Anionic Polymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Superbases

 ,
,  ,
,
Abstract
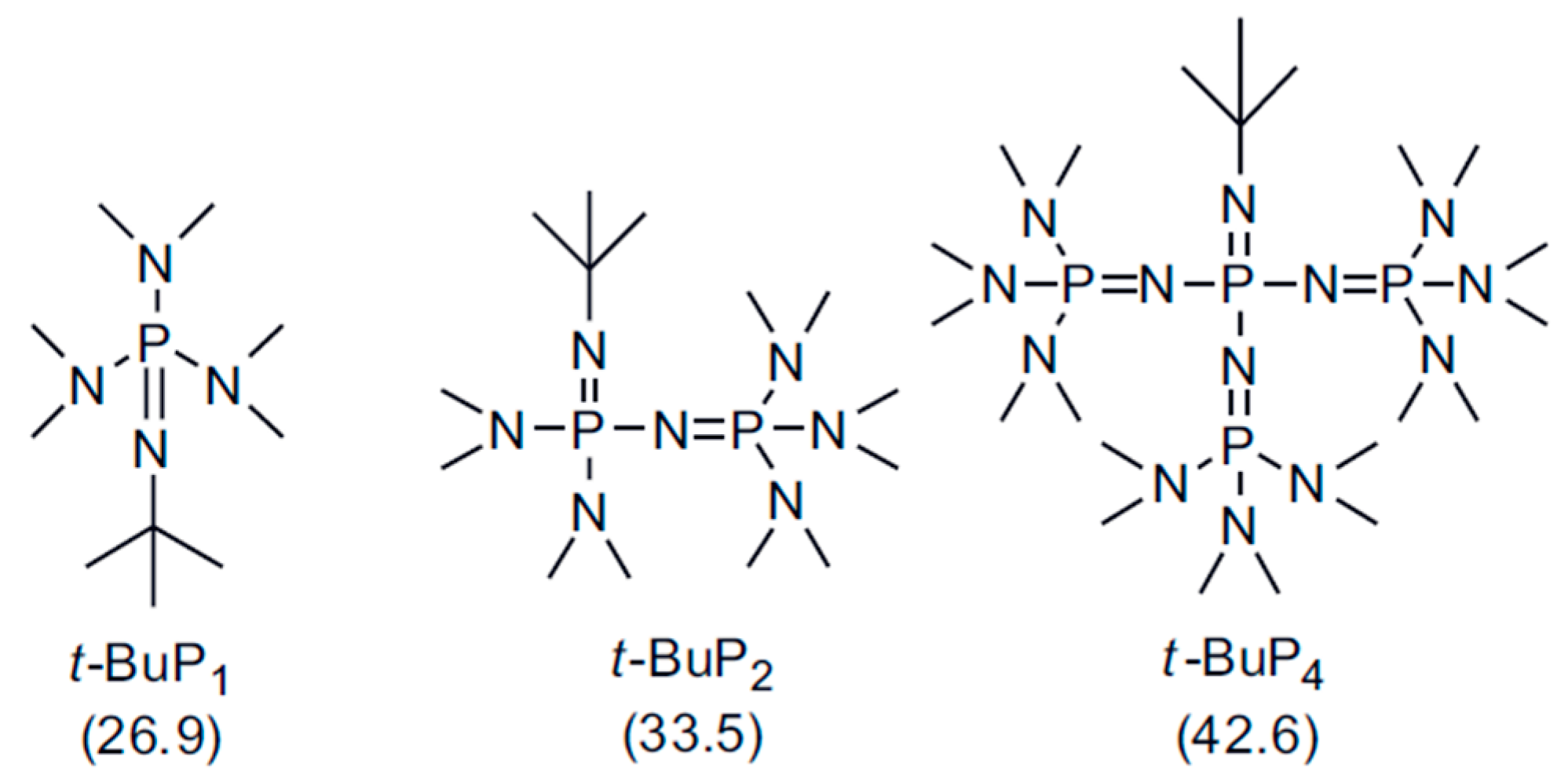
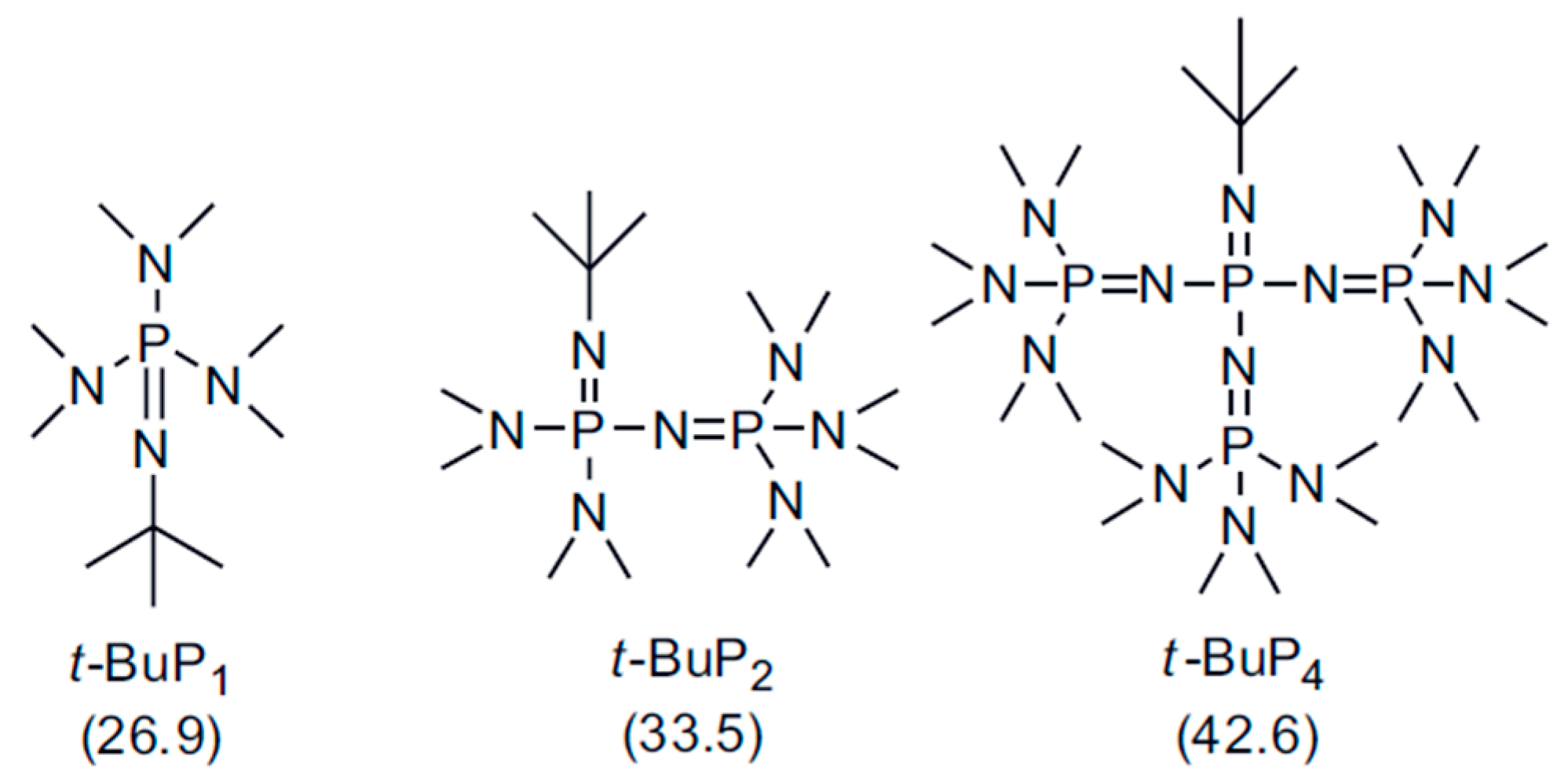
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instrumentation
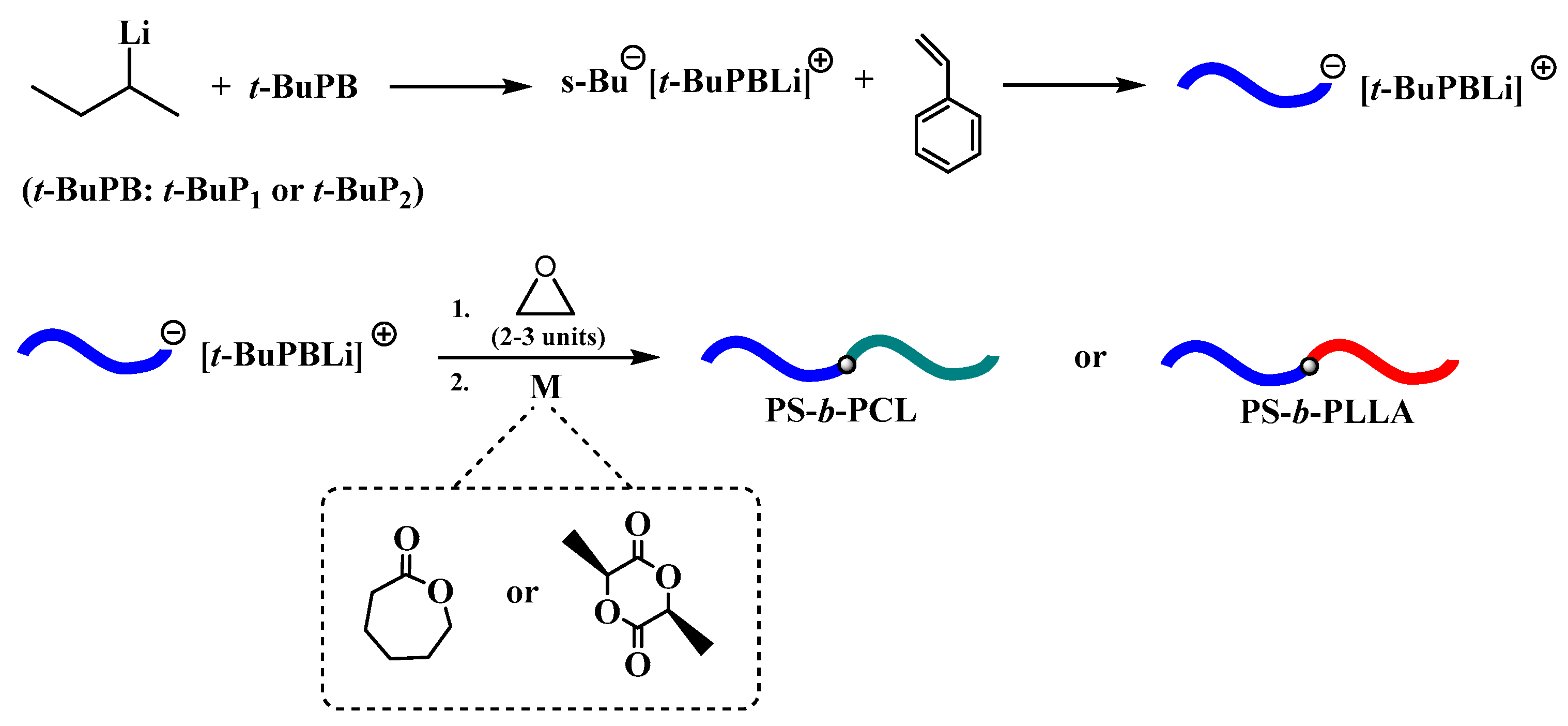
2.3. Homopolymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Bases (t-BuP4, t-BuP2, t-BuP1)
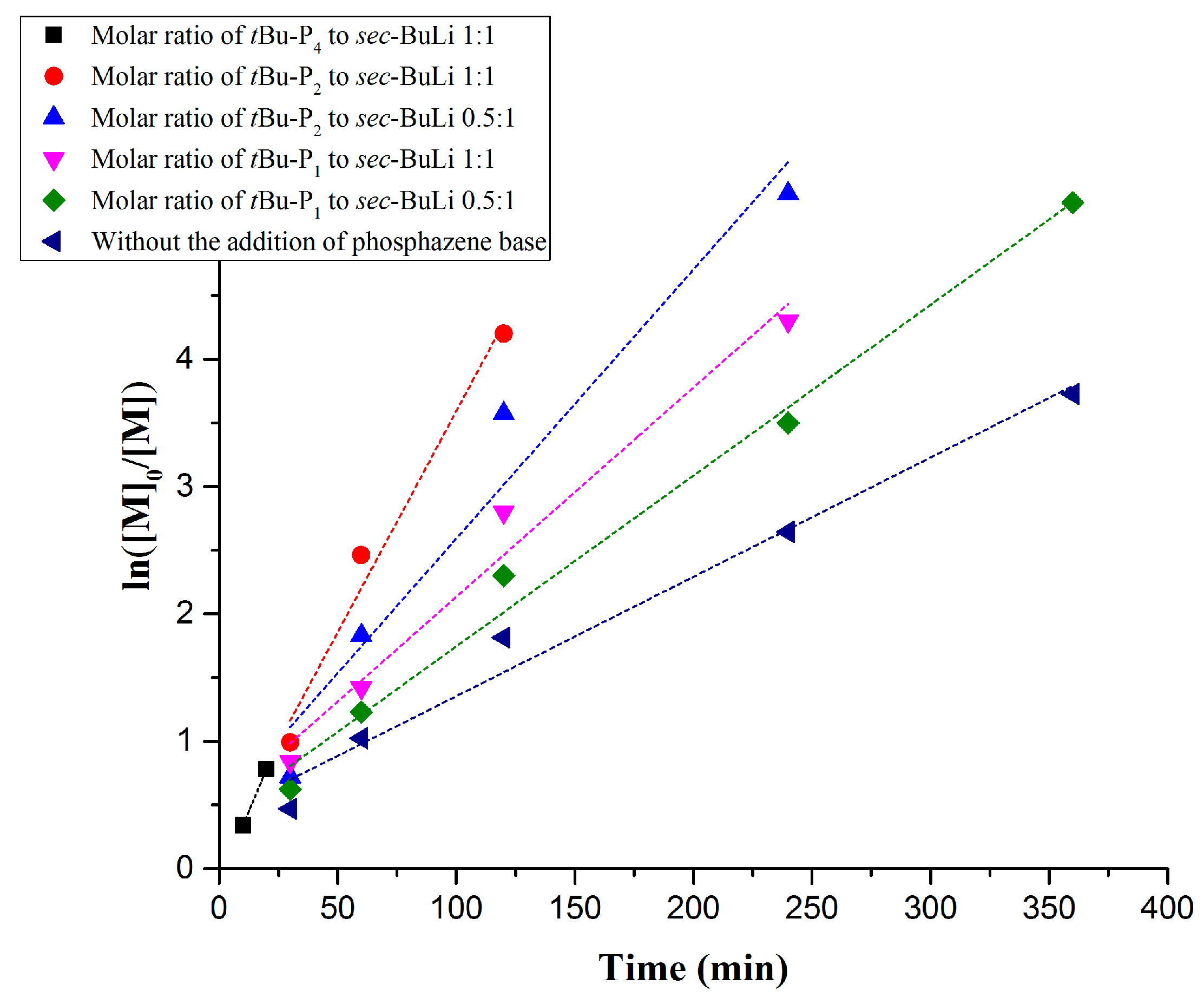
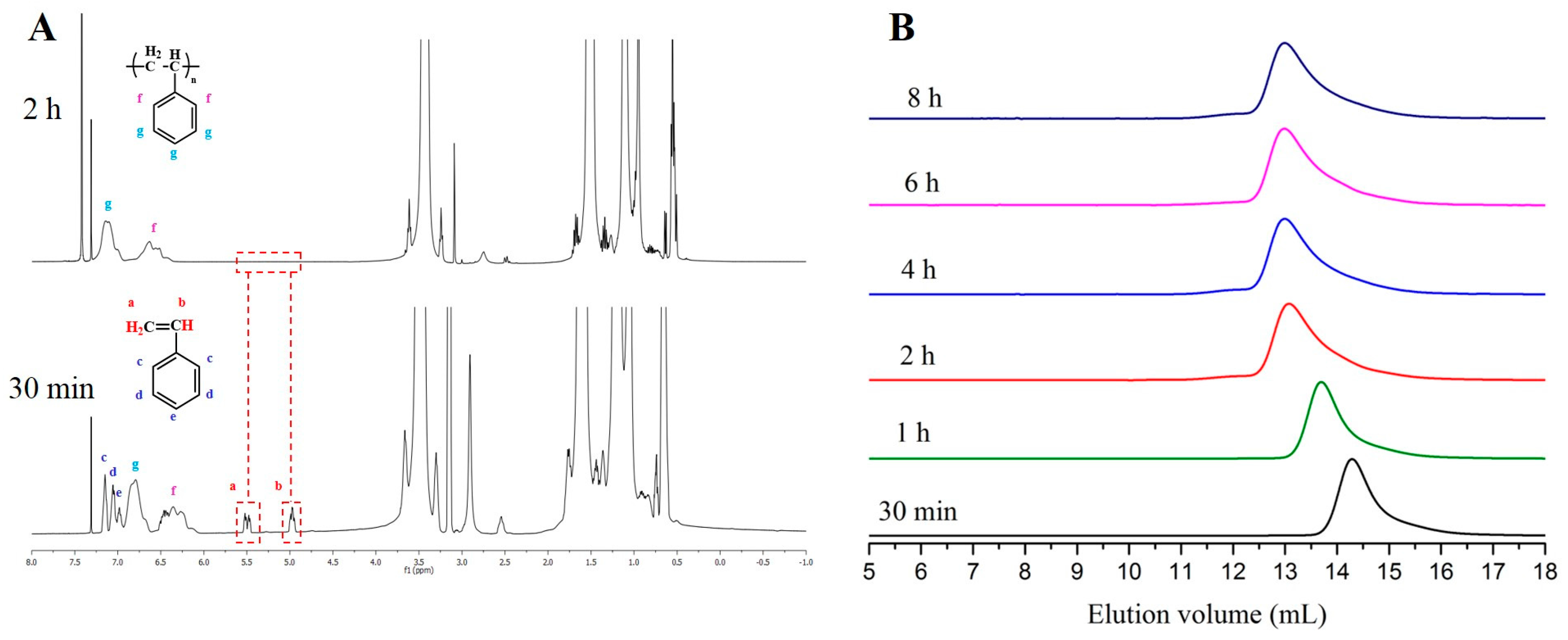
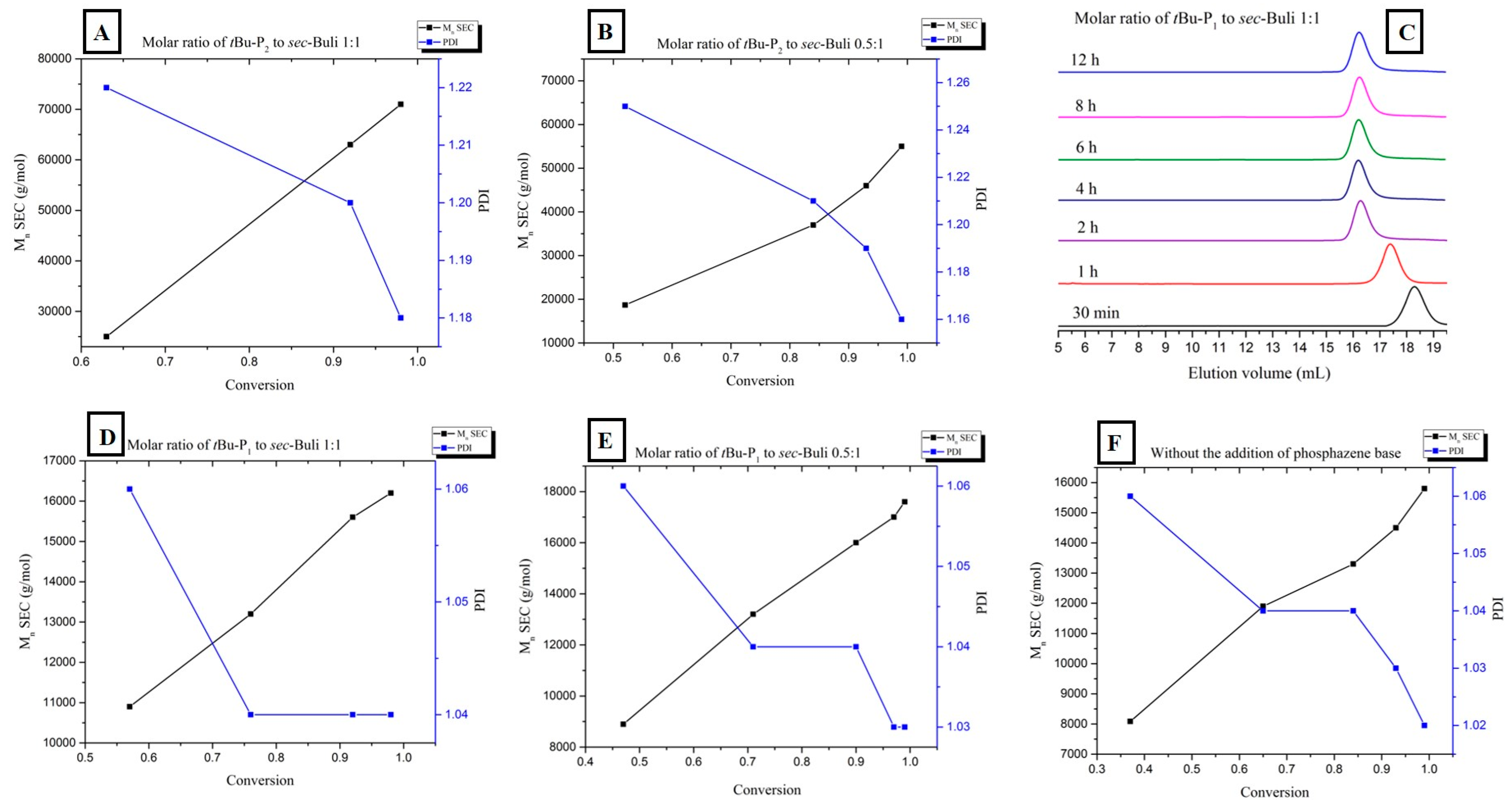
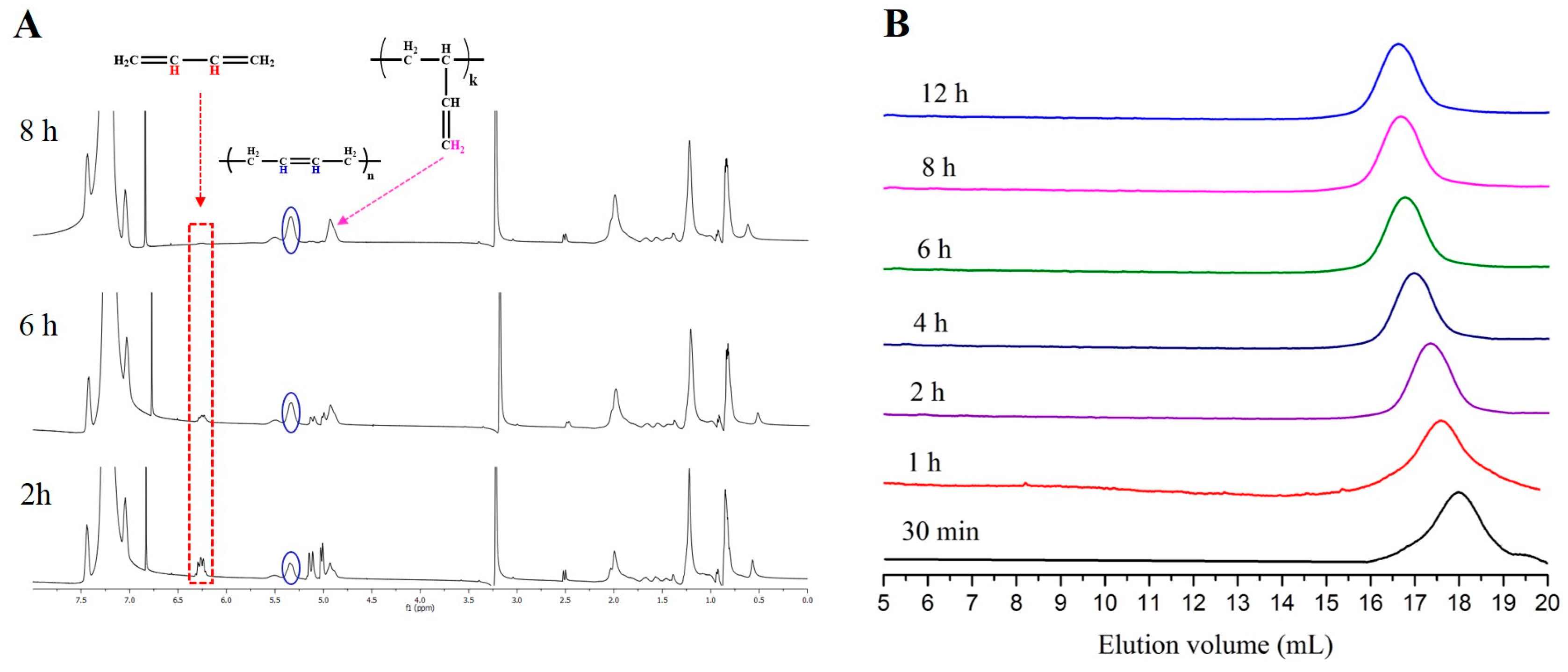
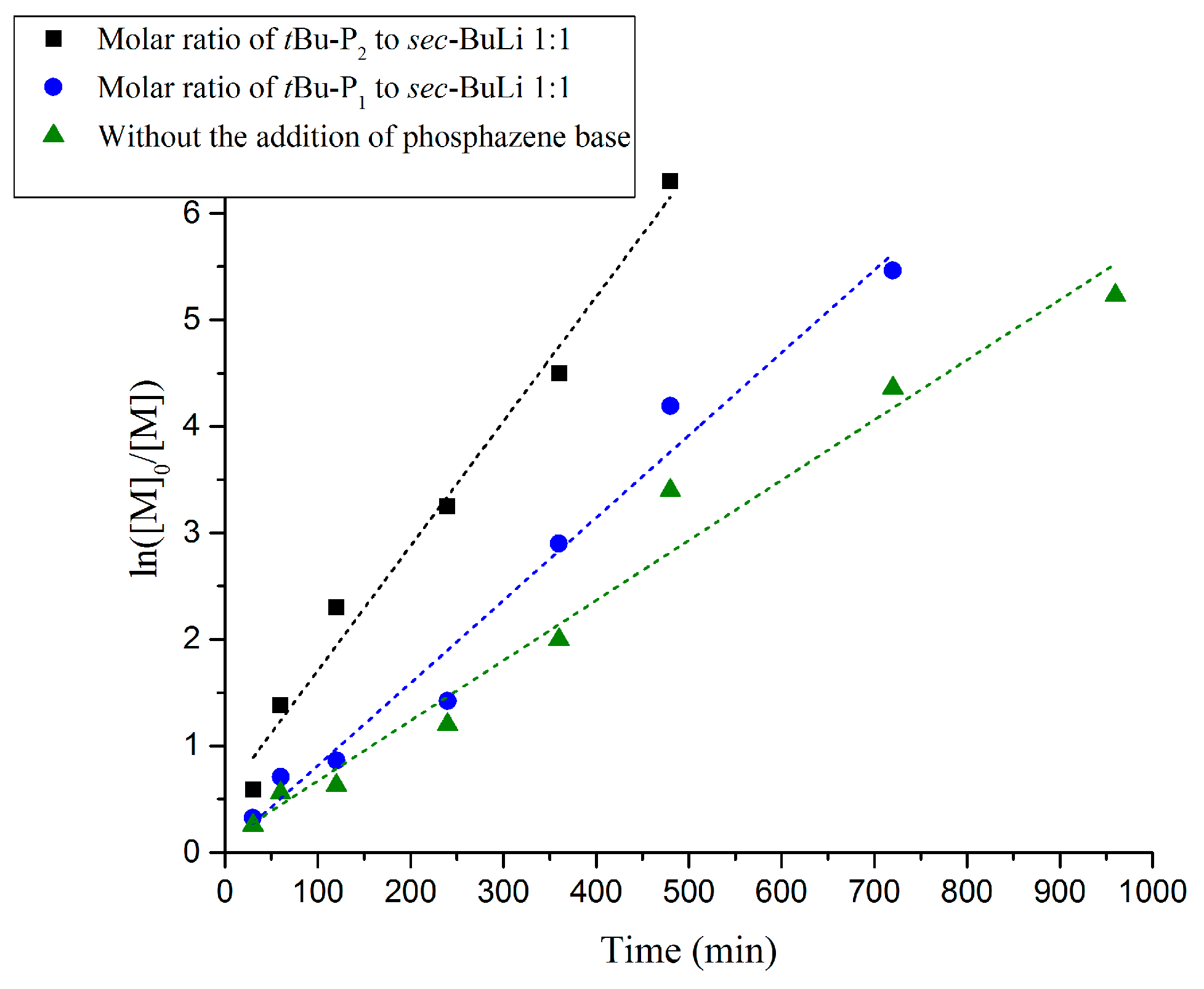
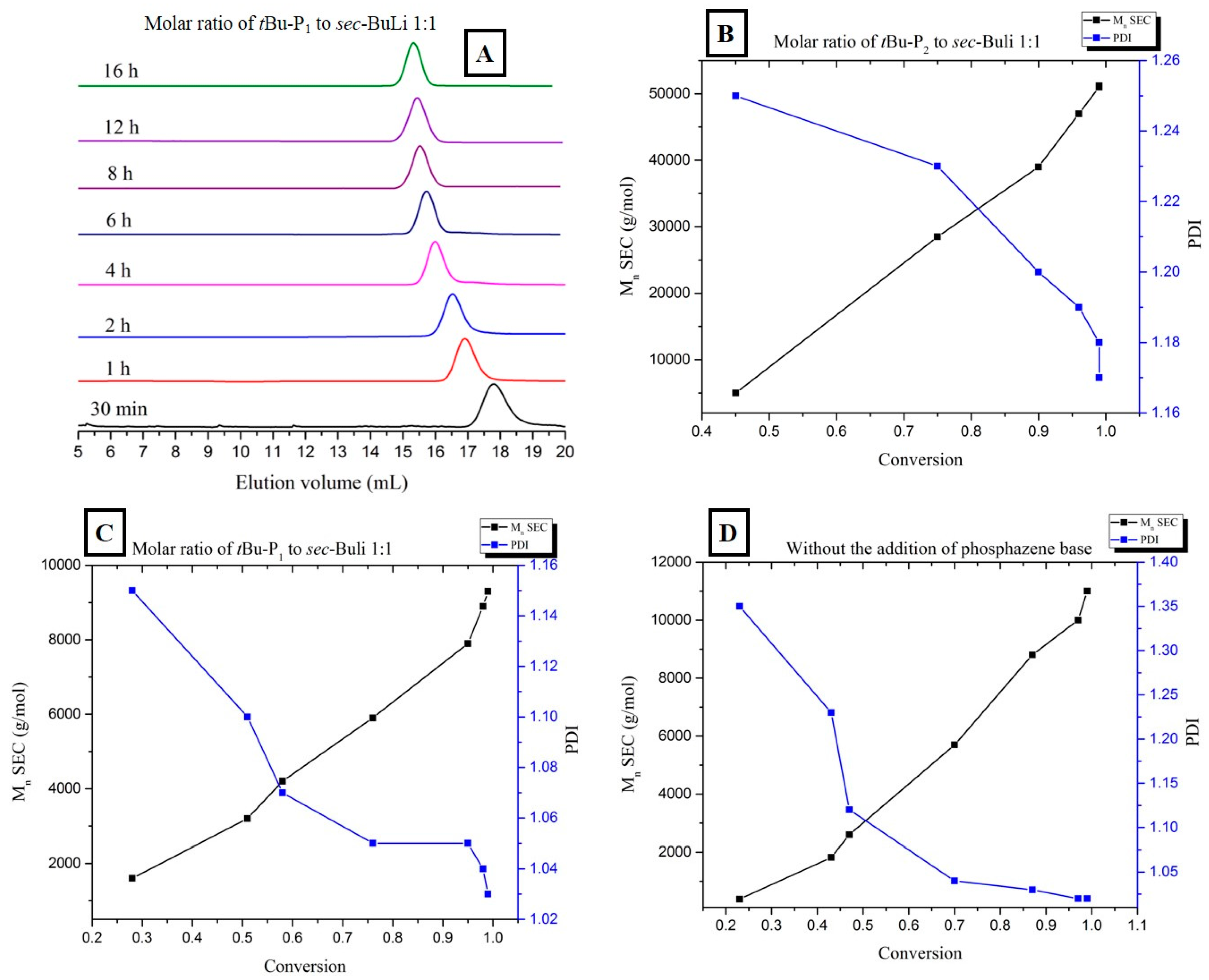
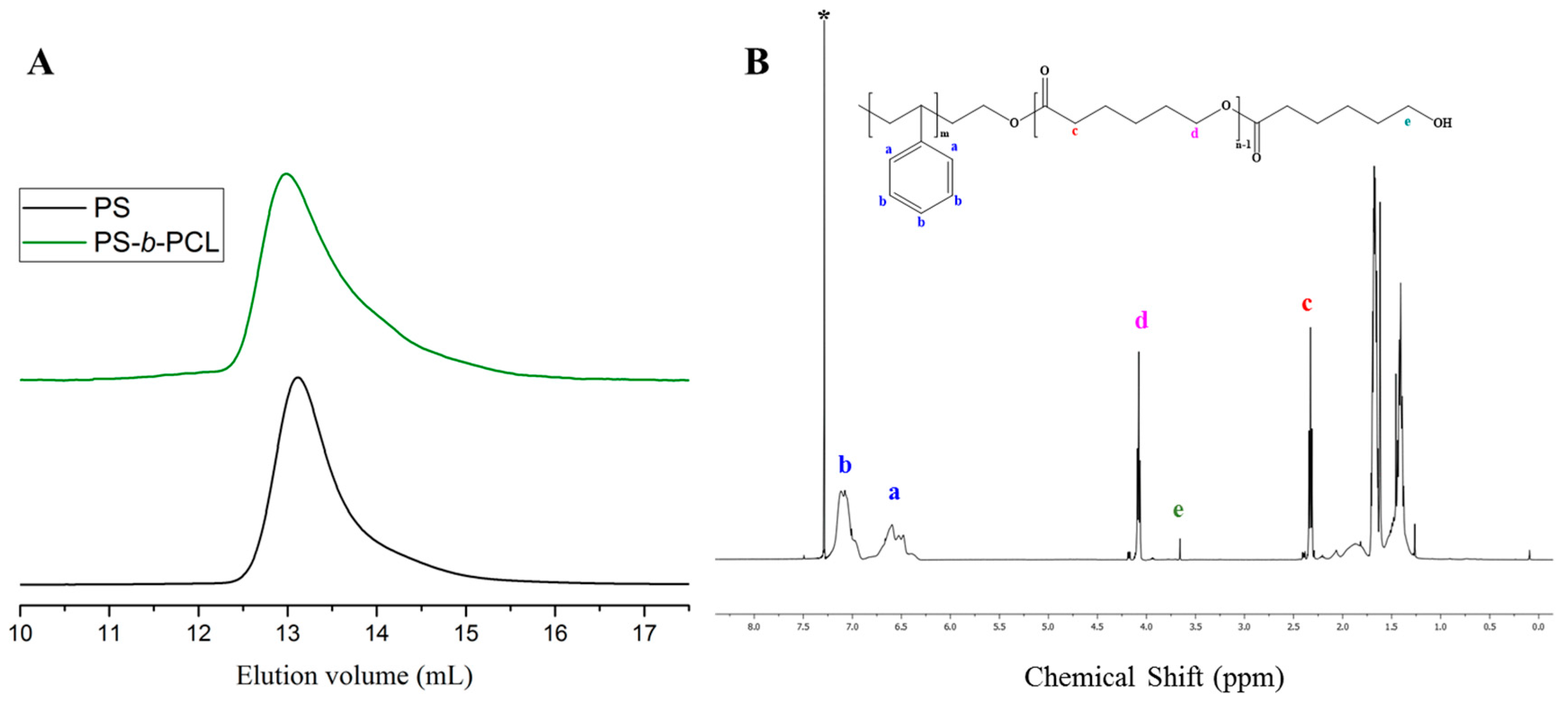
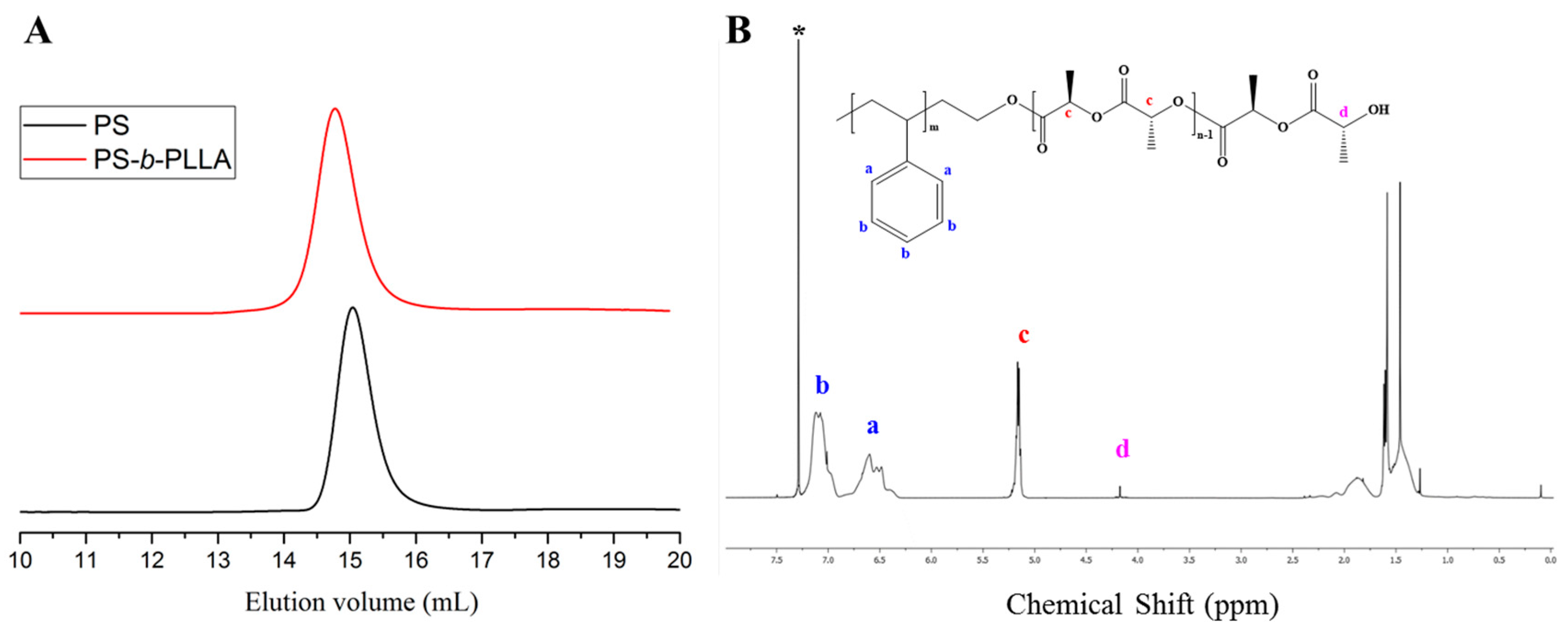
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ziegler, K.; Bähr, K. Über den vermutlichen mechanismus der polymerisationen durch alkalimetalle (Vorläufige Mitteilung). Ber. Dtsch. Chem. Ges. 1928, 61, 253–263. [Google Scholar] [CrossRef]
- Szwarc, M. ‘Living’ Polymers. Nature 1956, 178, 1168–1169. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Hirao, A. Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Hadjichristidis, N.; Pispas, S.; Floudas, G. Block Copolymers: Synthetic Strategies, Physical Properties, and Applications; Wiley-Interscience: Hoboken, NJ, USA, 2003; pp. 3–4. [Google Scholar]
- Polymeropoulos, G.; Zapsas, G.; Ntetsikas, K.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. 50th Anniversary Perspective: Polymers with Complex Architectures. Macromolecules 2017, 50, 1253–1290. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Quirk, R.P. Anionic Polymerization: Principles and Practical Applications; Marcel Dekker Inc.: New York, NY, USA, 1996. [Google Scholar]
