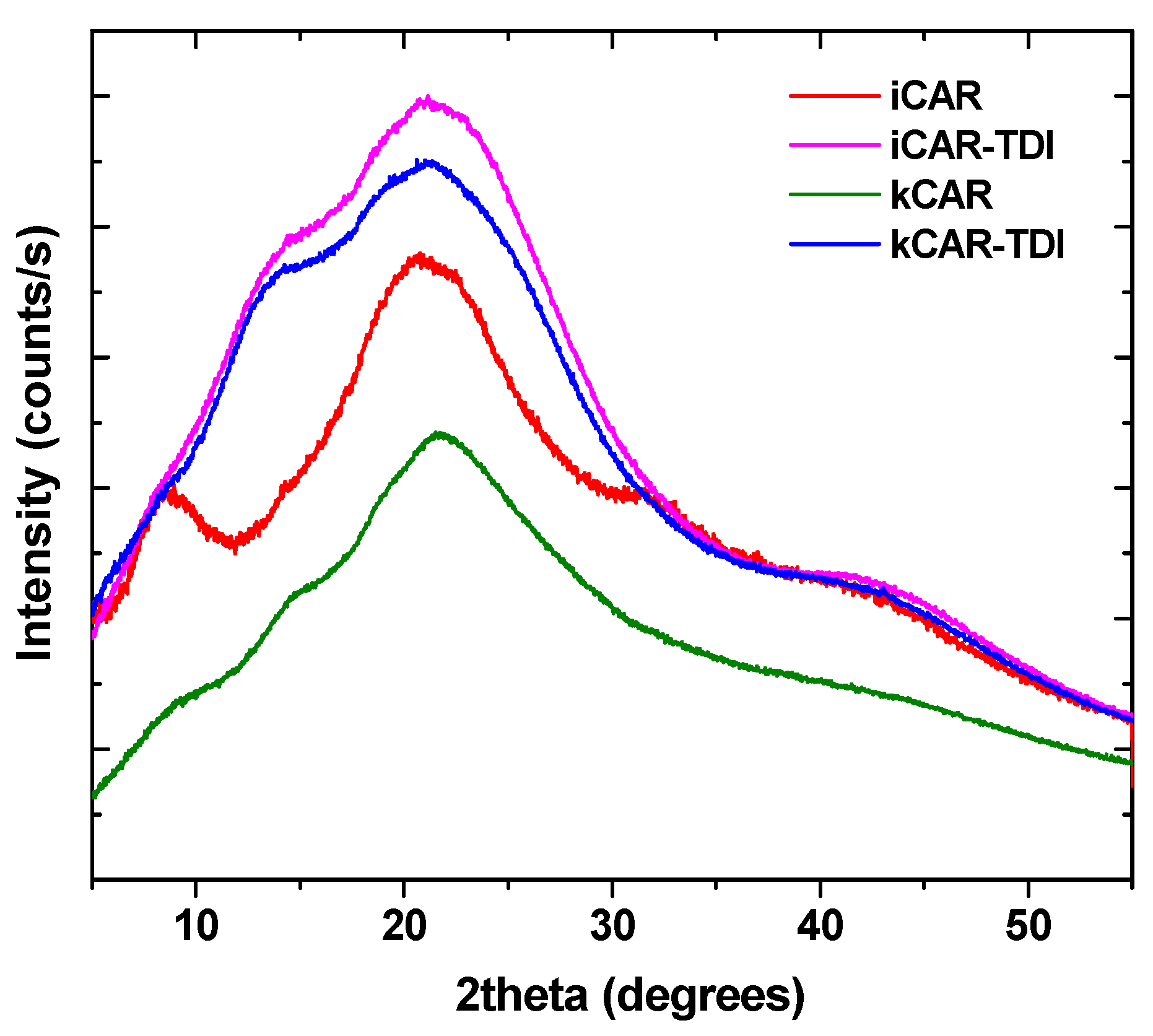
XRD was used to examine any changes to the physical state of carrageenans (
Figure 3). As was foundin a previous study [
33], carrageenans show a wide broad peak indicating their amorphous state. The addition of –NCO– groups did not affect the amorphous state and the resulting cross-linked carrageenans also had a wide broad peak.
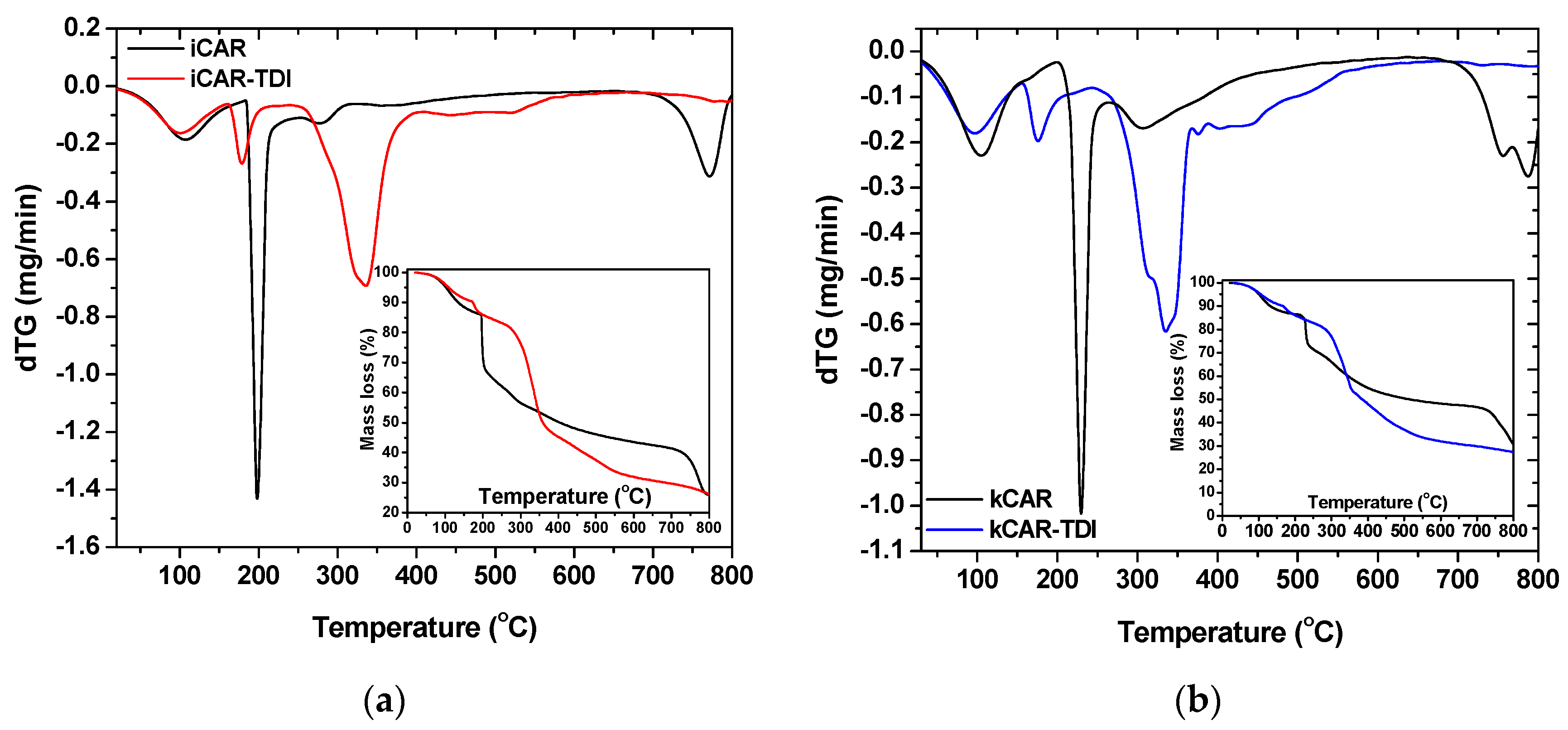
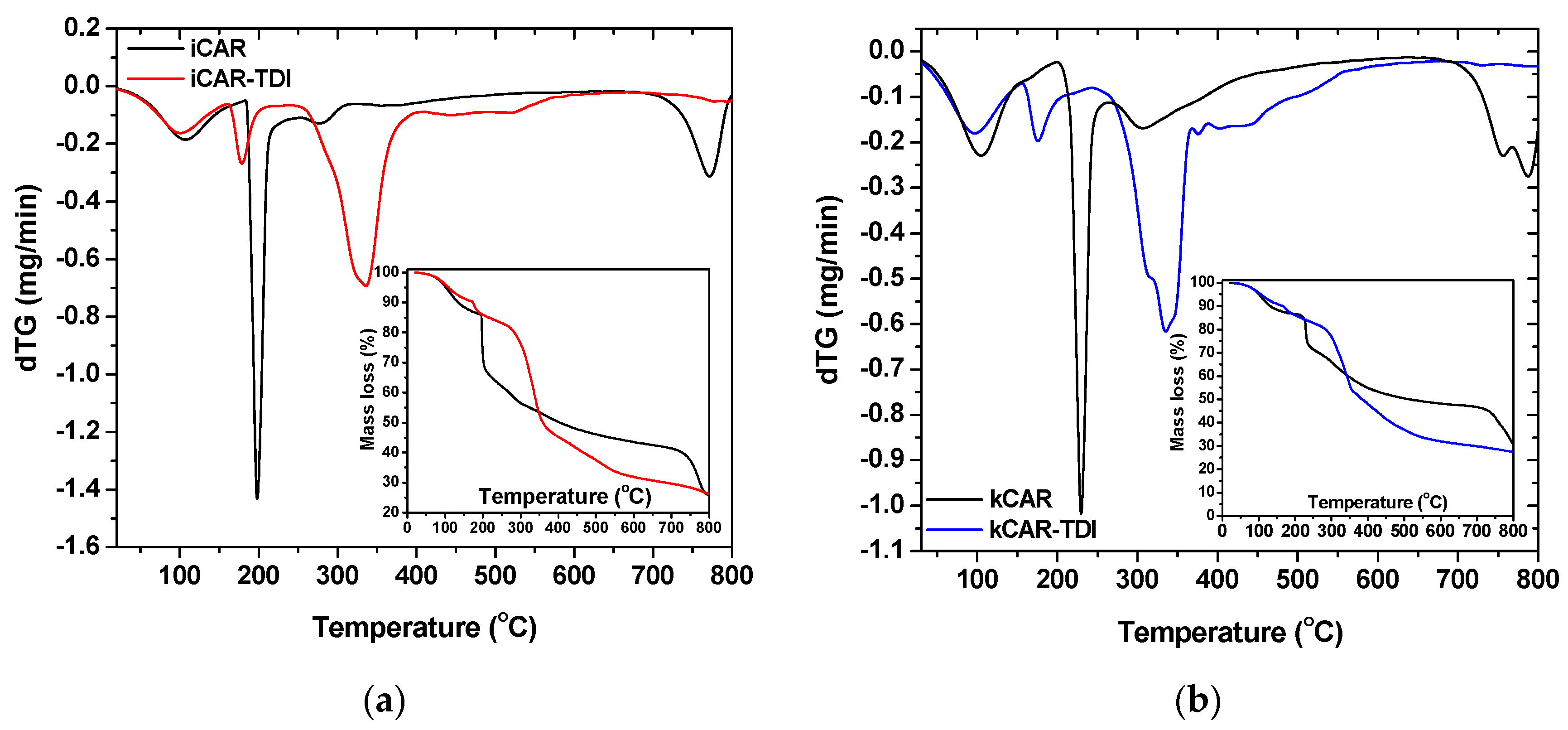
To assess the thermal stability of the modified carrageenans, TGA was carried out (
Figure 4). As can be seen, thermal degradation profile of –NCO– functionalized (TDI) carrageenan turned out to be quite different compared to that of neat carrageenans. The modified also showed reduced thermal degradation stability due to –NCO– groups. Similar observations were made in previous studies concerning cross-linked chitin and chitosan with HDMI [
33,
34].
Moreover, kCAR showed to be more resistant to thermal stability than iCAR. Both carrageenans showed three decomposition stages (black lines in insets of
Figure 4a,b): (i) an initial stage attributed to water evaporation and lasted at 100 °C; (ii) a main decomposition stage due to degradation of the saccharide structure of the molecule, to the dehydration of saccharide rings and decomposition of deacetylated carrageenan units; and (iii) a third stage owing to the acetylated part of the molecule. As was mentioned previously, all samples (modified and non-modified) showed an initial mass loss until 100 °C, which can be reasonably explained by the evaporation of water. DTG curves showed that decomposition for net carrageenans exhibited three stages.
On the other hand, four decomposition stages were observed for the modified carrageenans (red and blue lines in insets of
Figure 4a,b): (i) the initial stage of water evaporation; (ii) a second stage owing to –NCO– segments loss; (iii) a third stage of the degradation of the saccharide structure of molecule, the dehydration of saccharide rings and decomposition of deacetylated carrageenan units; and (iv) a fourth stage owed to the acetylated part of the molecule. As was previously reported, modified carrageenans showed reduced thermal stability with iota-modified carrageenan showing the lowest one. A closer observation at the third stage of DTG curves showed that two different decomposition processes might occur during decomposition.
