1. Introduction
Polyurethanes (PUs) are polymers mainly consisting of long chain oligodiol as the soft segment and diisocyanate as the hard segment, in which short chain diol or diamine is added, serving as a chain extender. Conventional synthesis methods involve organic solvents, leading to concerns of volatile organic compounds [
1]. In contrast, the process of synthesizing waterborne PU is more eco-friendly, and therefore, water-based PUs are more favorable compared to conventional PUs. The commonly-applied approach to uniformly disperse PU particles in an aqueous solution is the incorporation of an ionic chain extender into the chemical structure of PU [
2].
Amphiphilic copolymers, which contain both hydrophilic and hydrophobic parts, have drawn much attention for biomedical applications in the past few decades. One category of these block copolymers consists of polylactide and poly(ethylene glycol) (PEG). PLA presents outstanding biodegradability and biocompatibility, high mechanical strength and plasticity and is widely used in surgical repair, drug delivery and tissue engineering [
3]. PEG is highly hydrophilic and biocompatible and provokes less immune responses [
4]. Furthermore, PEG can reduce protein adsorption onto PLA segments [
4,
5]. In addition, PLA-PEG copolymers present better biodegradability than the PLA homopolymers [
6]. Therefore, copolymers combining PEG and PLA are promising for many biomedical applications, in particular as drug carrier [
7]. PEG is more likely to move close to the surface of the material since it has relatively lower surface energy. This surface rearrangement is obvious especially in aqueous media [
5]. Amphiphilic PLA-PEG copolymers would self-assemble to form micelles once placed in an aqueous environment. PLA segments are located in the interior of micelles, while PEG segments are at the surface. The solution of micelles may respond to the change of temperature, and gelation can occur. By changing the ratio of PLA to PEG, various kinds of environment responsive materials can be synthesized [
8].
Hydrogels responding to a changing environment can be used as drug or cell delivery carriers, scaffolds for tissue engineering and wound dressings [
9,
10,
11]. The cell-laden hydrogels have been used in regenerative medicine. However, some cells, such as mesenchymal stem cells, require adhesion onto the hydrogels to survive. Therefore, the viability and proliferation of MSCs need to be evaluated when the material is used for tissue regeneration [
12]. Among these, thermo-responsive hydrogels appear the most promising. In other words, a polymer solution at room temperature turns into a hydrogel after injection into the human body without adding toxic curing agents [
13]. Common examples are copolymers of PEG and poly(propylene glycol) (PPG) and poly(N-isopropylacrylamide) (PNIPAAm), which form gels when heated. However, PNIPAAm is not degradable in vivo [
14]. Hence, the focus of research moves to copolymers composed of PEG, PLA, poly(lactide-
co-glycolide) (PLGA) or poly(ε-caprolactone) (PCL) [
15]. Thermo-responsive gelation requires that the polymer be amphiphilic and have an appropriate hydrophilic hydrophobic balance. Because most of these copolymers are linear, in order to make them mechanically strong enough to be applied in the fields of biomedicine, higher molecular weight is preferred. Nonetheless, the length of PEG has a significant influence on the gelation temperature. The concentration at which the gelation occurs decreases as the portion of hydrophobic blocks increases, whereas the gelation temperature rises as the molecular weight of the hydrophobic blocks increases [
16].
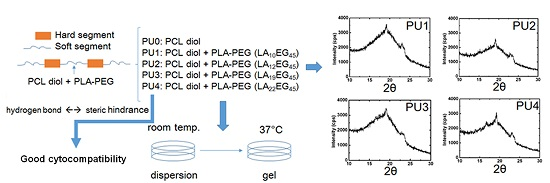
In this study, PUs are synthesized, which consisted of PCL as the main soft segment. Part of the soft segment structure (10%) is replaced with PLA-PEG diblock copolymers with different PLA block lengths to improve the hydrophilicity. By using dynamic light scattering (DLS) and transmission electron microscopy (TEM), the particle size and the surface charge were determined. The gelation properties were investigated through rheological analyses. Fourier transform-infrared spectroscopy (FTIR) and wide-angle X-ray diffraction (XRD) were performed to evaluate hydrogen bonding and crystallinity changes in order to elucidate the influencing factors on gelation. By combining the above analyses, we could determine how the PLA block length of PLA-PEG copolymers affects the thermo-responsive gelation properties of the PU nanoparticle dispersion.
2. Materials and Methods
2.1. Synthesis of PLA-PEG Block Copolymer
Four PLA-PEG diblock copolymers were synthesized by ring opening polymerization of
l-lactide using monomethoxy poly(ethylene glycol) (mPEG), as listed in
Table 1.
l-lactide and mPEG (
Mn = 2000) were supplied by Purac and Sigma, respectively. L-lactide and mPEG were first added into a flask at different molar ratios. Then, the catalyst zinc lactate (0.1 wt %) was added. The flask was degassed and sealed under vacuum, and the polymerization was conducted at 140 °C. After 3 days, the product was dissolved in dichloromethane (CH
2Cl
2), precipitated in diethyl ether and further dried under vacuum [
17]. The PLA-PEG diblock copolymers were named LA-EO diblock diol.
2.2. Synthesis of Waterborne PU
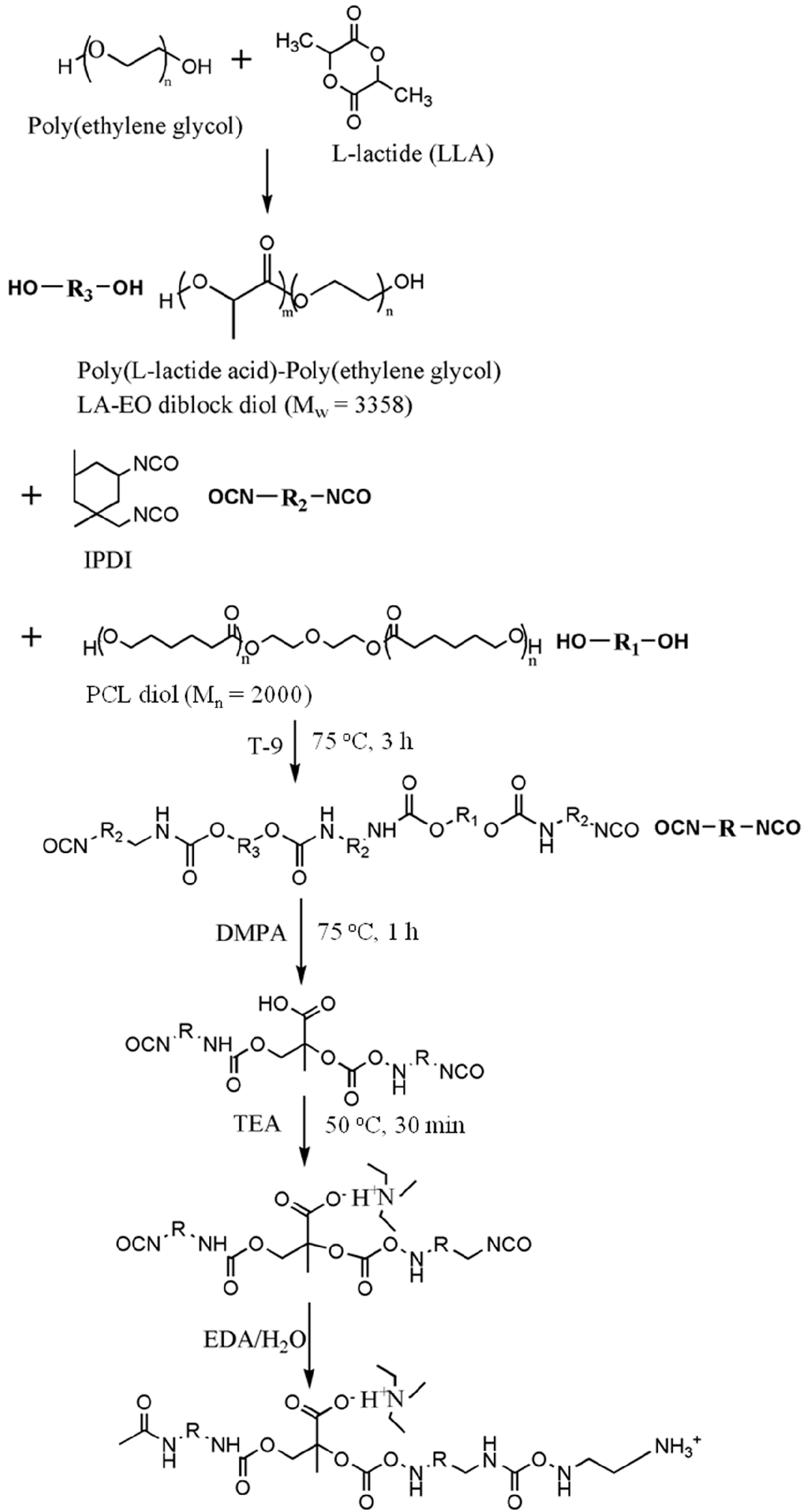
PU was prepared from a two-step reaction process previously described [
18]. The reaction scheme is shown in
Figure 1. The reactants were isophorone diisocyanate (IPDI, from Evonik Degussa GmbH, Essen, Nordrhein-Westfalen, Germany), ethylenediamine (EDA, from Tedia, Fairfield, OH, USA), 2,2-bis(hydroxymethyl)propionic acid (DMPA, from Sigma Aldrich, St. Louis, MI, USA), triethylamine (TEA, from RDH, Spring Valley, CA, USA), methyl ethyl ketone (MEK, from J.T. Baker, Phillipsburg, NJ, USA), and T-9. The reaction was performed in a 250-mL round-bottomed four-necked separable flask with a mechanical stirrer, a thermometer and a condenser with a drying tube and N
2 inlet. The flask was immersed in an oil bath in order to achieve a constant-temperature environment. First, PCL diol (
Mn = 2000 from Sigma Aldrich, St. Louis, MI, USA) and LA-EO diblock diol were added into the dried flask, followed by the addition of IPDI and T-9. The solution was stirred at 105 °C for 3 h. DMPA and MEK were then added with the temperature controlled at around 70 °C, and the solution was stirred for 1 h. The heater was turned off, and after the temperature of the mixture dropped to 50 °C, TEA was added and the mixture stirred for 30 min. An EDA solution in deionized water was then added, and the mixture was vigorously stirred for another 30 min. The stoichiometric ratio of IPDI, oligodiols, DMPA, EDA and TEA was 3.52:1:1:1.52:1. Residual MEK and TEA were removed by vacuum distillation. The obtained PU dispersion in water had a solid content of about 30%. The PU dispersion was either directly characterized or was cast onto Teflon plates to prepare films for further bulk characterization. All PU dispersions were characterized or cast in 3 days after synthesis to avoid aggregation.