- Young, R.N.; Quirk, R.P.; Fetters, L.J. Anionic polymerizations of non-polar monomers involving lithium. Adv. Polym. Sci. 1984, 56, 1–90. [Google Scholar]
- Nuyken, O.; Pask, S.D. Ring-Opening Polymerization-An Introductory Review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef]
- Zhu, W.; Zhong, M.; Li, W.; Dong, H.; Matyjaszewski, K. Clickable Stars by Combination of AROP and Aqueous AGET ATRP. Macromolecules 2011, 44, 1920–1926. [Google Scholar] [CrossRef]
- Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S. Living ring-opening polymerizations of heterocyclic monomers. Prog. Polym. Sci. 2007, 32, 247–282. [Google Scholar] [CrossRef]
- Pedersen, C.J. Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 1967, 89, 7017–7036. [Google Scholar] [CrossRef]
- Boileau, S. Anionic ring-opening polymerization: Epoxides and episulfides. In Comprehensive Polymer Science; Eastmond, G.C., Ledwith, A., Russo, S., Sigwalt, P., Eds.; Pergamont Press: Oxford, UK, 1989; Volume 3, pp. 467–487. [Google Scholar]
- Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S. Living ring-opening polymerization of heterocyclic monomers. In Controlled and Living Polymerizations; Müller, A.H.E., Matyjaszewski, K., Eds.; Wiley: Weinheim, Germany, 2009; pp. 241–296. [Google Scholar]
- Ganachaud, F.; Boileau, S. Siloxane-containing polymers. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.M., Eds.; Wiley: Weinheim, Germany, 2009; pp. 65–95. [Google Scholar]
- Boileau, S. Use of cryptates in anionic polymerization of heterocyclic compounds. In Ring-Opening Polymerization Kinetics, Mechanisms and Synthesis; Mc Grath, J.E., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1981; pp. 283–305. [Google Scholar]
