Phase Transformation of Adefovir Dipivoxil/Succinic Acid Cocrystals Regulated by Polymeric Additives


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
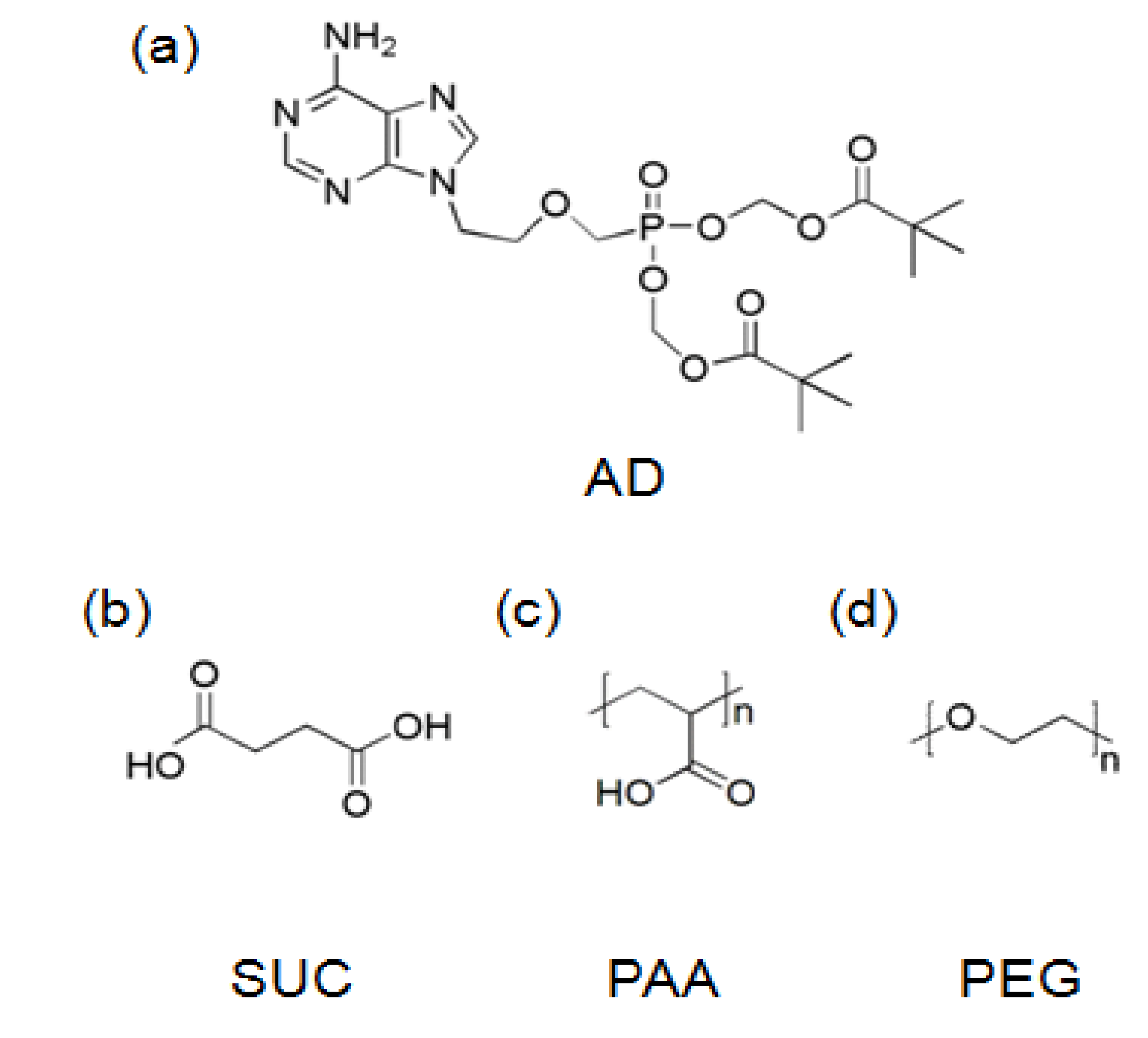