2.3. Physico-Chemical Characterization
The PU dispersion was diluted with distilled water to bring down the solid content to 0.3 wt %. The hydrodynamic diameter (Dh) and zeta potential of the PU nanoparticles were measured by the DelsaTM Nano C Particle Analyzer (Beckman Coulter, Brea, CA, USA).
The water contact angle of the PU film surface was measured by a contact angle analyzer (FTA-1000 B, First Ten Angstrom Company, Portsmouth, VI, USA) at room temperature. Water droplets were around 5 μL in volume for each measurement. Values of water contact angles were obtained at various spots on the film and averaged. The attenuated total reflection-infrared (ATR-IR) spectroscopy was used to characterize the surface chemistry of the films. The spectra between wavenumbers of 4000 and 650 cm−1 were collected by using a spectrophotometer (Spectrum 100, Perkin-Elmer, Waltham, MA, USA).
The tensile mechanical properties were determined by a universal testing instrument (HT-8504, Hung Ta Co., Nantun District, Taichung, Taiwan) following the ASTM standard protocol (ASTMD638.10). The samples were tailored into a size of 25 mm (length) × 5 mm (width) × 1 mm (thickness), and the stretching rate was 100 mm·min−1.
The thermogravimetric analysis (TGA) was conducted with a thermogravimetric analyzer (TA Instrument, New Castle, DE, USA). Each sample (5 mg) was placed in an alumina cubicle, heated at a rate of 10 °C per min under N2. Differential scanning calorimetry (DSC, Pyris 6, Perkin-Elmer, Waltham, MA, USA) was used to determine the glass transition temperature (Tg) and the melting temperature (Tm). The heating rate was 10 °C per min, and the measurement was operated in a temperature range of −70–150 °C. The weight average molecular weight was estimated by gel permeation chromatography (GPC, Waters, Milford, MA, USA) in dimethylacetamide using polystyrene standards.
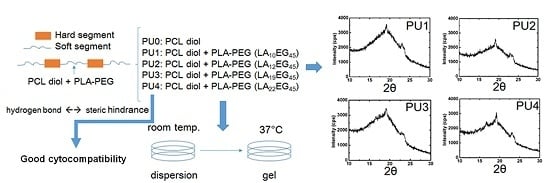
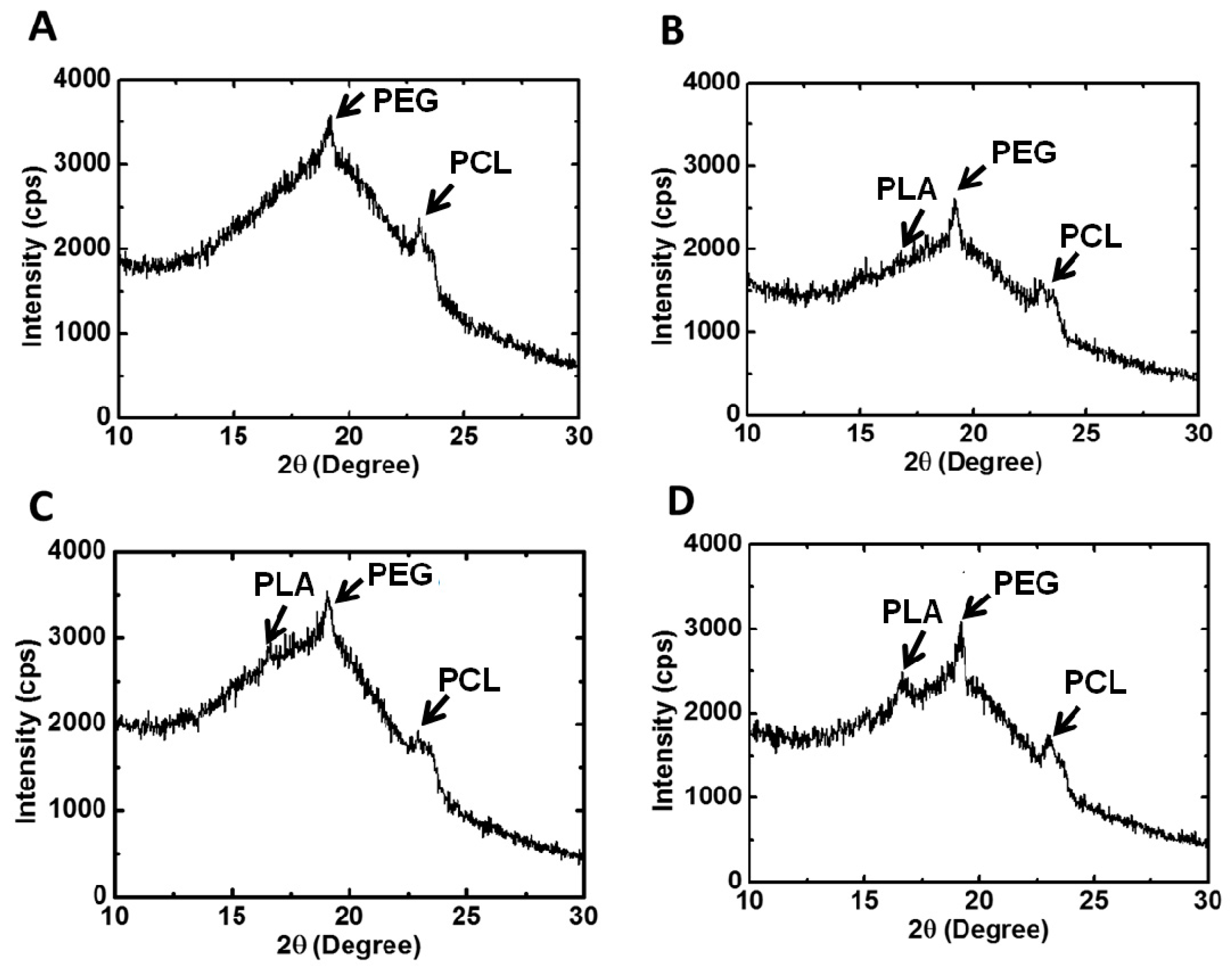
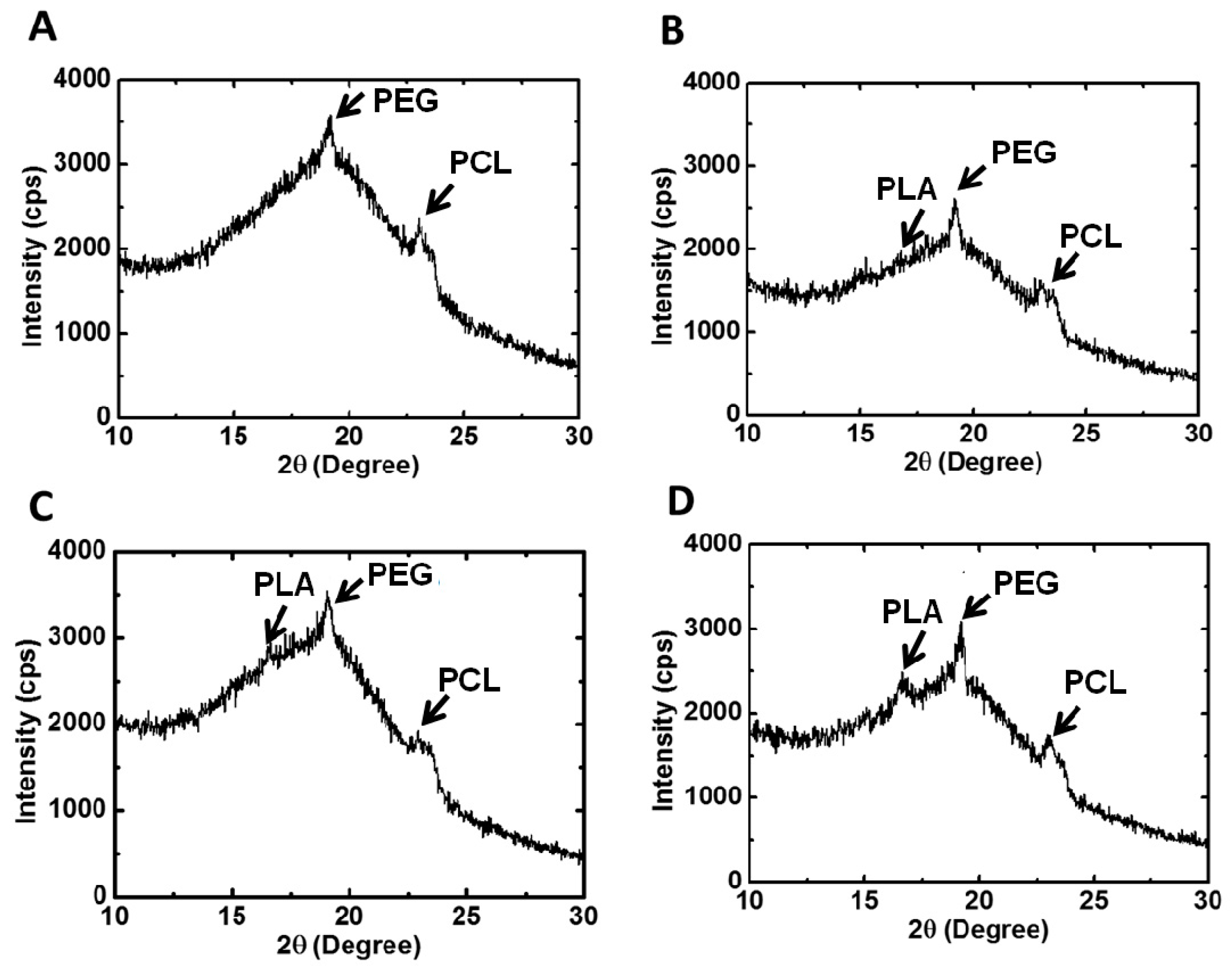
XRD was used to examine the crystallization characteristics of the PU films. The equipment was operated at a power of 2 kW, and measurements were made in the scattering angle range of 10°–30°. The degree of crystallinity was calculated from the integration area under characteristic peaks.
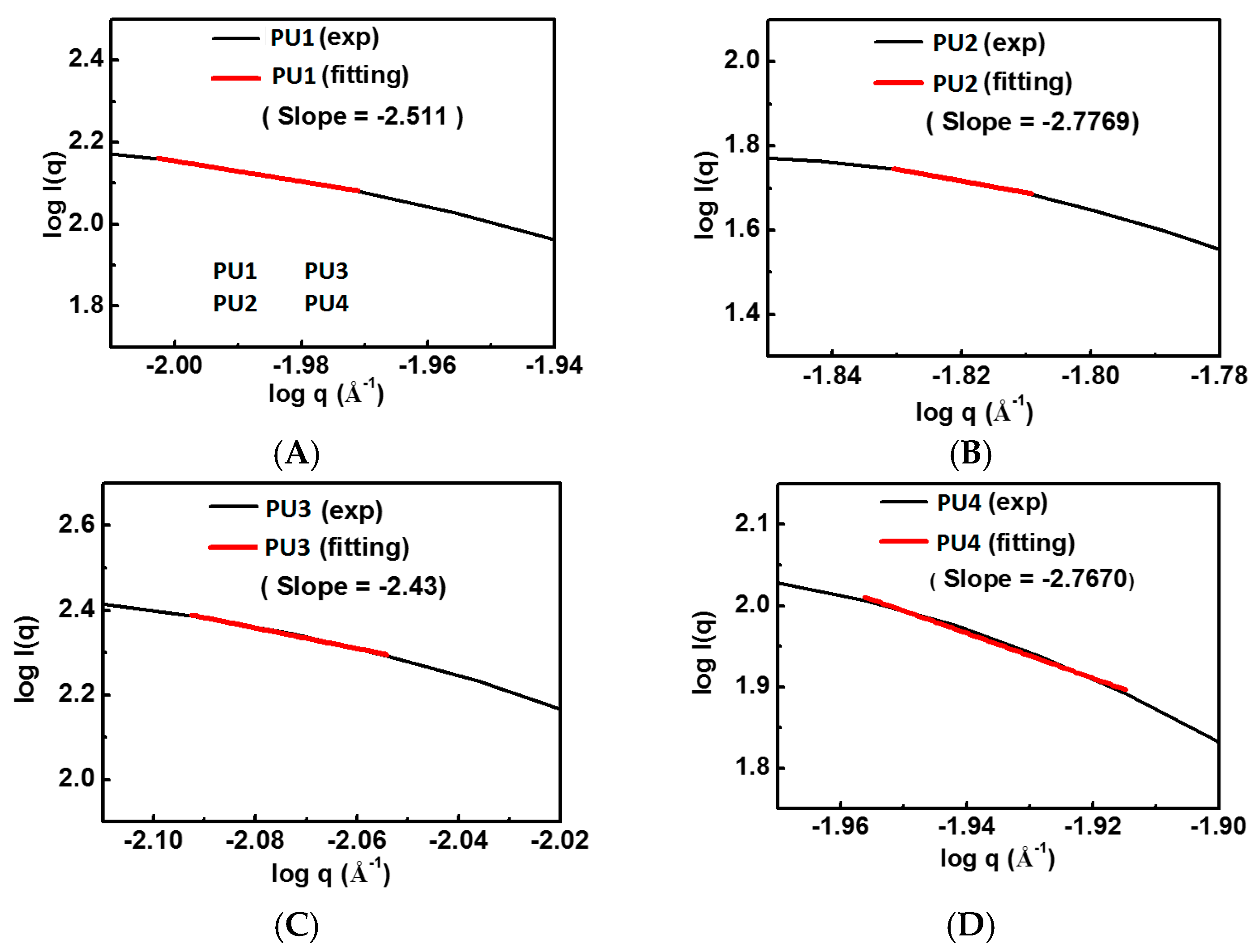
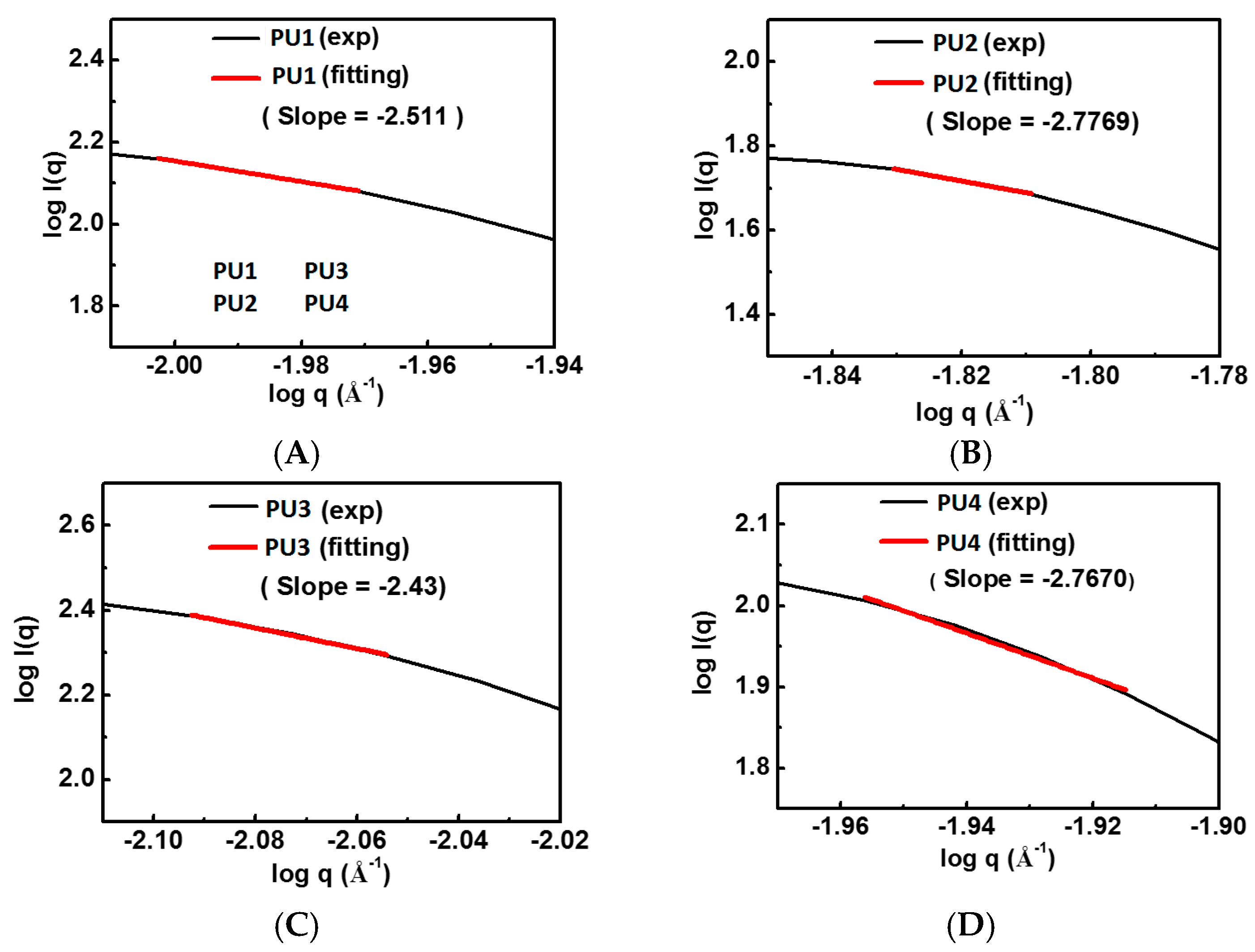