- Cheng, T.C.; Halasa, A.F. Anionic polymerization. IV. Effect of crown ethers on alkylsodium initiator. J. Polym. Sci. Part A Polym. Chem. 1976, 14, 583–589. [Google Scholar] [CrossRef]
- Boileau, S.; Kaempf, B.; Lehn, J.M.; Schué, F. Use of cryptates in anionic polymerization. I. Anionic activation. J. Polym. Sci. Part C Polym. Lett. 1974, 12, 203–209. [Google Scholar] [CrossRef]
- Alev, S.; Schué, F.; Kaempf, B. Use of dicyclohexyl-18-crown-6 in anionic polymerization. I. Solutions of alkali metals in benzene and in tetrahydrofuran. J. Polym. Sci. Part C Polym. Lett. 1975, 13, 397–400. [Google Scholar] [CrossRef]
- Varshney, S.K.; Jerome, R.; Bayard, P.; Jacobs, C.; Fayt, R.; Teyssie, P. Anionic polymerization of (meth)acrylic monomers. 7. Macrocyclic crown ethers as promoters of the living polymerization of methyl methacrylate using monofunctional initiators. Macromolecules 1992, 25, 4457–4463. [Google Scholar] [CrossRef]
- Boileau, S.; Illy, N. Activation in anionic polymerization: Why phosphazene bases are very exciting promoters. Prog. Polym. Sci. 2011, 36, 1132–1151. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H. Peralkylated Polyaminophosphazenes—Extremely Strong, Neutral Nitrogen Bases. Angew. Chem. Int. Ed. 1987, 26, 1167–1169. [Google Scholar] [CrossRef]
- Schwesinger, R.; Hasenfratz, C.; Schlemper, H.; Walz, L.; Peters, E.-M.; Peters, K.; von Schnering, H.G. How Strong and How Hindered Can Uncharged Phosphazene Bases Be? Angew. Chem. Int. Ed. 1993, 32, 1361–1363. [Google Scholar] [CrossRef]
- Weideman, I.; Pfukwa, R.; Klumperman, B. Phosphazene base promoted anionic polymerization of n-butyraldehyde. Eur. Polym. J. 2017, 93, 97–102. [Google Scholar] [CrossRef]