3.3. Optimization of D-SPE Methodology by Central Composite Design
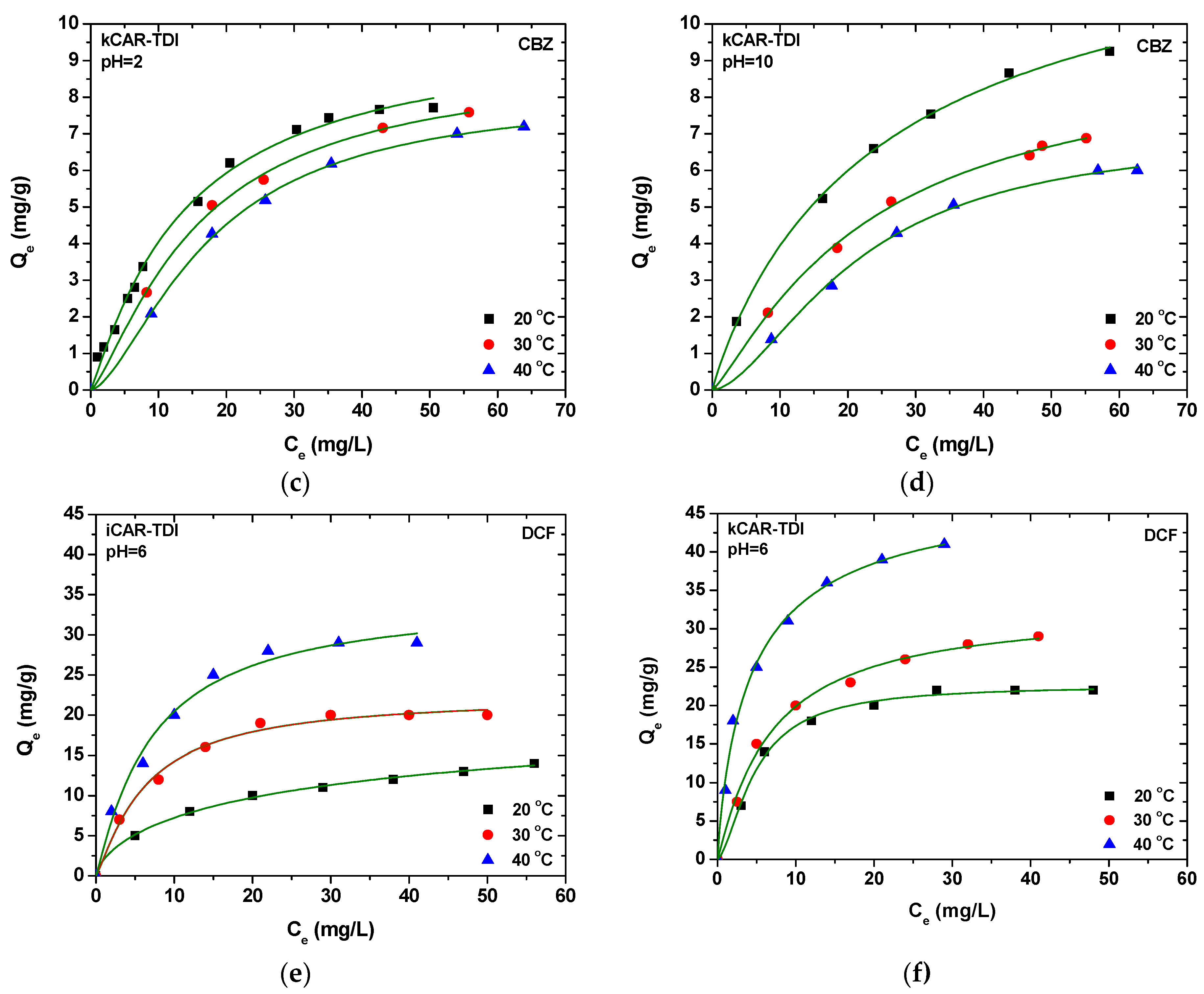
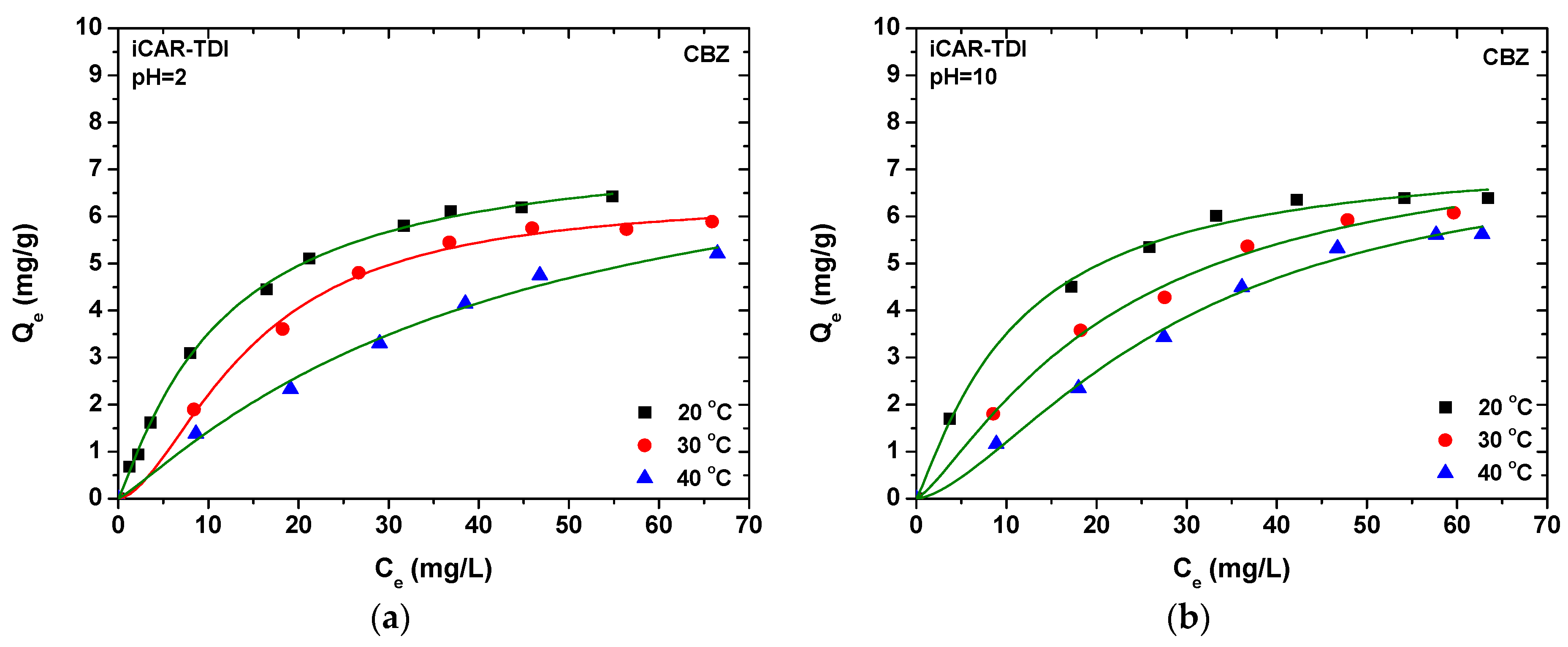
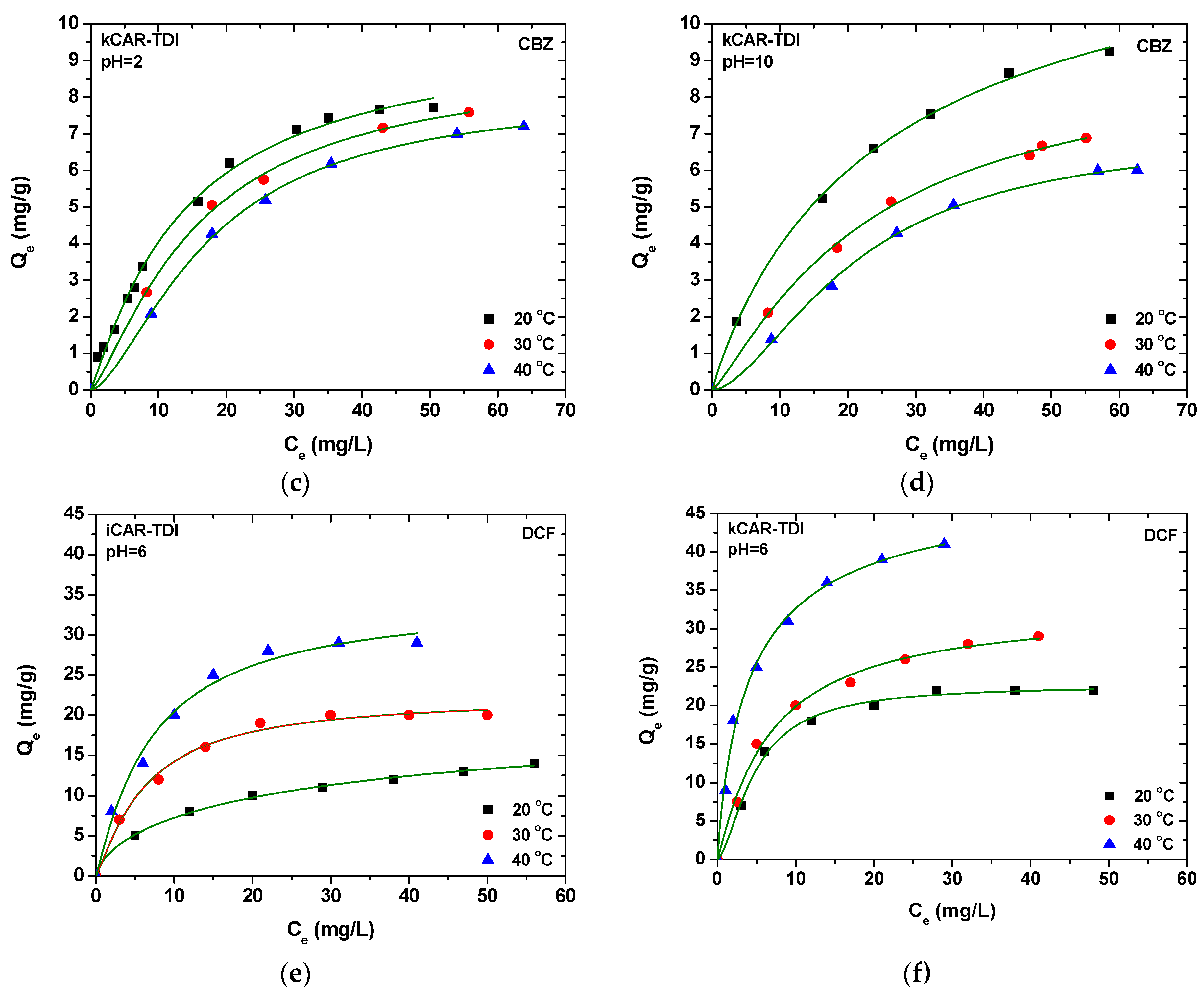
In extraction procedures, the optimization step is very important to increase the extraction efficiency. In the present study, the pH and the temperature of the solution have been well studied for the adsorption experiments. Desorption solvents were selected primarily using one variable at a time, while, for the other variables, a response surface methodology (RSM) using a central composite design (CCD) was applied for searching the optimal experimental conditions for both analytes. RSM is a multivariate optimization procedure which helps us to find out the optimized condition with the least number of experiments. Based on the higher adsorption capacity for both compounds, kCAR-TDI material was used for the development and optimization of the D-SPE analytical methodology.
3.3.1. Desorption Solvents
To obtain reliable and reproducible analytical results and a high enrichment factor, the eluent for the D-SPE procedure must have high affinity towards the target analytes than the sorbent. In this study, three eluents including methanol, acetonitrile and acetone were tested for elution of DCF and CBZ from the sorbents, according to the principles of green chemistry, to avoid halogenated solvents. When 1 mL of these desorption solvent was evaluated, it was observed that methanol and acetonitrile resulted in higher recoveries than acetone. Ultimately, methanol was chosen as the eluting solvents due to its better extraction efficiency (increase of 5–10% compared to ACN), lower toxicity and cost.
3.3.2. Central Composite Design
The approach of studying one factor at a time cannot describe adequately the importance of certain factors on the extraction process, because interactions between factors are not considered. In this light, to evaluate the combined effects and interactions of extraction time (A), sorbent mass (B) and eluent volume (C), the D-SPE process was further assessed by experimental design and RSM. The second order response surface, which is modeled on the resultsobtained from CCD experiments,can be expressed as:
Following to fitting the second order polynomial equation with the actual data of both responses of interest (i.e., DCF (
R1) and CBZ (
R2)), two multiple regression analyses were separately proposed in terms of all the 17 possible combinations of three independent criteria. To test the significance and adequacy of the model, the analysis of variance (ANOVA) was performed for each response and the obtained results are given in
Table 4. The statistical significance of the model equations was evaluated by the F-test and
p-value. According to the results, the high F-values (55.55 for DCF and 15.34 for CBZ) and small
p-values (<0.0001 for DCF and 0.0008 for CBZ value, both
p-values < 0.01) suggested that the regression models obtained are highly significant. The goodness of fit of regression model was carried out by determination coefficient (
R2) and adjusted determination coefficient (
R2adj).