For a better understanding of the microstructure of PU nanoparticles, SAXS was conducted at the beamline 23A of National Synchrotron Radiation Research Center (Hsinchu Science Park, Hsinchu, Taiwan). The photon energy of about 10 eV was applied. According to the Guinier method [ln(I) = ln(I0) − q2Rg2/3], by plotting the logarithm of the intensity versus the square of scattering vector, the fractal dimension (df) was obtained from the negative value of slope of the plot. The radius of gyration (Rg) was estimated under the circumstance that qRg < 1.3.
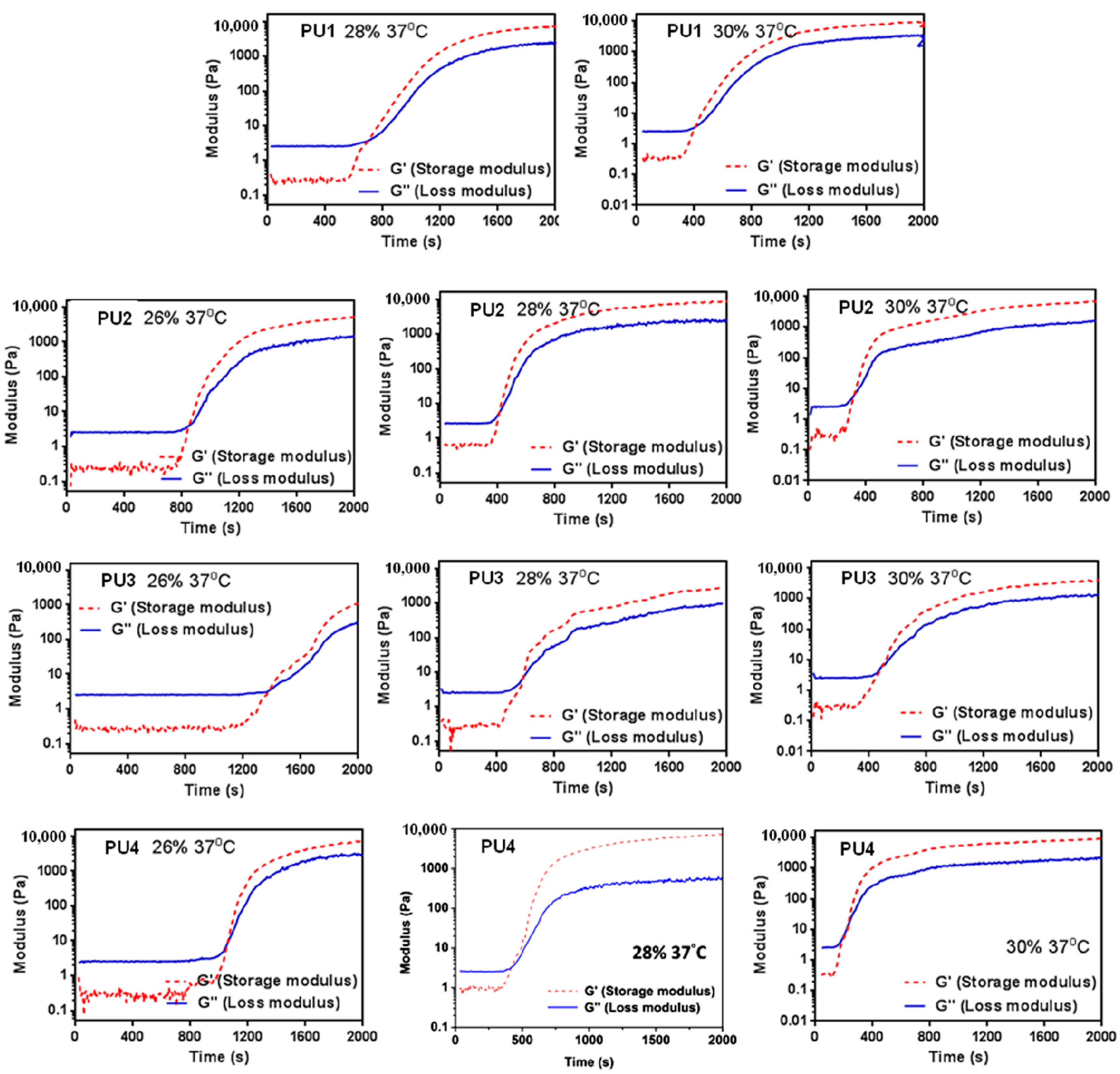
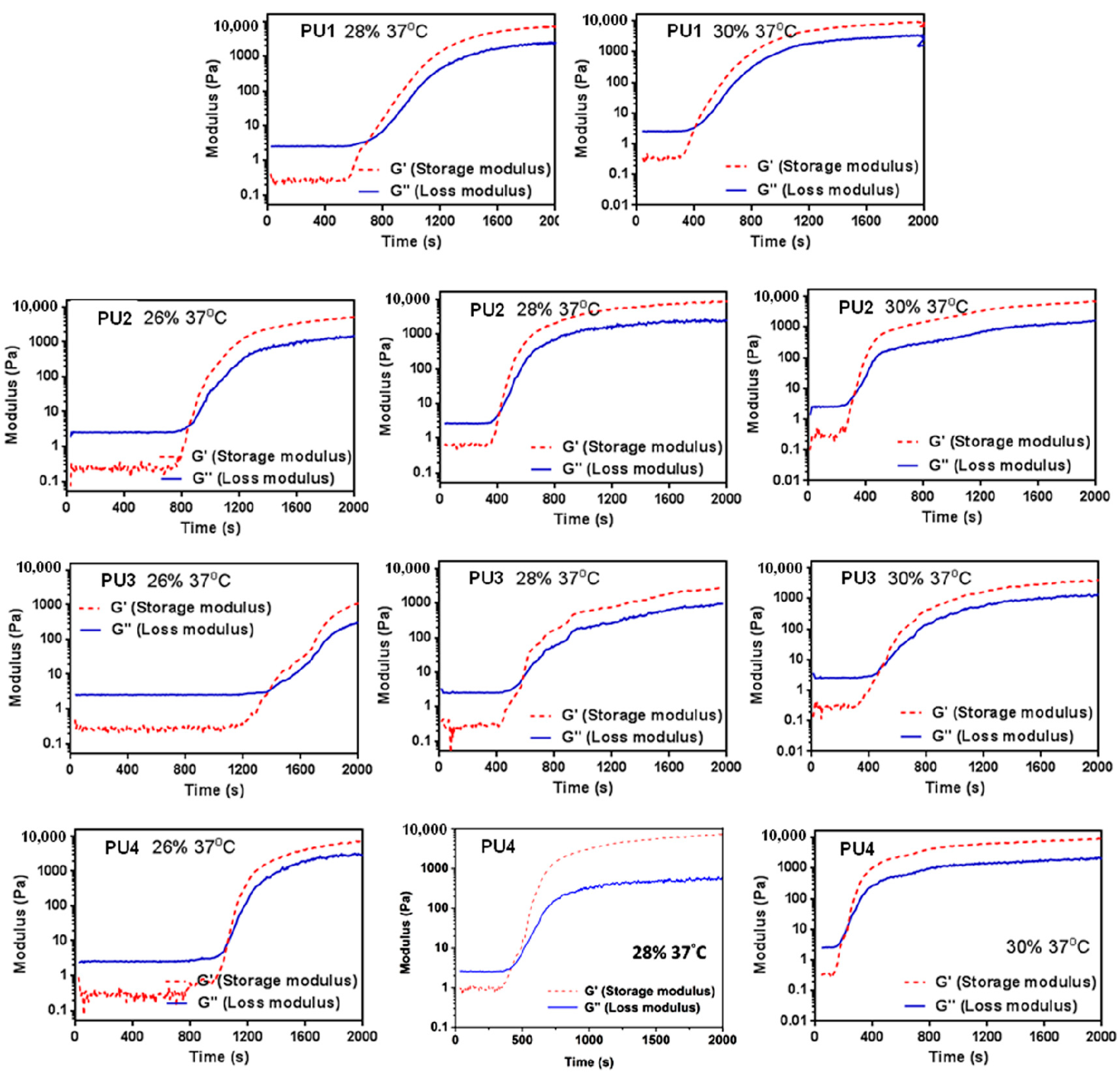
The rheological properties of the PU dispersions were measured in 3 days after synthesis. Storage beyond 2 weeks could significantly shorten the gelation time and increase the gel modulus. The rheological analysis was carried out using a rheometer (RS-5, TA Instruments, New Castle, DE, USA) with a cone and plate geometry at a frequency of 1 Hz and a deformation of 1%. The time-dependent dynamic storage modulus (G’) and loss modulus (G’’) were measured for each PU. The point where G’ = G’’ was defined as the point of sol-gel transition.