- Esswein, B.; Steidl, N.M.; Möller, M. Anionic polymerization of oxirane in the presence of the polyiminophosphazene base t-Bu-P4. Macromol. Rapid Commun. 1996, 17, 143–148. [Google Scholar] [CrossRef]
- Schlaad, H.; Kukula, H.; Rudloff, J.; Below, I. Synthesis of α,ω-heterobifunctional poly(ethylene glycol)s by metal-Free anionic ring-opening polymerization. Macromolecules 2001, 34, 4302–4304. [Google Scholar] [CrossRef]
- Misaka, H.; Sakai, R.; Satoh, T.; Kakuchi, T. Synthesis of high molecular weight and end-functionalized poly(styrene oxide) by living ring-opening polymerization of styrene oxide using the alcohol/phosphazene base initiating system. Macromolecules 2011, 44, 9099–9107. [Google Scholar] [CrossRef]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: Synthesis, novel polymer architectures and bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef] [PubMed]
- Molenberg, A.; Möller, M. A fast catalyst system for the ring-opening polymerization of cyclosiloxanes. Macromol. Rapid Commun. 1995, 16, 449–453. [Google Scholar] [CrossRef]
- Pibre, G.; Chaumont, P.; Fleury, E.; Cassagnau, P. Ring-opening polymerization of decamethylcyclopentasiloxane initiated by a superbase: Kinetics and rheology. Polymer 2008, 49, 234–240. [Google Scholar] [CrossRef]
- Zhang, L.; Nederberg, F.; Messman, J.M.; Pratt, R.C.; Hedrick, J.L.; Wade, C.G. Organocatalytic stereoselective ring-opening polymerization of lactide with dimeric phosphazene bases. J. Am. Chem. Soc. 2007, 129, 12610–12611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nederberg, F.; Pratt, R.C.; Waymouth, R.M.; Hedrick, J.L.; Wade, C.G. Phosphazene bases: A new category of organocatalysts for the living ring-opening polymerization of cyclic esters. Macromolecules 2007, 40, 4154–4158. [Google Scholar] [CrossRef]