R2 values computed as 0.9862 for DCF and 0.9517 for CBZ, indicating the goodness-of-fit of the proposed models. Similarly, the values of Adj
R2 for both
R1 and
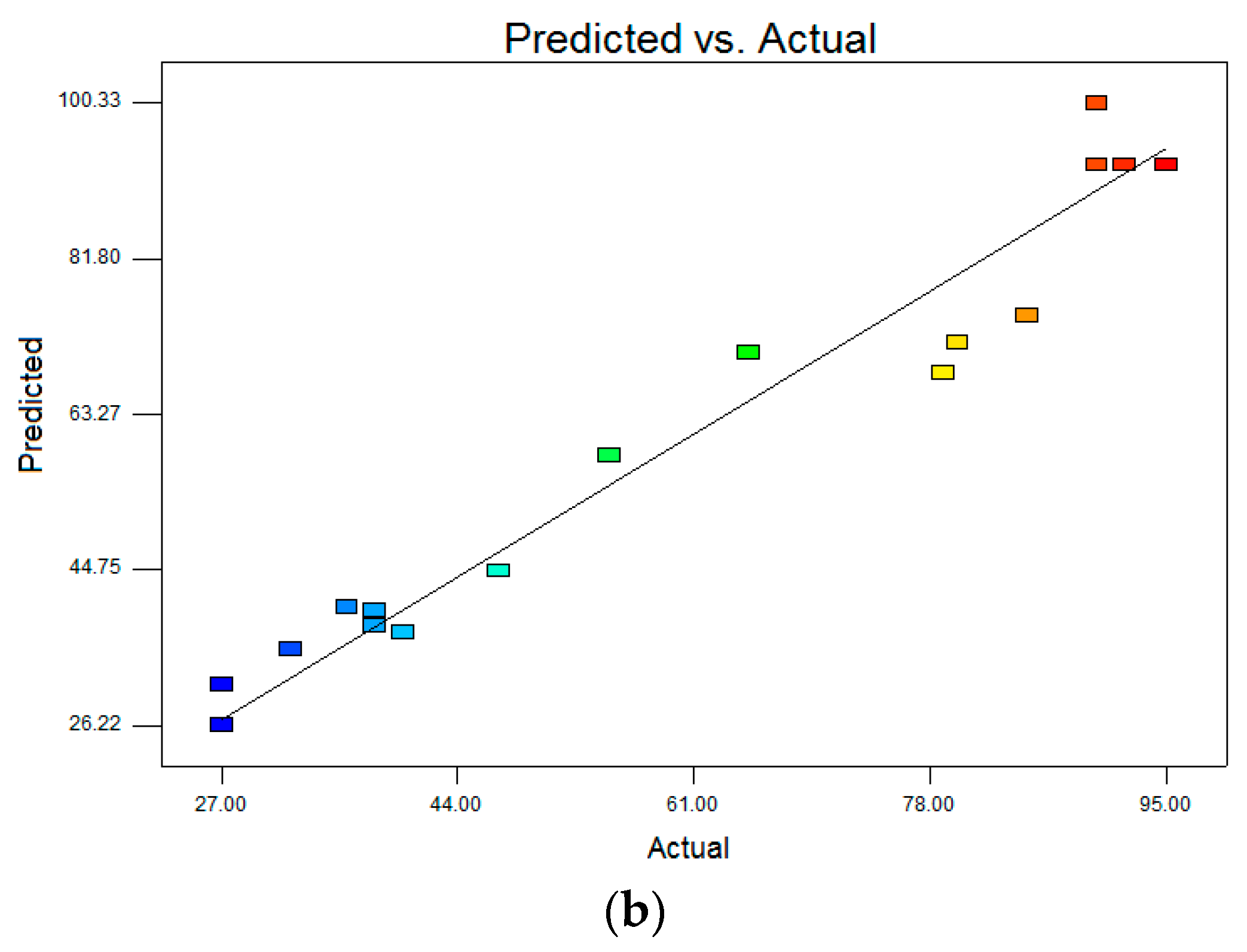
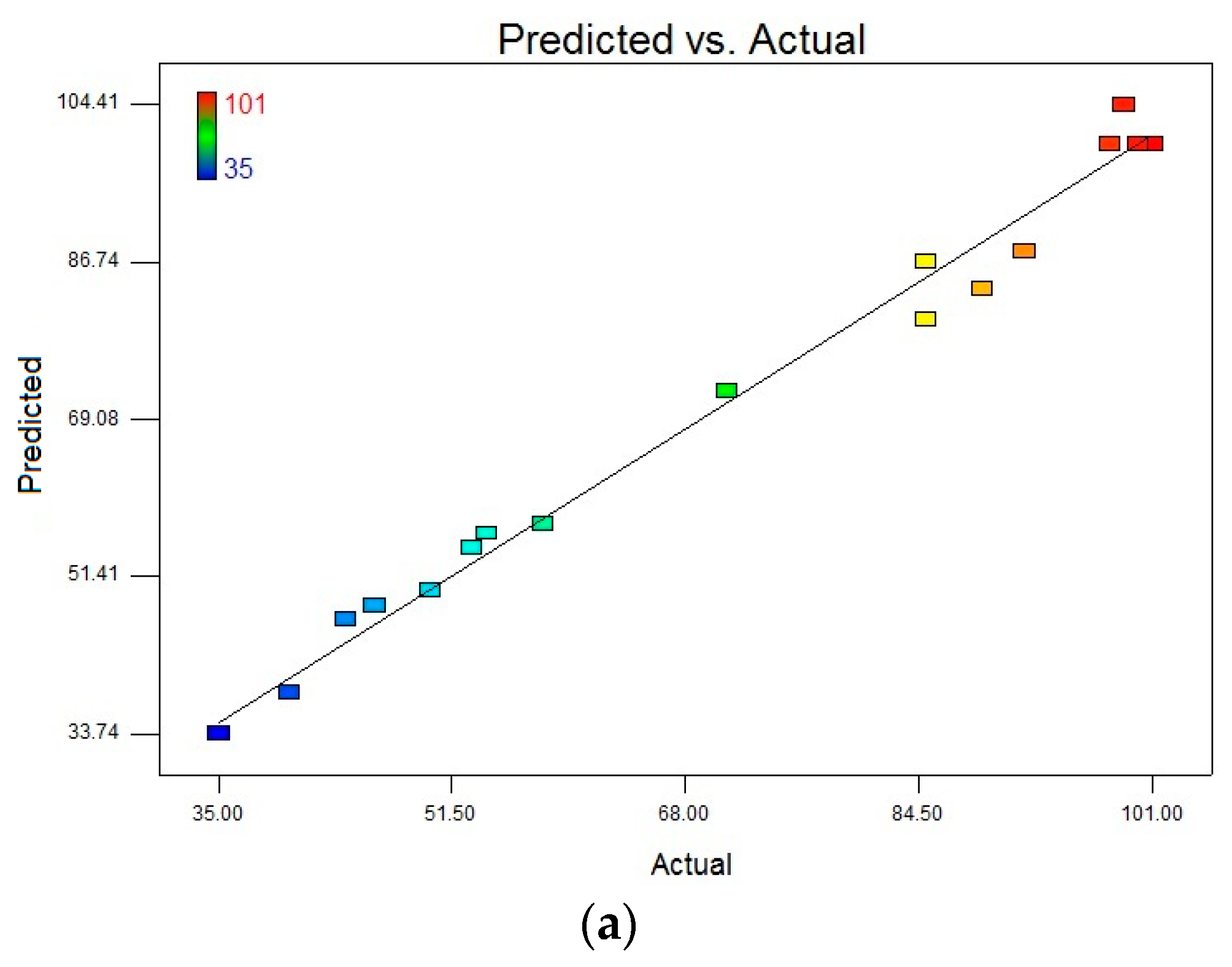
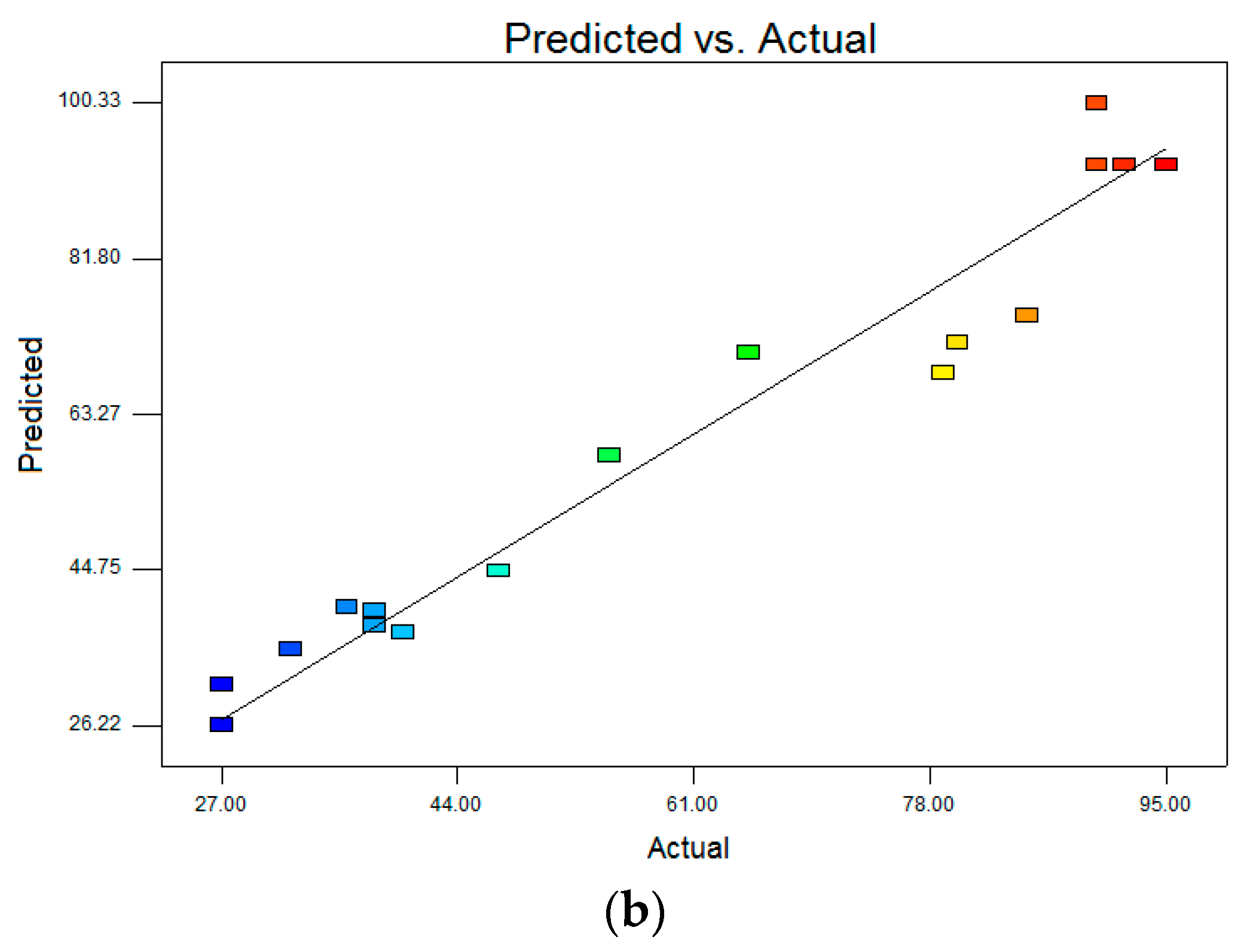
R2 were reasonably close to 1 (0.9684 for DCF and 0.8897 for CBZ), corroborating a high degree of correlation between the experimental and predicted values. Moreover, the high values of Adequate precision (DCF 21.578 and CBZ 11.323), which measures the signal to noise of the model, indicated a very high degree of precision and a good deal of reliability of the experimental values. This means that the model can be used to navigate the design space. The correlation between observed and predicted values is given in
Figure 8. The points are placed very closely to the diagonal line, indicating low discrepancies between them.
Results from the CCD experiments, as shown in
Table 4 and
Table 5, indicate that the extraction of both analytes depends significantly on all the studied variables, having either negative or positive effects. In particular, the linear terms of extraction time (A), sorbent mass (B) and eluent volume (C), and their quadratic terms A
2, B
2 and C