2.4. Cell Culture
Human umbilical cord-derived mesenchymal stem cells (hMSCs) obtained from BIONET Corp (Neihu District, Taipei, Taiwan) were cultured in T150 tissue-culture flasks (Falcon, BD Bioscience, San Jose, CA, USA). The culture medium was composed of low-glucose Dulbecco’s Modified Eagle Medium (LG-DMEM), 10% fetal bovine serum (FBS; SAFC Biosciences, St. Louis, MI, USA), 1% penicillin-streptomycin (Caisson, Logan, UT, USA) and 0.4% gentamicin (Gibco, Waltham, MA, USA). The incubation of the cells took place in a humidified incubator with 5% CO2 at 37 °C. Cells of the eleventh to thirteenth passage were used in the analyses.
2.5. Cell Labeling
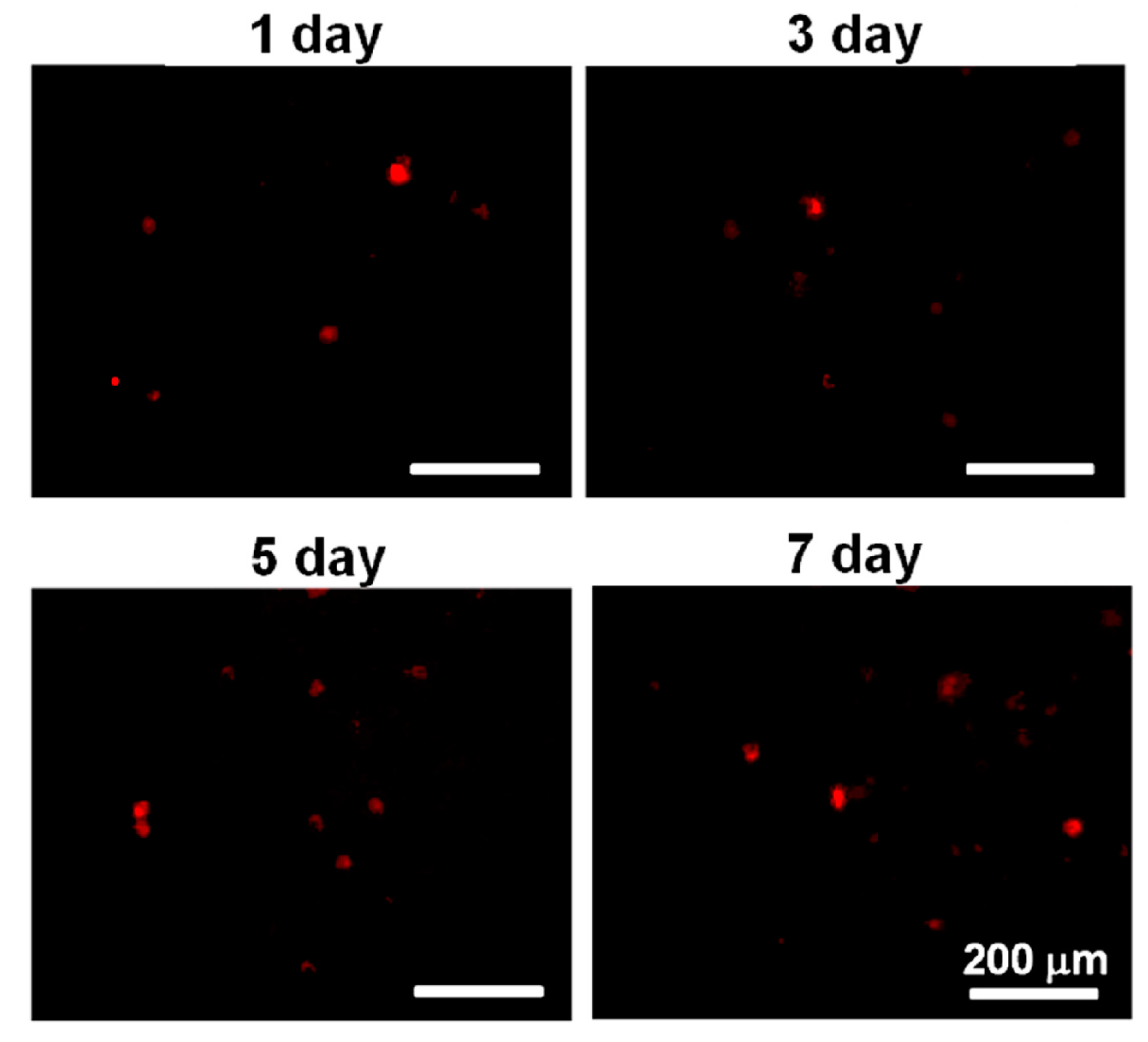
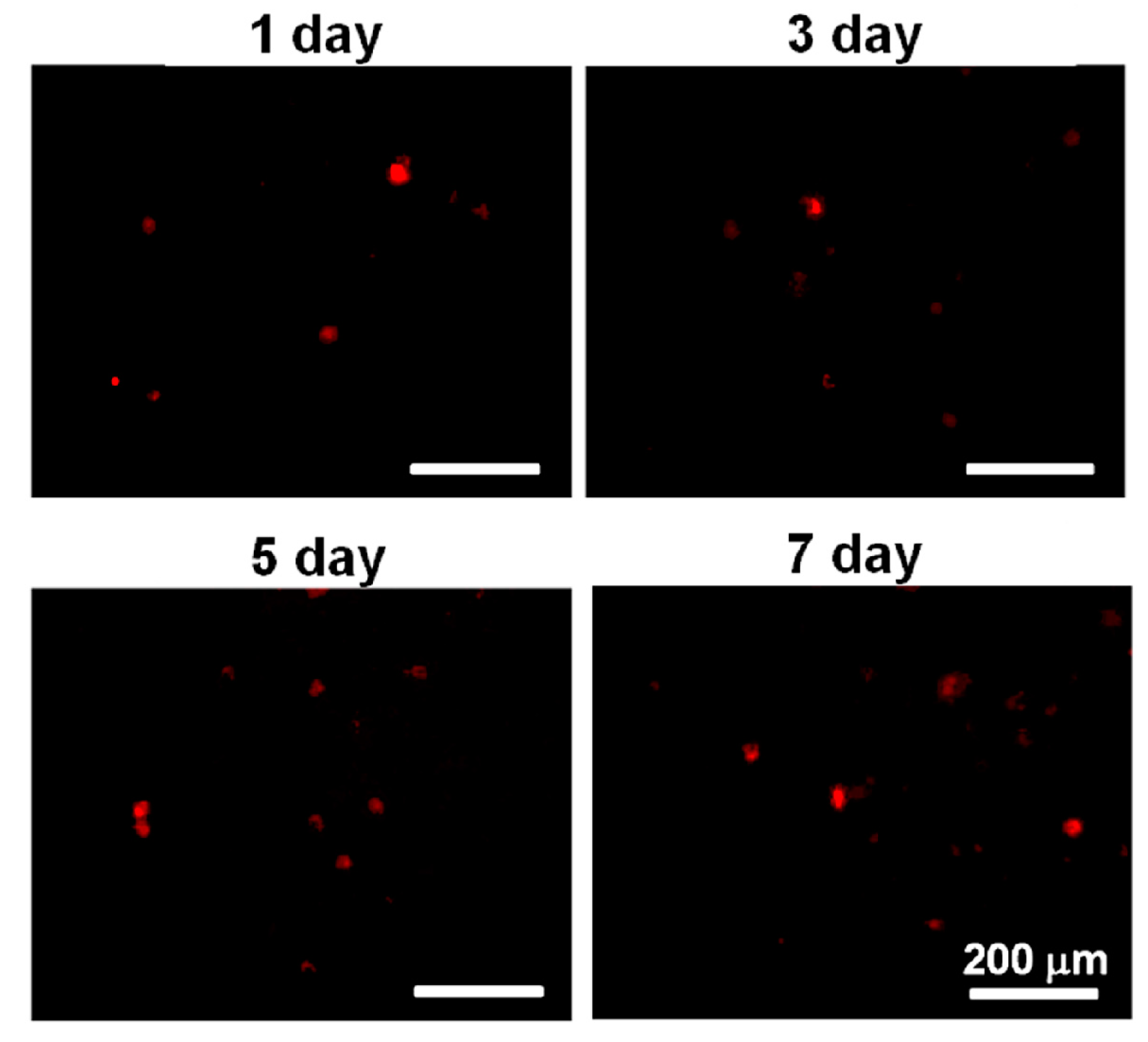
Before being mixed with the PU4 dispersion with a solid content of 26% and gelation at 37 °C, the cells were stained with a red fluorescent dye PKH26 (Sigma-Aldrich, St. Louis, MI, USA). The dye could be stably introduced into the cell membrane, thus labeling the cells. Cells with a density of 2 × 106 cells/mL were mixed with PKH26 with a concentration of 2 × 10−6 M, and the labeling was stopped with the complete medium. Lastly, the cells were washed to be ready for use.
2.6. Cell Viability and Vitality
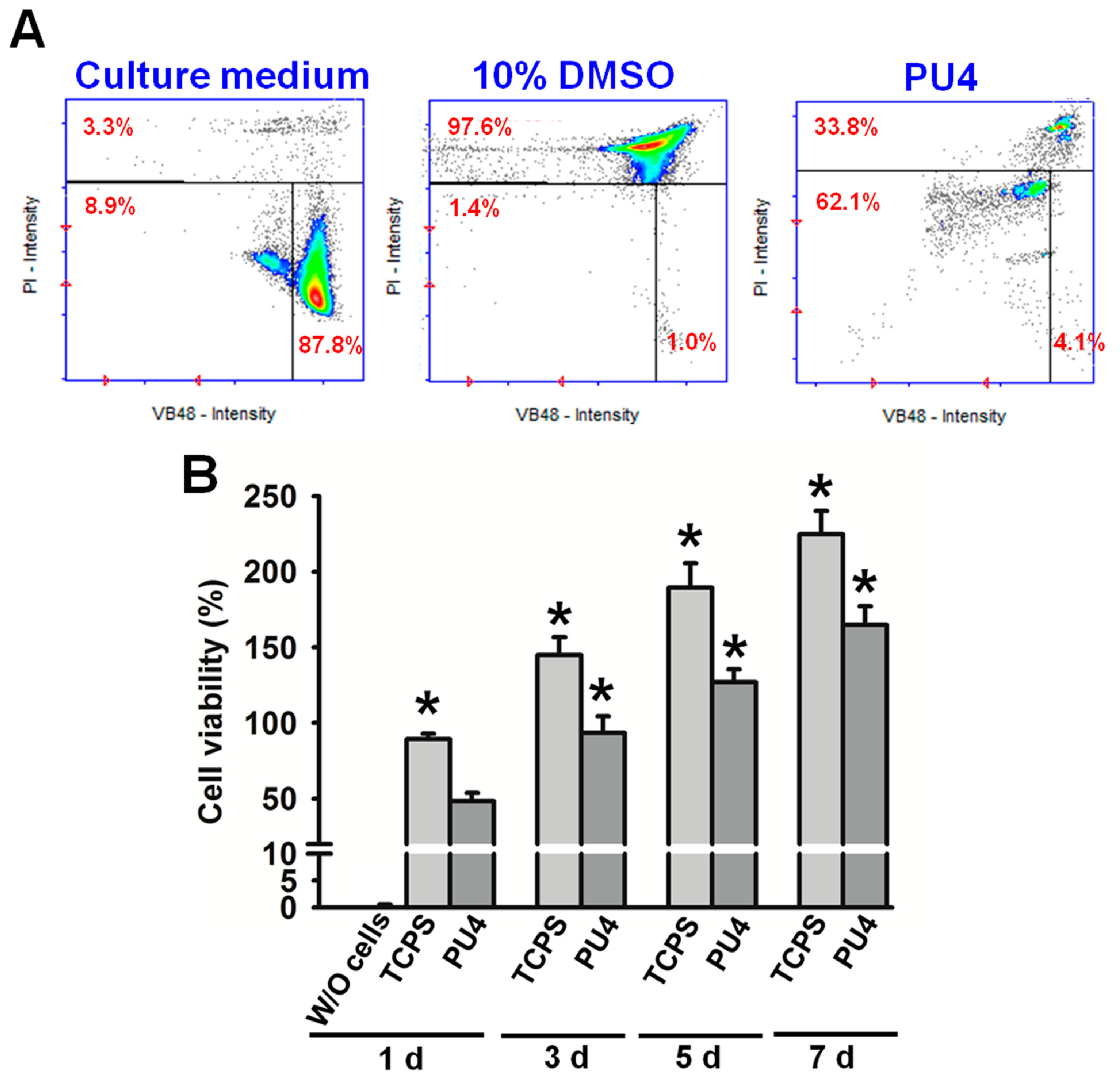
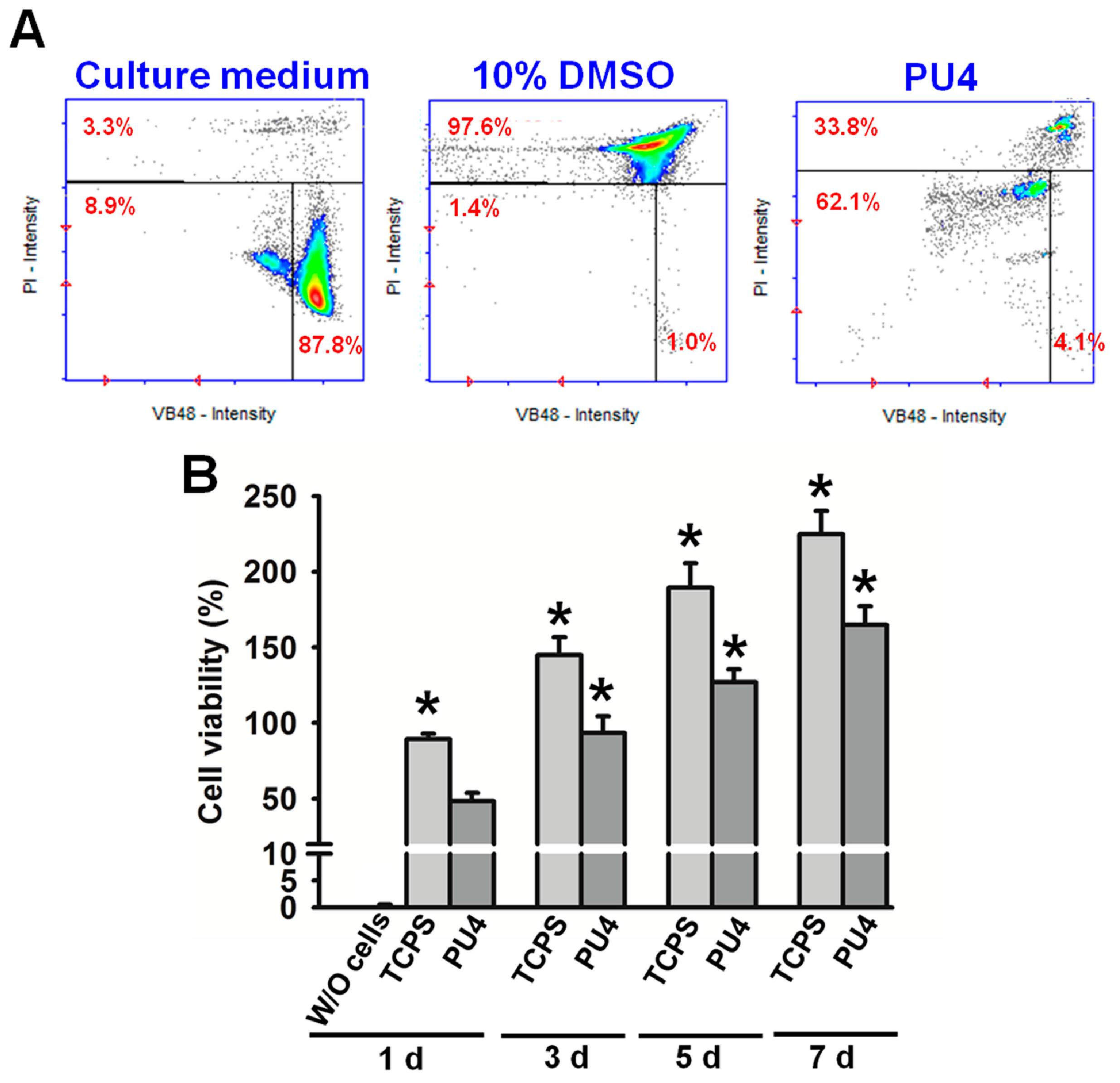
Cell viability was measured by staining the cells in the dispersion with Solution 5, which contained propidium iodide (PI) and acridine orange, marking the dead cells and serving as a counterstain, respectively, and a patented fluorophore VB-48™, which showed fluorescence only when bound to reduced thiols, could be used to analyze the cell health condition. Therefore, the distribution of live cells and the intensity of thiol levels in cells could be obtained using the NucleoCounter® NC-3000™ system (ChemoMetec, Davis, CA, USA). Specifically, the reduced thiol type in the cells was glutathione (GSH), whose decrease in concentration could be related to apoptosis. Thus, the lower fluorescent intensity, which indicating the lower level of GSH, implied the reduced healthiness of the cells. The healthiness of hMSCs was compared to those in culture medium with or without dimethyl sulfoxide (DMSO, 10 wt %).
2.7. Cell Proliferation