- Alamri, H.; Zhao, J.; Pahovnik, D.; Hadjichristidis, N. Phosphazene-catalyzed ring-opening polymerization of ε-caprolactone: Influence of solvents and initiators. Polym. Chem. 2014, 5, 5471–5478. [Google Scholar] [CrossRef]
- Ladelta, V.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. Ring-opening polymerization of ω-pentadecalactone catalyzed by phosphazene superbases. Polym. Chem. 2017, 8, 511–515. [Google Scholar] [CrossRef]
- Yang, H.; Yan, M.; Pispas, S.; Zhang, G. Synthesis of poly[(ethylene carbonate)-co-(ethylene oxide)] copolymer by phosphazene-catalyzed ROP. Macromol. Chem. Phys. 2011, 212, 2589–2593. [Google Scholar] [CrossRef]
- Zhao, J.; Pahovnik, D.; Gnanou, Y.; Hadjichristidis, N. Phosphazene-promoted metal-free ring-opening polymerization of ethylene oxide initiated by carboxylic acid. Macromolecules 2014, 47, 1693–1698. [Google Scholar] [CrossRef]
- Zhao, J.; Pahovnik, D.; Gnanou, Y.; Hadjichristidis, N. A “Catalyst Switch” strategy for the sequential metal-free polymerization of epoxides and cyclic esters/carbonate. Macromolecules 2014, 47, 3814–3822. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, S.; Zhao, J.; Zhang, G. Phosphazene-catalyzed alternating copolymerization of dihydrocoumarin and ethylene oxide: Weaker is better. Macromolecules 2017, 50, 4198–4205. [Google Scholar] [CrossRef]
- Yang, H.; Xu, J.; Pispas, S.; Zhang, G. Hybrid copolymerization of ε-caprolactone and methyl methacrylate. Macromolecules 2012, 45, 3312–3317. [Google Scholar] [CrossRef]
- Alamri, H.; Hadjichristidis, N. One-pot synthesis of well-defined polyether/polyester block copolymers and terpolymers by a highly efficient catalyst switch approach. Polym. Chem. 2016, 7, 3225–3228. [Google Scholar] [CrossRef]