2, have significant effects on the extraction efficiency of both analytes at 5% significance level (95% confidence interval) as
p-value for all these terms is less than 0.05. On the other hand, only one considerable interaction effect was established between the extraction time and the sorbent mass for both analytes. The remaining two interaction terms are non-significant as
p-values for them are more than 0.05%. However, the non-significant terms were not eliminated from the model, since we are interested for the overall effect of the coefficients on the response surface.
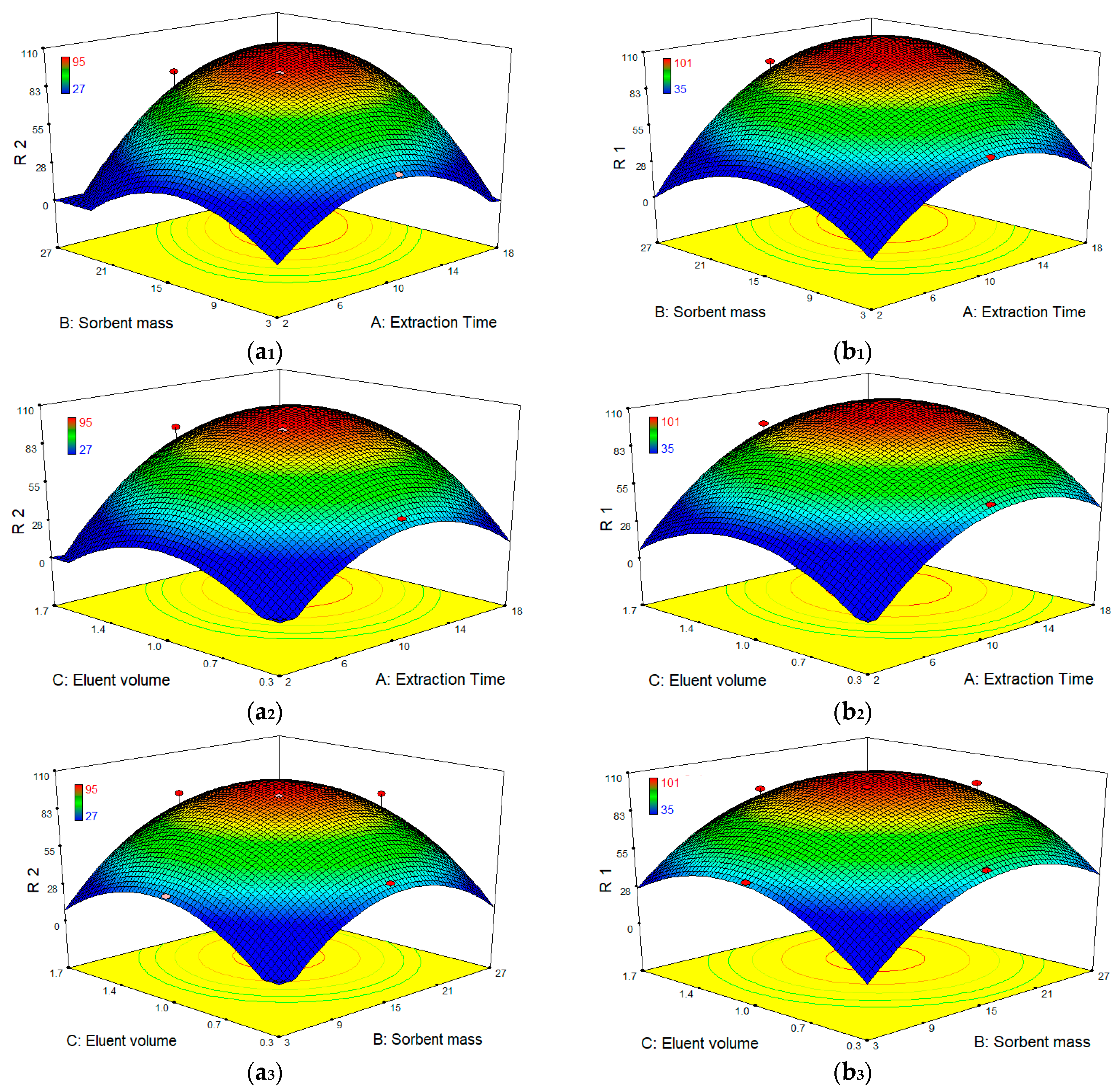
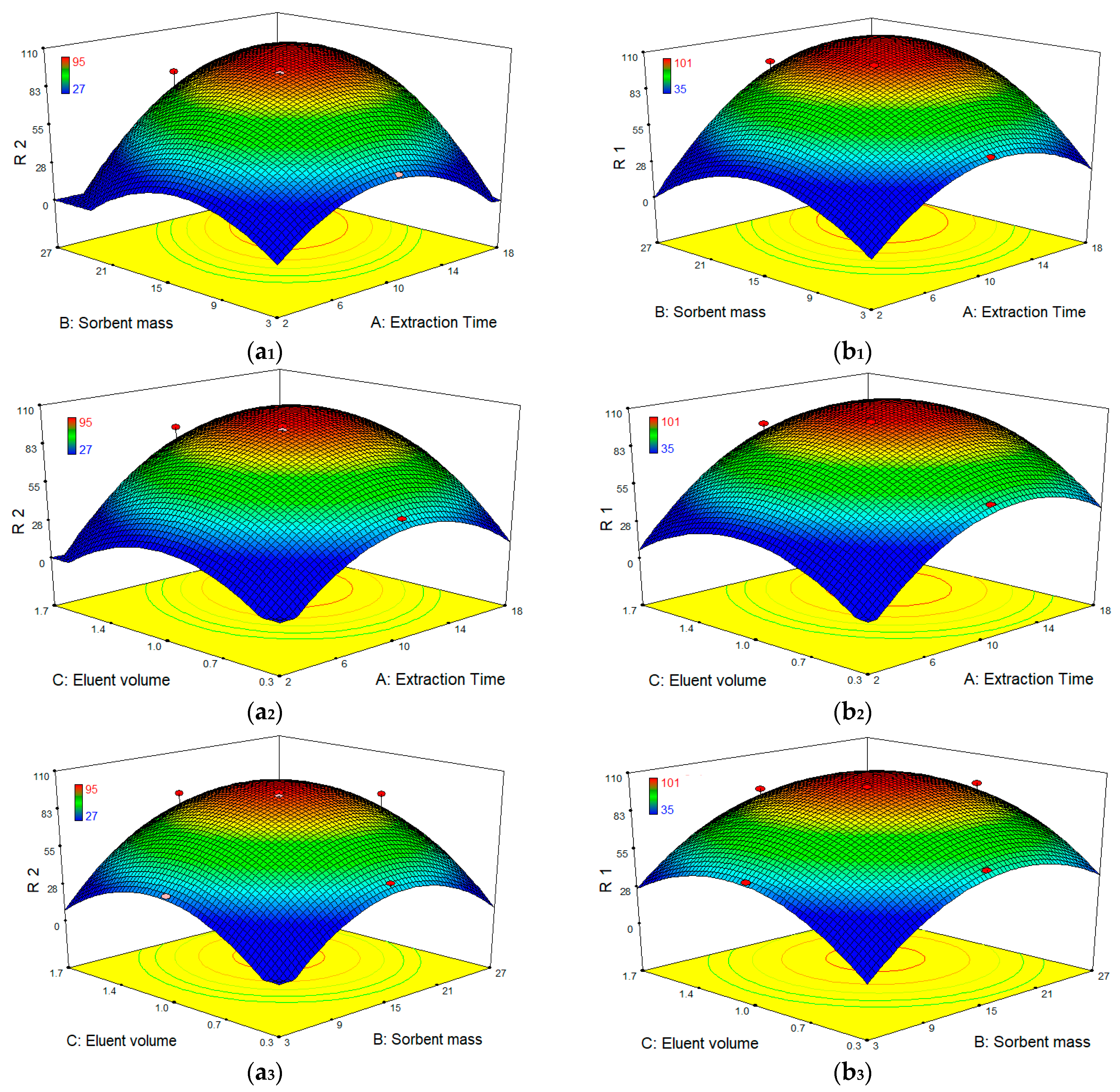
After identifying the most significant parameters, RSM was used to find optimum condition for the best extraction efficiency. Three-dimensional surface plots (
Figure 9) show the interactive effects of extraction time, sorbent mass and eluent volume on the extraction efficiency of both compounds. These plots depict the influence of any two independent variables on the response and the maximum value for each variable can be obtained. According to the results, when eluent volume is fixed at level 0, the extraction efficiency of both compounds is increased by increasing the sorbent amount, reaching a peak of ~100% for DCF and 95% for CBZ at the point of ~15 mg of the sorbent. Such increment in extraction efficiency appears to be because of the accessibility of higher surface areas together with abundant sorption sites. Meanwhile, sorbent addition beyond an adequate quantity to entirely sorb the accessible molecules of the analytes had no significant impact on further improvement in extraction efficiency, demonstrating unsaturated surface active sites of the polymer sorbent. In addition, an increase of extraction efficiency was observed as the extraction time increased up to 10–12 min and then reached a plateau. On the other hand, the extraction efficiency was nearly associated with an enhancement of elution volume from 0.3 to 1 mL and then remained almost constant.
Hence, the optimum working conditions to obtain the best response were as: 1 mL for volume of eluting solvent; 15 mg for sorbent amount; and 15 min extraction time. To confirm the model adequacy for predicting maximum extraction efficiency, three replicate experiments were performed at optimal conditions. A mean value of 97 ± 5.0% for DCF and 92 ± 8.0% for CBZ, obtained from real experiments, demonstrate the suitability of the fitted response surface model.