The MTT assay was applied to measure cell proliferation. The cell-laden hydrogels were washed in phosphate-buffered saline (PBS), followed by the addition of 200 μL of 0.1 mg/mL tetrazolium dye (Sigma-Aldrich, St. Louis, MI, USA) and incubation for 4 h at 37 °C. After incubation, the tetrazolium dye was removed. The reduced product of tetrazole, the purple formazan, was dissolved by DMSO and detected at 570 nm using a microplate reader (SpectraMax® M5, Molecular Devices LLC, Sunnyvale, CA, USA). The optical density was normalized to that measured for cells (cell density: 2 × 106 cells/mL) before mixing with hydrogels (100% viability). The viabilities were measured at the 1st, 2nd, 3rd and 7th day of culture. The medium without cells and cells in a blank well (tissue culture polystyrene (TCPS)) were used as control groups, where the MTT assay was performed for cells on 24-well plates (cell density: 3 × 104 cells/wells), and the viabilities were normalized to those measured for cells before the culture. The results were proven to be reproducible in at least three independent experiments. Through one-way analysis of variance, p-values < 0.05 indicated significant statistical differences.
4. Discussion
All of the PU nanoparticles in the study had very small hydrodynamic diameters and negative zeta potentials, demonstrating that they were stably dispersed in aqueous solutions. The negative charges on the surfaces of the particles may be attributed to the COO
− functional group on the hard segment. The hydrodynamic diameters and diameters of gyration of the PU nanoparticles with amphiphilic PLA-PEG blocks decreased while the zeta potentials increased. The decrease in nanoparticle size might be due to the tight package of the PLA blocks. The increase in zeta potentials was probably a result of surface enrichment of the PEG segment. Based on the ratio of
Dg over
Dh, which was 0.775 for spherical particles and 1.54 for random coils [
19], PU2 and PU4 were more spherical than PU1 and PU3.
By dividing the molecular weight of a single nanoparticle by the molecular weight of a single polymer chain, the number of polymer chains in one PU nanoparticle was estimated. The number of polymer chains in a single PU nanoparticle was reduced significantly in all PU nanoparticles containing various PLA-PEG blocks (<200 polymer chains in one nanoparticle), compared to the PCL-based PU (>300 polymer chains in one nanoparticle).
The contact angles of the PU films containing various PLA-PEG blocks were much lower than the PCL-based PU, indicating that the surface was much more hydrophilic. The changes in contact angles might be associated with the more hydrophilic PEG blocks in the structure [
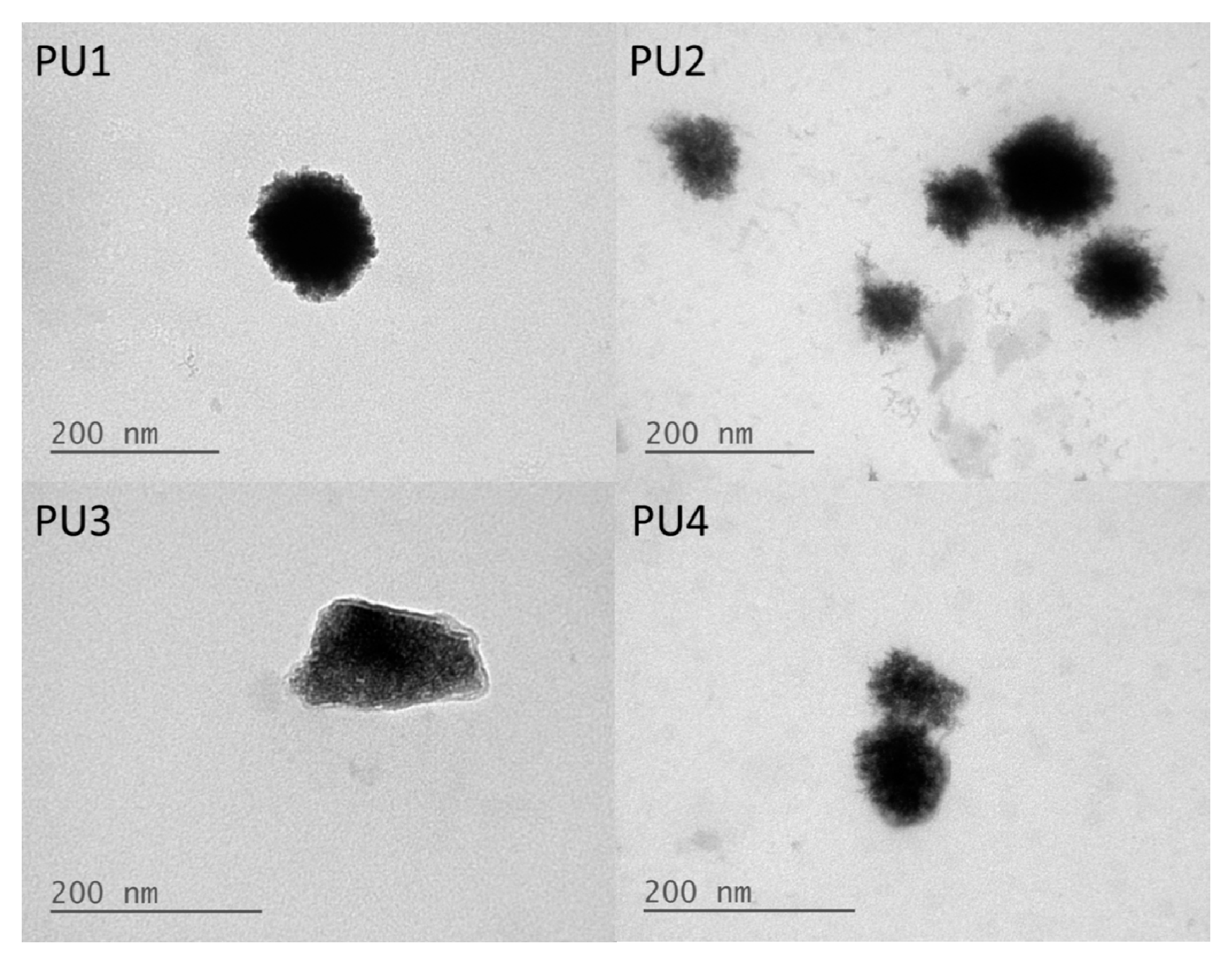
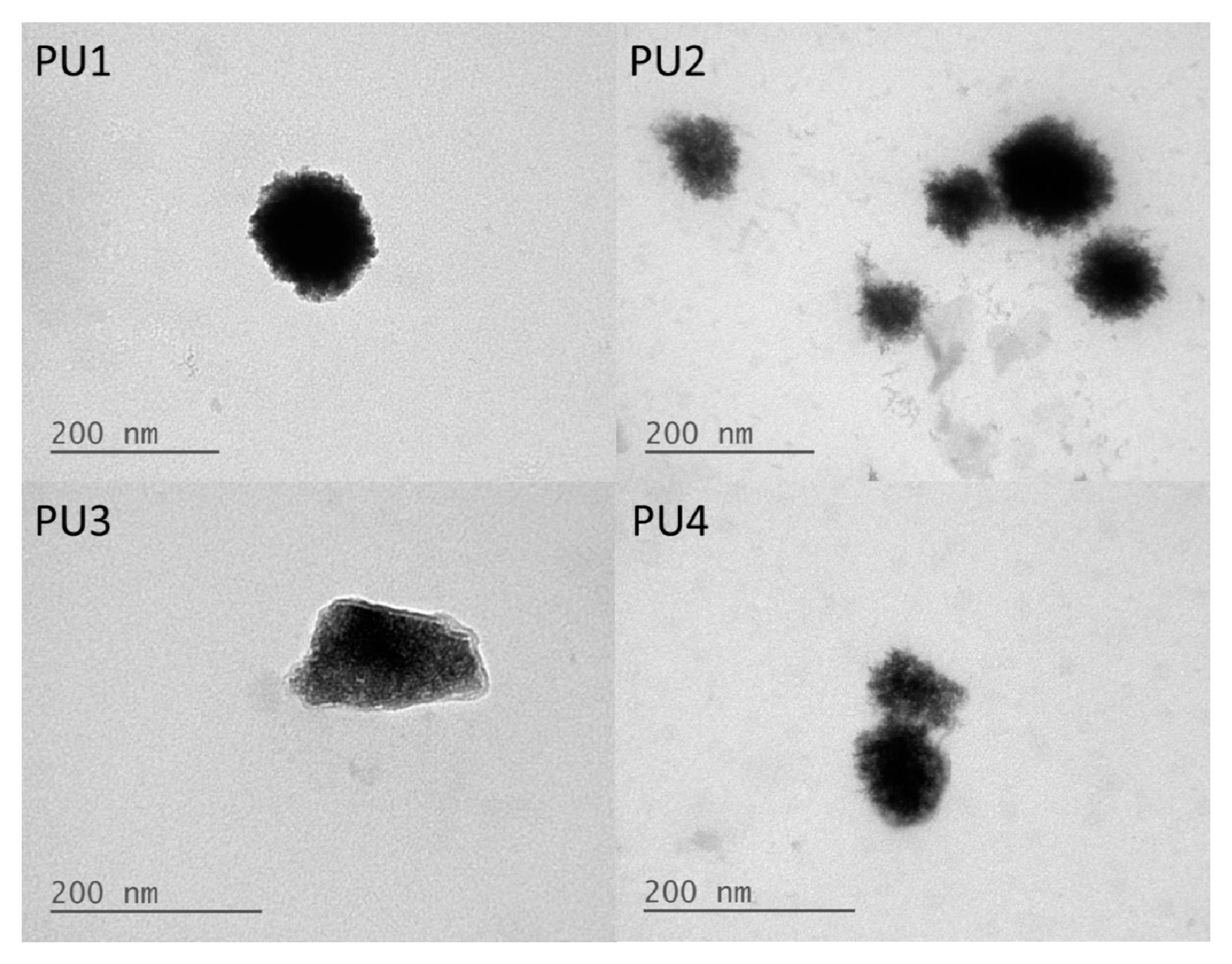