- Esswein, B.; Möller, M. Polymerization of ethylene oxide with alkyllithium compounds and the phosphazene base “t-BuP4”. Angew. Chem. Int. Ed. Engl. 1996, 35, 623–625. [Google Scholar] [CrossRef]
- Förster, S.; Krämer, E. Synthesis of PB-PEO and PI-PEO block copolymers with alkyllithium initiators and the phosphazene base t-BuP4. Macromolecules 1999, 32, 2783–2785. [Google Scholar] [CrossRef]
- Hassouna, L.; Illy, N.; Guegan, P. Phosphazene/triisobutylaluminum-promoted anionic ring-opening polymerization of 1,2-epoxybutane initiated by secondary carbamates. Polym. Chem. 2017, 8, 4005–4013. [Google Scholar] [CrossRef]
- Hinman, J.G.; Lough, A.J.; Morris, R.H. Properties of the polyhydride anions [WH5(PMe2Ph)3]− and [ReH4(PMePh2)3]− and periodic trends in the acidity of polyhydride complexes. Inorg. Chem. 2007, 46, 4392–4401. [Google Scholar] [CrossRef] [PubMed]
- Worsfold, D.J.; Bywater, S. Anionic polymerization of isoprene. Can. J. Chem. 1964, 42, 2884–2892. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Xu, Z.; Fetters, L.J.; Roovers, J. The characteristic ratios of stereoirregular polybutadiene and polyisoprene. J. Polym. Sci. Part B Polym. Phys. 1982, 20, 743–750. [Google Scholar] [CrossRef]
- Hofmann, A.; Alegrıa, A.; Colmenero, J.; Willner, L.; Buscaglia, E.; Hadjichristidis, N. Secondary and segmental relaxation in polybutadienes of varying microstructure: Dielectric relaxation results. Macromolecules 1996, 29, 129–134. [Google Scholar] [CrossRef]
- Huckstadt, H.; Gopfert, A.; Abetz, V. Influence of the block sequence on the morphological behavior of ABC triblock copolymers. Polymer 2000, 41, 9089–9094. [Google Scholar] [CrossRef]