2.1. Materials
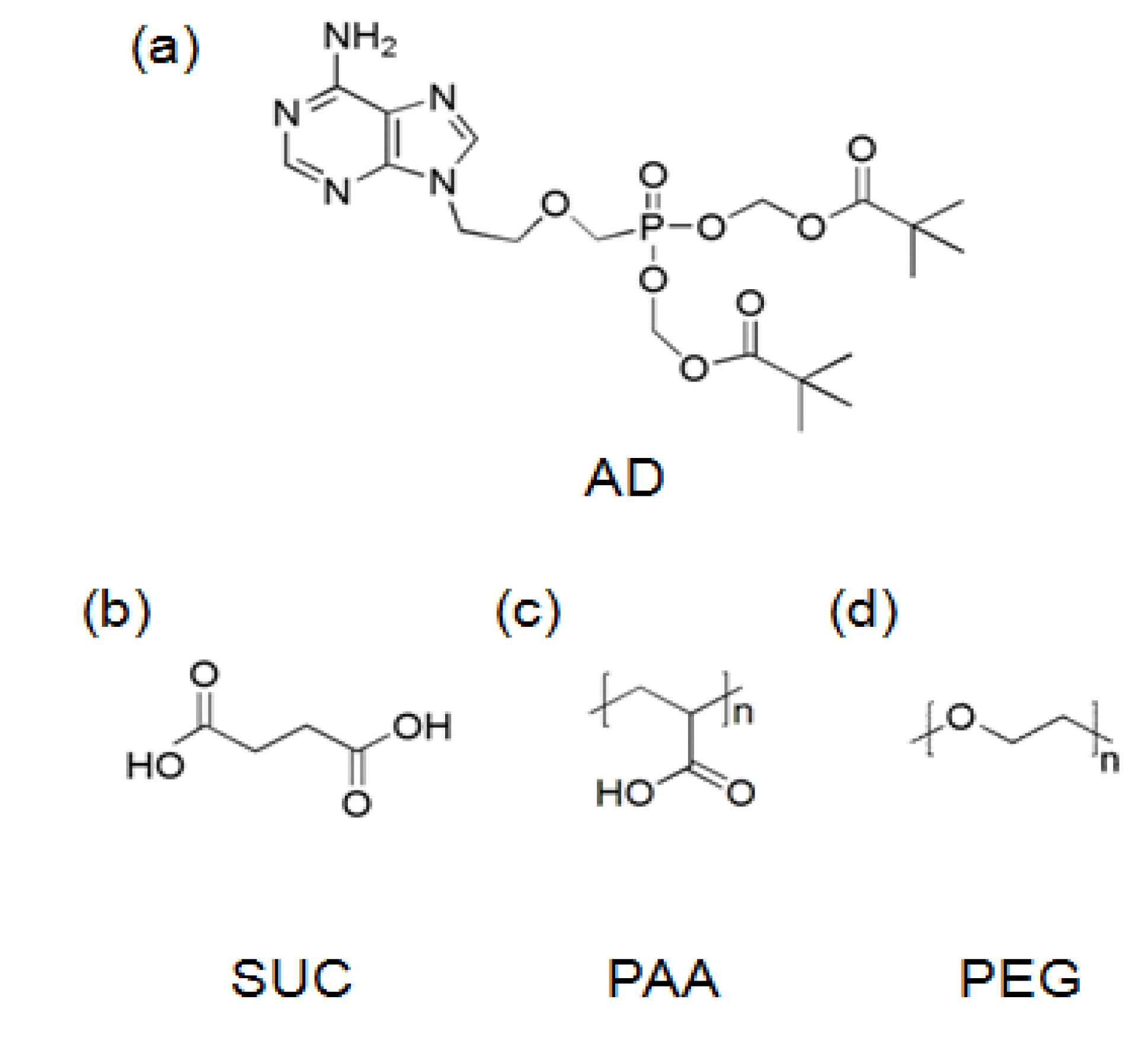
2.2. Formation of