20]. Furthermore, the TEM images showed that the PU nanoparticles were covered in a relatively brighter layer of presumably the PEG block. These data were consistent with the less negative zeta potential.
The ATR-IR spectra of PU did not demonstrate any peaks around 2260–2280 cm−1 (–NCO) or 3200–3600 cm−1 (–OH), indicating that all monomers reacted completely. On the other hand, the absorption peaks showed up around 1060–1250 cm−1 in PUs containing PLA-PEG blocks, which were associated with the PEG segment. The appearance of this band agreed with the greater hydrophilicity of the surface containing the amphiphilic blocks.
All PU NP dispersions in this study had low viscosities below 37 °C and therefore could be mixed well with cells. Upon the temperature rise to 37 °C, gelation occurred in a short period of time (~min) in 30% dispersions. At a lower solid content (26%), as the PLA chain length increased, the gelation time also increased. This could be attributed to the decreased PCL content, leading to less hydrogen bonding and smaller interaction between NPs [
21], preventing the coacervation of PU NPs. On the other hand, when the solid content was 28% or 30%, the gelation time of PU NP dispersions became shorter, and the particles were aggregated more readily. The gel modulus (
G’) increased with time at 37 °C. Besides, PU1, PU2 and PU4 gels at higher solid contents had large
G’. In contrast, PU3 had a significantly lower G’ compared to the other gels, which could be associated with the lower tendency of the crystallization of the sample [
22]. The obtained fractal dimension from the rheological analysis was around 2.4–2.8 for each gel, which is close to the one (~2.5) calculated from the theory of the percolation cluster. Moreover, the commonly-used biological polymer gelatin also has a value of the fractal dimension of 2.5 [
23]. All PLA-PEG block-containing PUs (PU1–PU4) showed relatively good gel strength at 37 °C without any crosslinking agent.
The gelation of PU hydrogels was related to the interaction between soft segments, demonstrated by their crystallization [
24]. However, the crystallization of wet hydrogels was difficult to detect due to the high water content. Therefore, PU emulsion deprived of water was used to prepare PU films, in which the degree of crystallization was measured. With the incorporation of PLA-PEG blocks into PU, the steric hindrance tended to affect the arrangement of polymer chains, causing the amorphous PCL-based PU to crystallize. The PLA blocks could facilitate the crystallization of PEG and PCL segments [