- Neumann, C.; Abetz, V.; Stadler, R. Indication of an order-order-transition by a partial disordering in ABC-triblock copolymers. Polym. Bull. 1996, 36, 43–50. [Google Scholar] [CrossRef]
- Halasa, A.F.; Lohr, D.F.; Hall, J.E. Anionic polymerization of high vinyl polybutadiene. J. Polym. Sci. Part A Polym. Chem. 1981, 19, 1357–1360. [Google Scholar] [CrossRef]
- Lecomte, P.; Jérôme, C. Recent Developments in Ring-Opening Polymerization of Lactones. Adv. Polym. Sci. 2012, 245, 173–218. [Google Scholar]
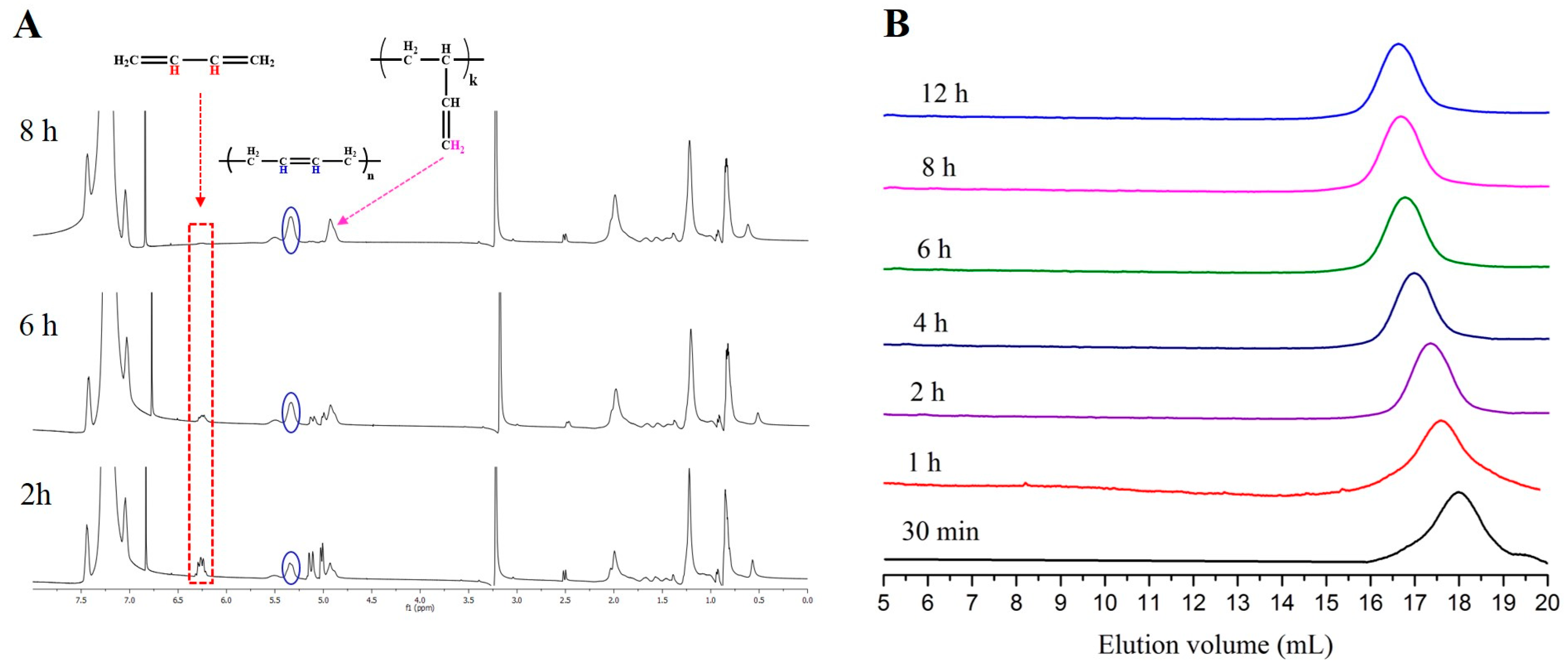
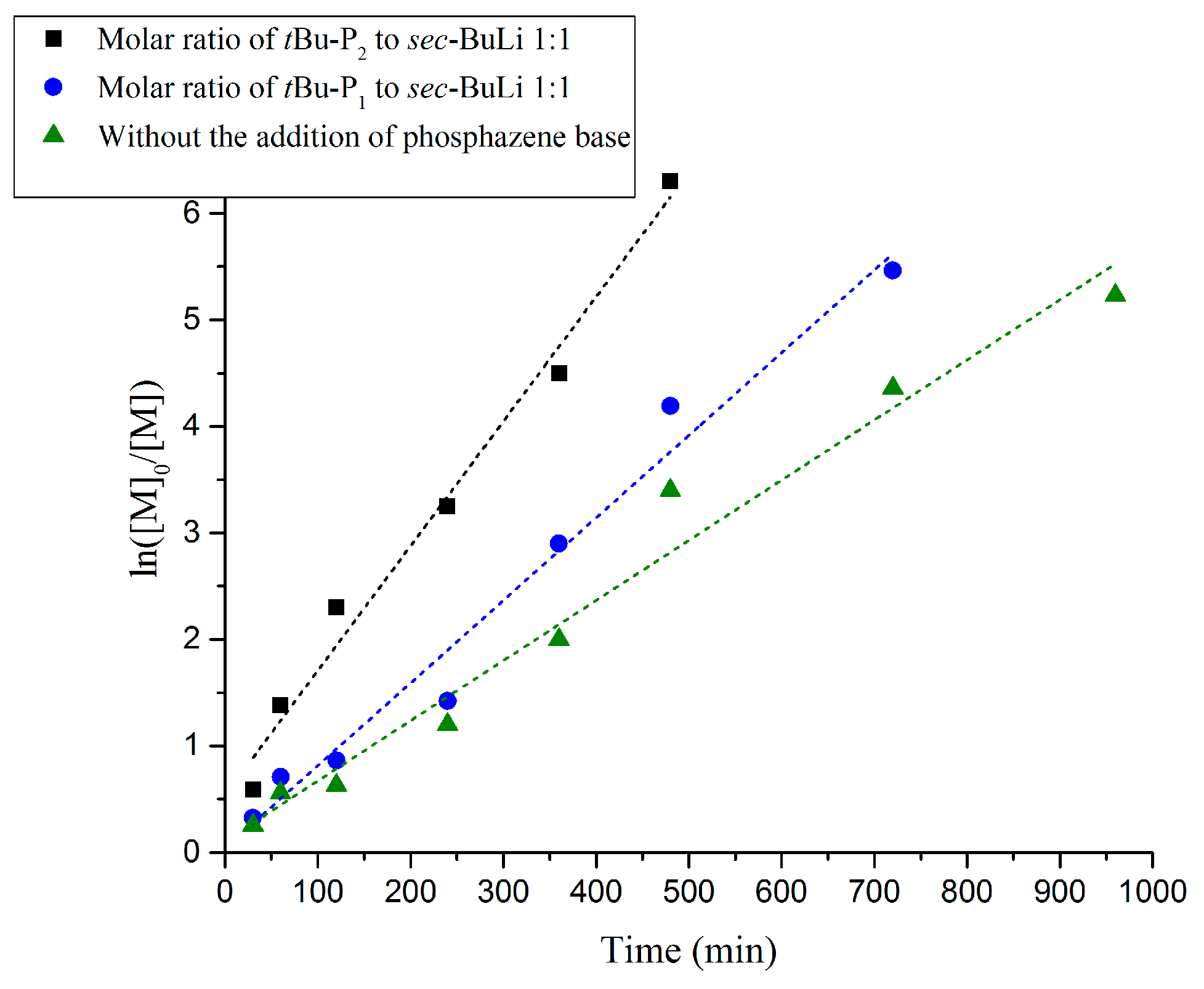

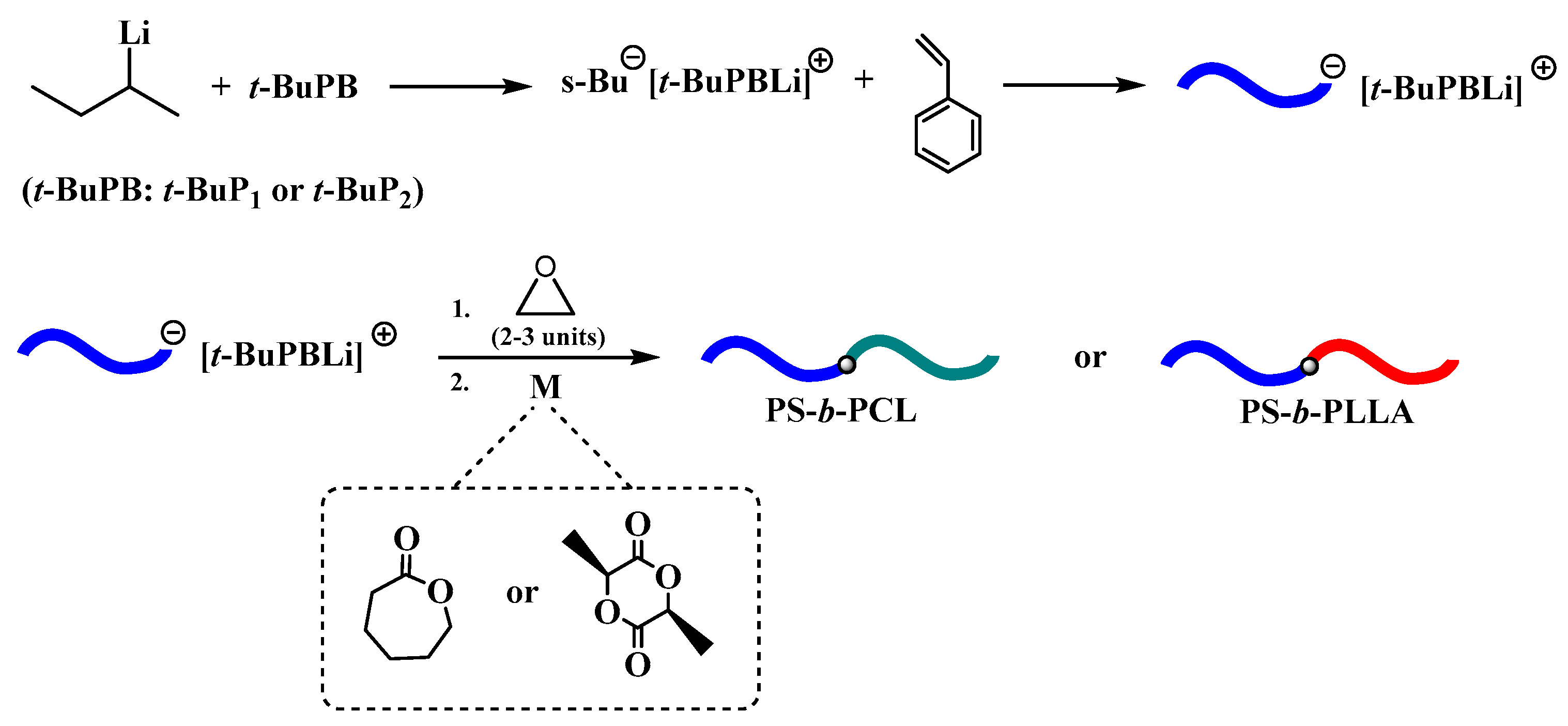
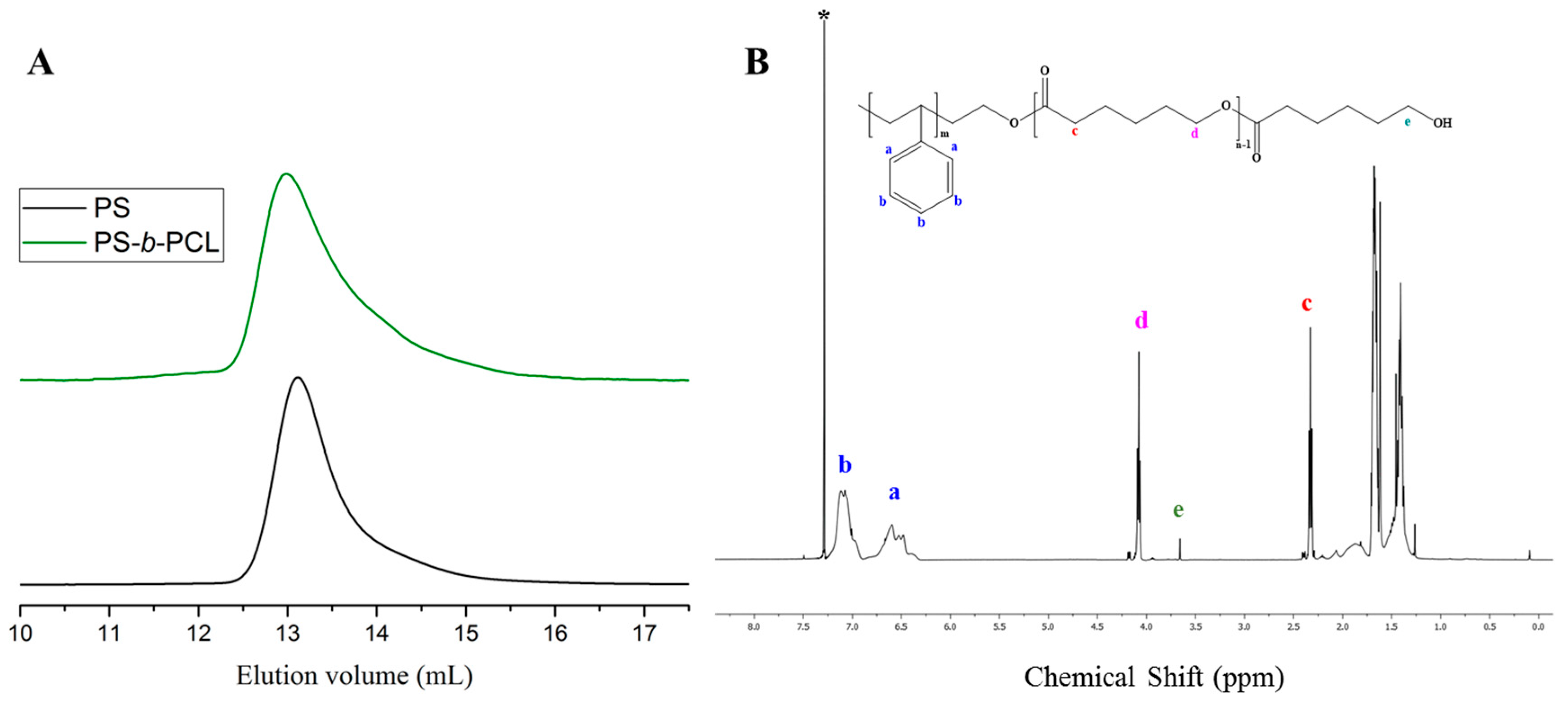

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
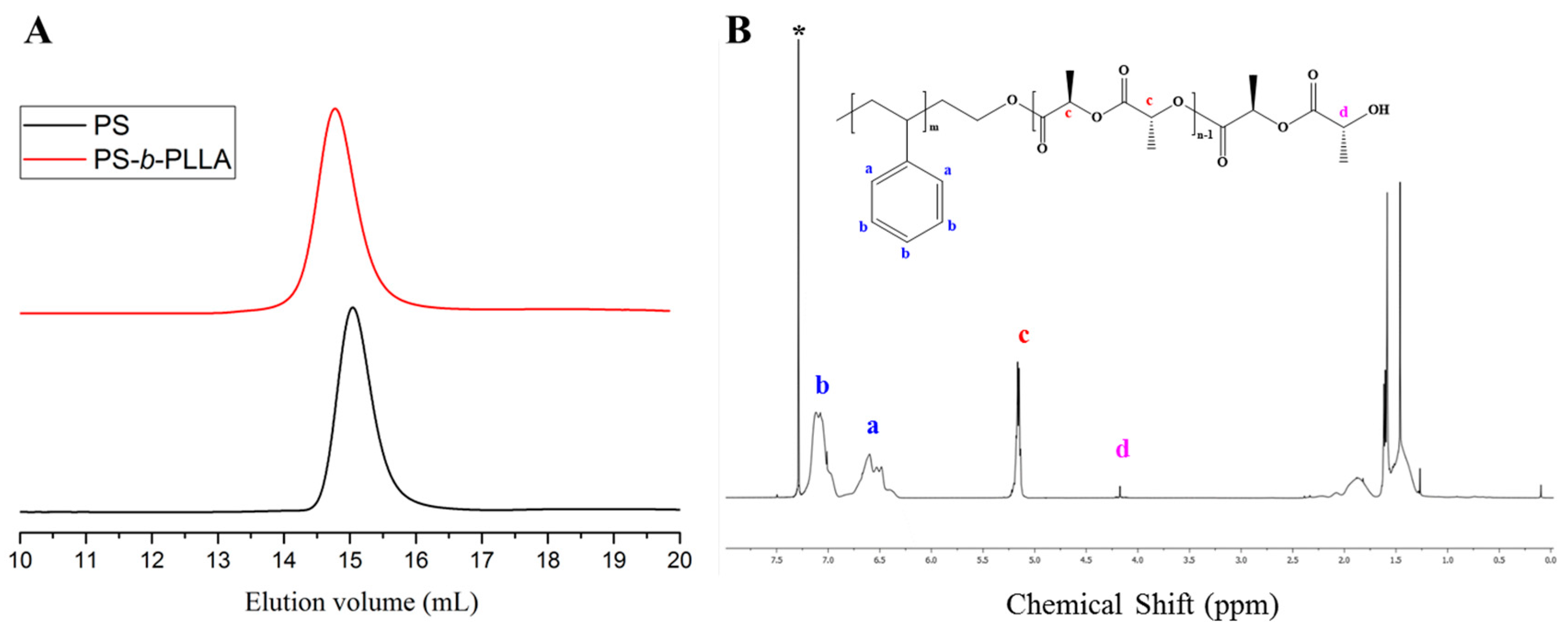
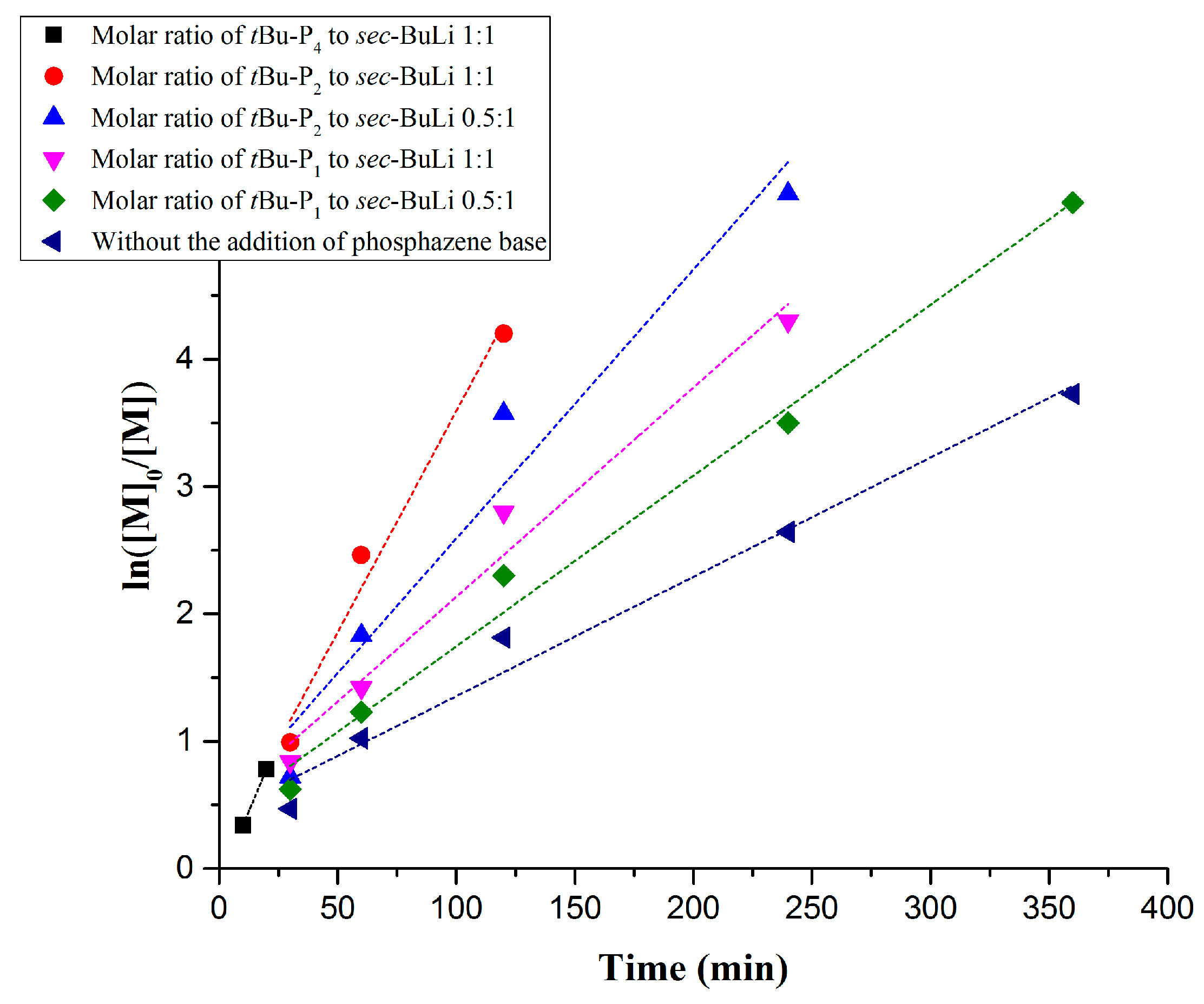
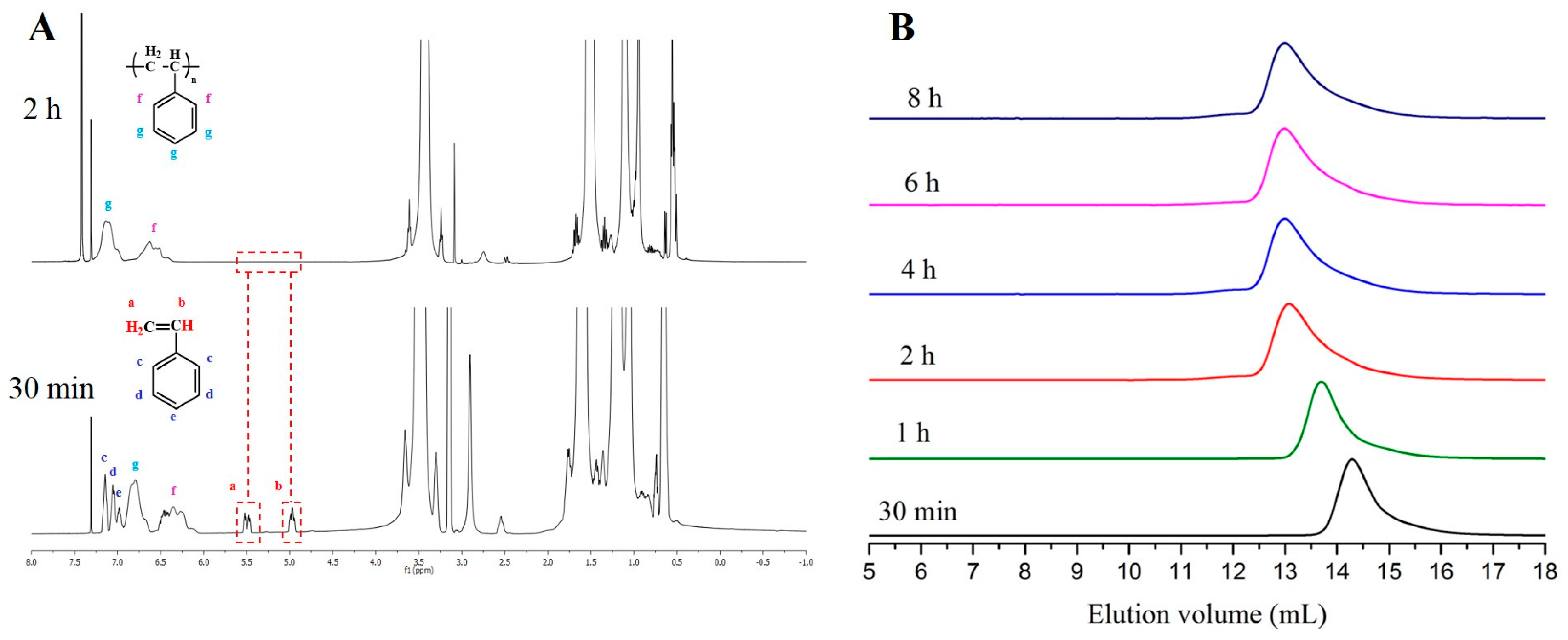
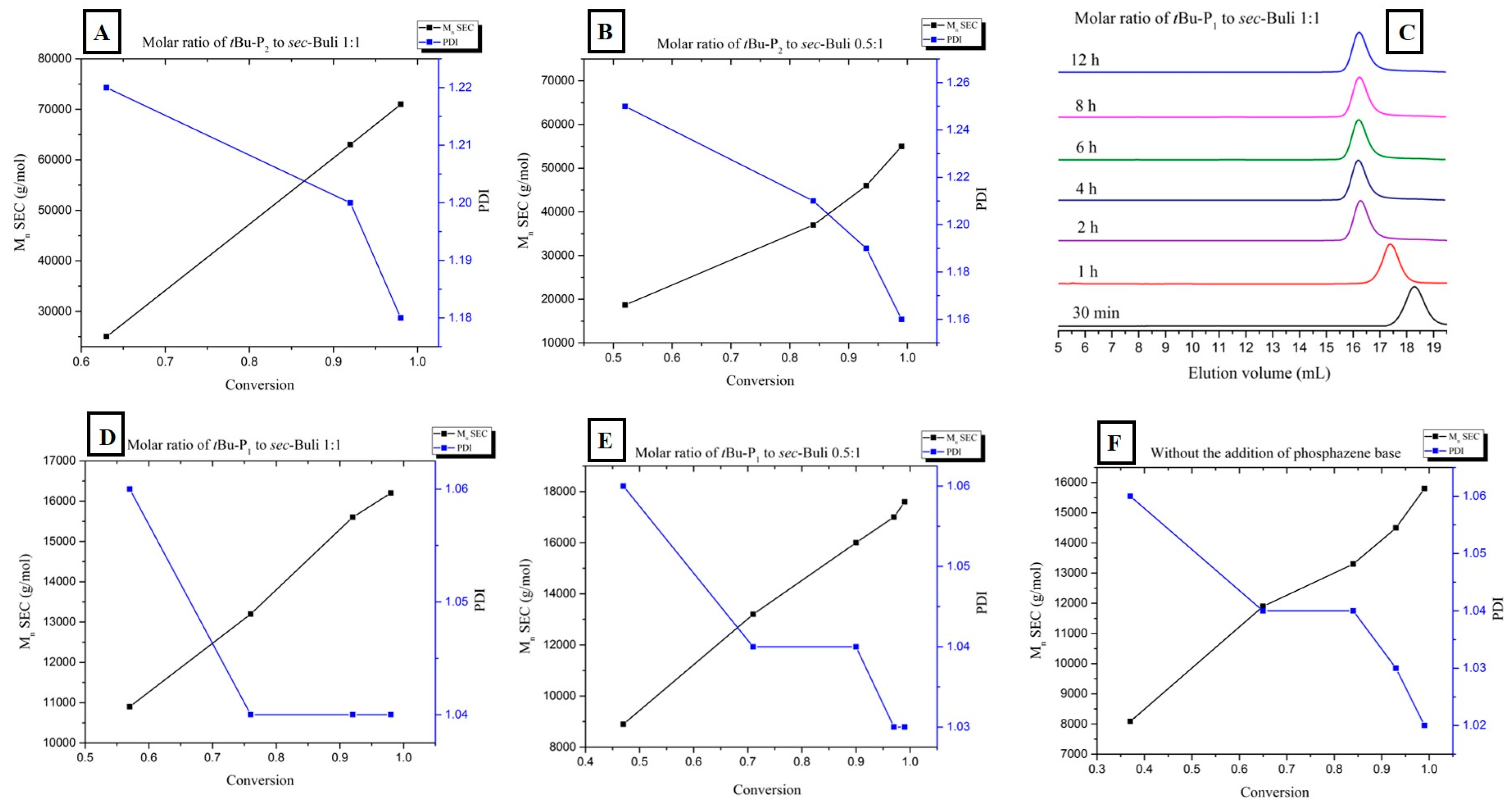
| Entry | Monomer | Phosphazene Base (PB) | PB/sec-BuLi a | Time (h) | Conv. b (%) | Mn Targeted c (g/mol) | Mn SEC d (g/mol) | Ð d |
|---|---|---|---|---|---|---|---|---|
| 1 | Styrene | t-BuP4 | 1:1 | 0.3 | 71.4 | 25,000 | 297,000 | 1.9 |
| 2 | Styrene | t-BuP2 | 1:1 | 2 | 100 | 25,000 | 72,000 | 1.19 |
| 3 | Styrene | t-BuP2 | 0.5:1 | 4 | 100 | 25,000 | 55,000 | 1.16 |
| 4 | Styrene | t-BuP1 | 1:1 | 4 | 100 | 15,000 | 16,500 | 1.04 |
| 5 | Styrene | t-BuP1 | 0.5:1 | 6 | 100 | 15,000 | 17,500 | 1.03 |
| 6 | Styrene | -- | -- | 6 | 100 | 15,000 | 15,800 | 1.02 |
| 7 | Butadiene | t-BuP2 | 1:1 | 8 | 100 | 15,000 | 51,000 | 1.17 |
| 8 | Butadiene | t-BuP1 | 1:1 | 12 | 100 | 8000 | 9300 | 1.03 |
| 9 | Butadiene | -- | -- | 16 | 100 | 10,000 | 11,300 | 1.02 |
| Entry | Macro-Initiator a | Monomer | Phosphazene Base (PB) | PB/Sec-BuLi b | Time (h) | Conv. c (%) | Mn SEC d (g/mol) | Ð d |