AD/SUC Cocrystals
2.3. Characterization
3. Results and Discussion
3.1. Microscopic Observation
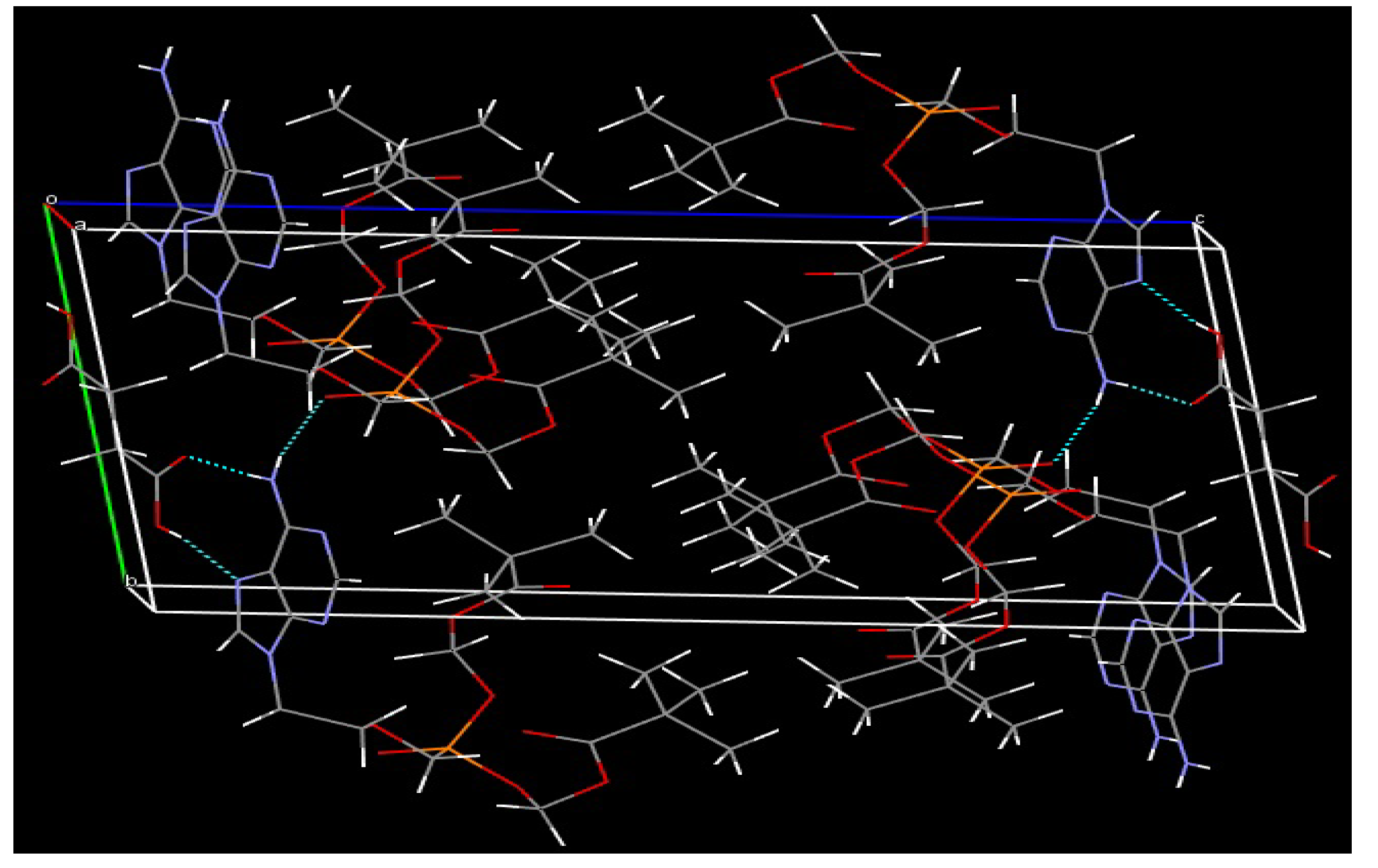

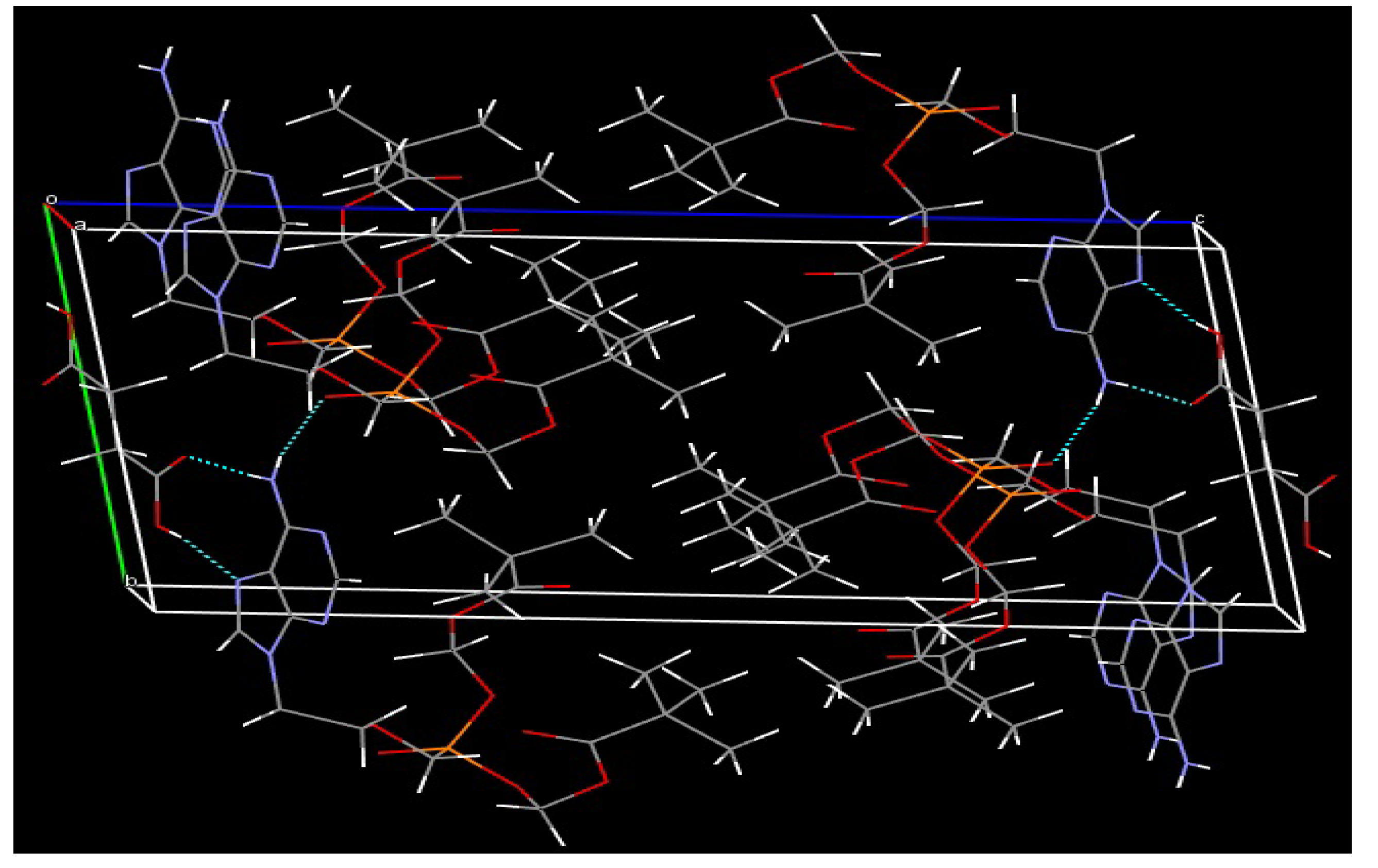