25]. Nonetheless, as the LA chain length increased to a certain degree, namely in PU3 and PU4, the effect became less obvious. As a result, the degree of crystallinity reached a maximum at PU2 and then decreased. Judging from the crystallization peaks in XRD, the crystallization of PU1, PU2 and PU3 was mainly attributed to PEG and PCL segments, while PLA blocks contributed a fair amount of crystallization in PU4 in comparison with PEG and PCL segments.
To investigate whether the crystallization affected the mechanical properties accordingly, the tensile properties of PU films were analyzed. For the PU films, the decrease of tensile strength in PUs containing PLA-PEG blocks might be associated with the lower PCL content of the PUs. PCL is more likely to form hydrogen bonds between polymer chains, and thus, a lower PCL content is linked to lower tensile strength [
24]. PU2 and PU4 films had relatively larger tensile strength and Young’s modulus, which may be ascribed to the higher degree of crystallinity. The elongation of PUs containing PLA-PEG blocks increased significantly to over 600%, suggesting the more elastic behavior of the resulting PU films.
Regarding the thermal properties, the
Tg of the PU films was mainly attributed to the crystallization of PLA blocks [
26]. The pyrolytic temperatures
Tonset and
Td both increased as the PLA chain lengths increased, but decreased abruptly in PU4, indicating that excessive PLA could make the polymer susceptible to decomposition.
The viability of hMSCs upon the initial contact with the PU4 hydrogel was only around 66%, and the health condition of most live cells was affected by the presence of hydrogel. However, the cells proliferated significantly after three days of culture, and the growth continued at the seventh day of culture. The cell viability reached 170% of the initially seeded cells. These data indicated that the hydrogel could support the proliferation of hMSCS regardless of the negative influence on the cell health at the early stage of culture.
In summary, the nanoparticle dispersions of PUs containing PLA-PEG blocks could be stably dispersed in aqueous solutions due to the hydrophilicity of the NPs. Upon the temperature rise to 37 °C, gelation occurred quickly in all samples. The fractal dimension of each PU gel was close to 2.5, similar to that of a percolation cluster. Taken together, these materials were fast thermos-responsive hydrogels with tunable gel strength and properties. Furthermore, PU4 with a solid content of 26% could support the proliferation of hMSCs.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}