|---|---|---|---|---|---|---|---|---|
| 1 | (PS−Li+)72k | LLA | t-BuP2 | 1:1 | 16 | -- | 72,000 | 1.20 |
| 2 | (PS−Li+)72k | ε-CL | t-BuP2 | 1:1 | 16 | -- | 72,000 | 1.20 |
| 3 | (PS−Li+)16.5k | LLA | t-BuP1 | 1:1 | 16 | -- | 16,500 | 1.04 |
| 4 | (PS−Li+)16.5k | ε-CL | t-BuP1 | 1:1 | 16 | -- | 16,500 | 1.04 |
| 5 | (PSO−Li+)72k | LLA | t-BuP2 | 1:1 | 0.3 | 100 | 87,000 | 1.22 |
| 6 | (PSO−Li+)72k | ε-CL | t-BuP2 | 1:1 | 17 | 70 | 82,000 | 1.23 |
| 7 | (PSO−Li+)16.5k | LLA | t-BuP1 | 1:1 | 50 | 75 | 28,000 | 1.07 |
| 8 | (PSO−Li+)16.5k | ε-CL | t-BuP1 | 1:1 | 70 | 25 | 20,000 | 1.15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ntetsikas, K.; Alzahrany, Y.; Polymeropoulos, G.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. Anionic Polymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Superbases. Polymers 2017, 9, 538. https://doi.org/10.3390/polym9100538
Ntetsikas K, Alzahrany Y, Polymeropoulos G, Bilalis P, Gnanou Y, Hadjichristidis N. Anionic Polymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Superbases. Polymers. 2017; 9(10):538. https://doi.org/10.3390/polym9100538
Chicago/Turabian StyleNtetsikas, Konstantinos, Yahya Alzahrany, George Polymeropoulos, Panayiotis Bilalis, Yves Gnanou, and Nikos Hadjichristidis. 2017. "Anionic Polymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Superbases" Polymers 9, no. 10: 538. https://doi.org/10.3390/polym9100538
APA StyleNtetsikas, K., Alzahrany, Y., Polymeropoulos, G., Bilalis, P., Gnanou, Y., & Hadjichristidis, N. (2017). Anionic Polymerization of Styrene and 1,3-Butadiene in the Presence of Phosphazene Superbases. Polymers, 9(10), 538. https://doi.org/10.3390/polym9100538