3.3.3. Performance Characteristics Measured in D-SPE Method Validation
The analytical performance of the developed D-SPE procedure using the novel isocyanate modified carrageenan polymer sorbent was evaluated for the pre-concentration of DCF and CBZ. Analytical figures of merit of the method are summarized in
Table 6 and
Table 7.
The calculated calibration curves gave a high level of linearity, yielding coefficients (r2) > 0.991 for both compounds. LOD and LOQ data of analytes for all water samples are in the ranges of 0.042–0.090 μg/L and 0.137–0.298 μg/L, respectively. The precision of the method was accessed by determining relative standard deviations (RSDs) of intra-day and inter-day at three different spiked levels. The results show that the RSDs of intra-day precision are 2.8–13.5%, while that of inter-day precision are 8.5–17.5%.
Uncertainty of the analytical method was also estimated based on in-house validation data according to EURACHEM/CITAC and GUM guide for both compounds at two spiking levels, as was explained in previous works. The relative expanded uncertainty was lower than 40% for both compounds in all matrices.
To demonstrate the reliability and versatility of the proposed methodology coupled to LC-MS system for the analysis of real samples, five categories of aqueous samples, including distilled water (DW), river water (RW), sea water (SW), lake water (LW), and influent and effluent wastewaters (WWI and WWE) samples, were analyzed. Recoveries ranged between 70% and 108% for all the matrices, demonstrating the suitability of the proposed method (
Figure 10).

 ,
, 





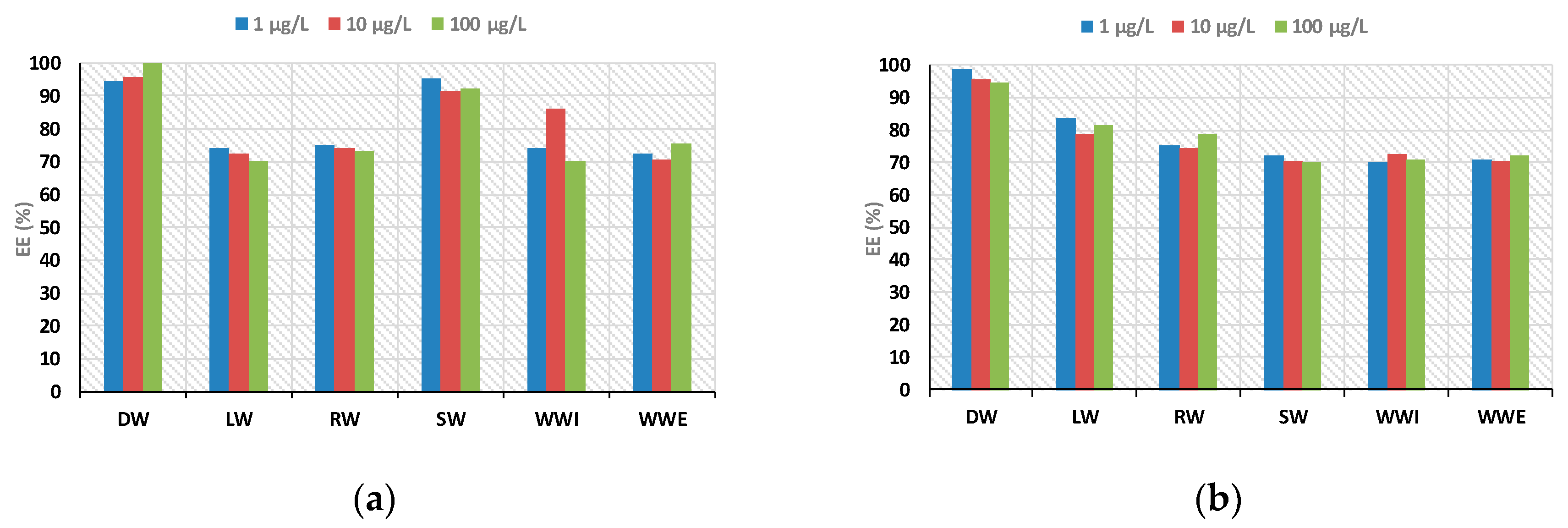

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
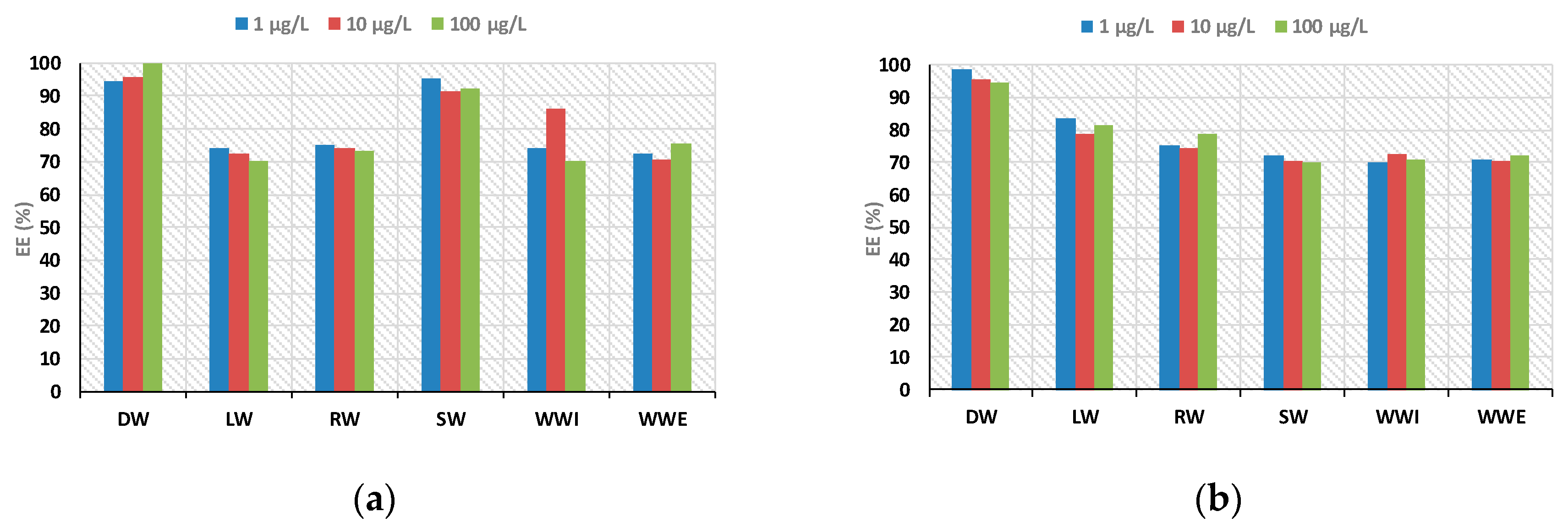{kind=link}