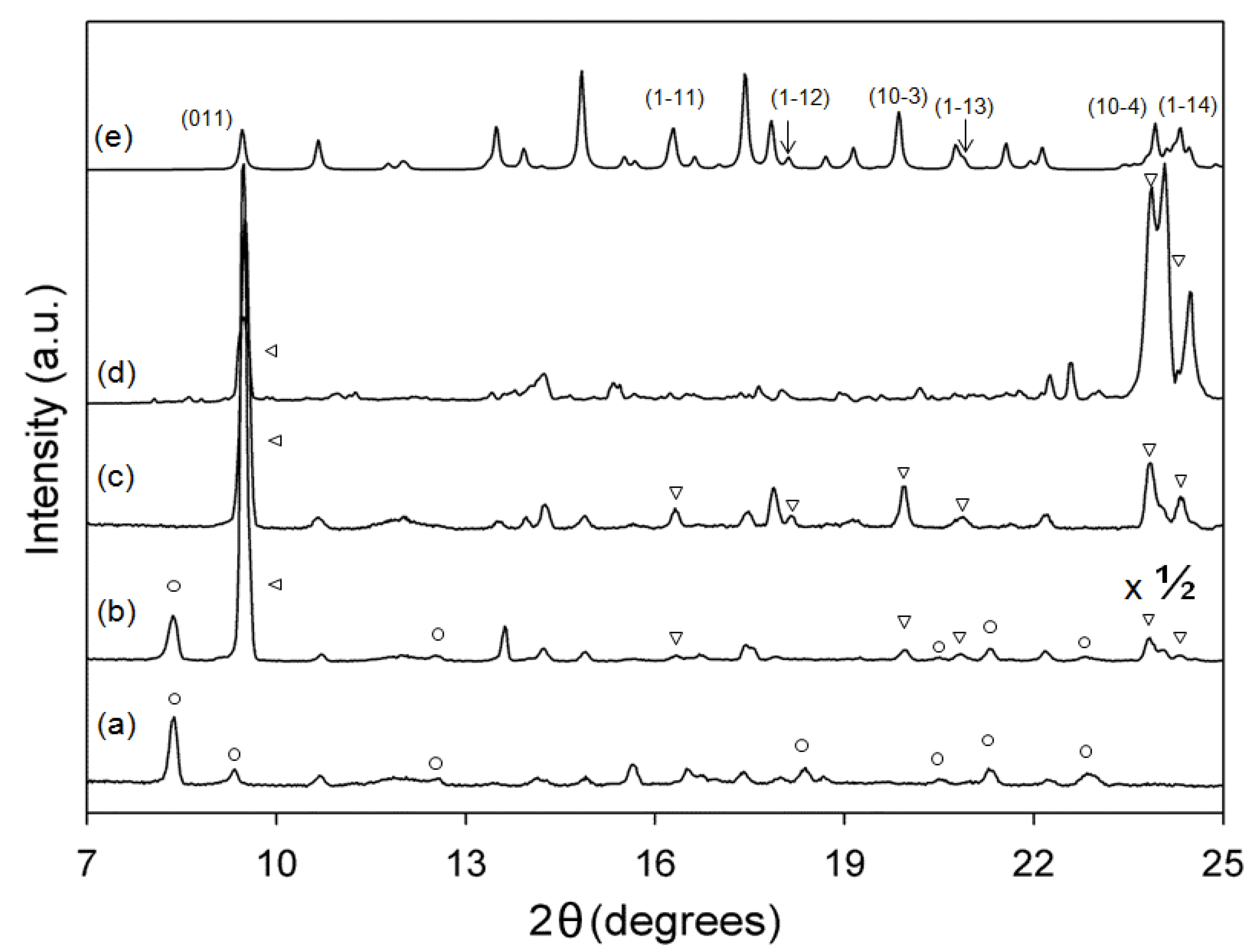
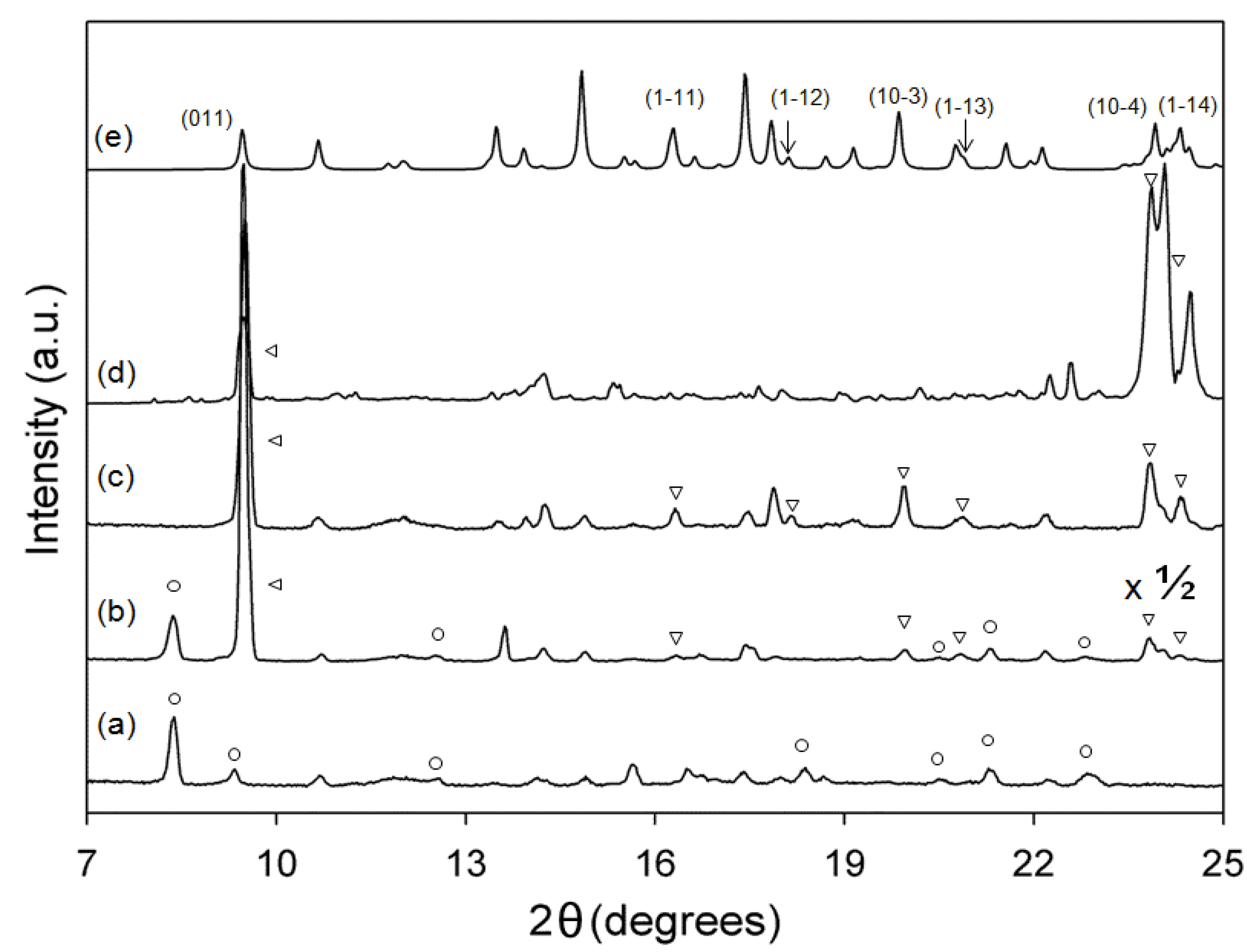
3.2. XRD Analysis
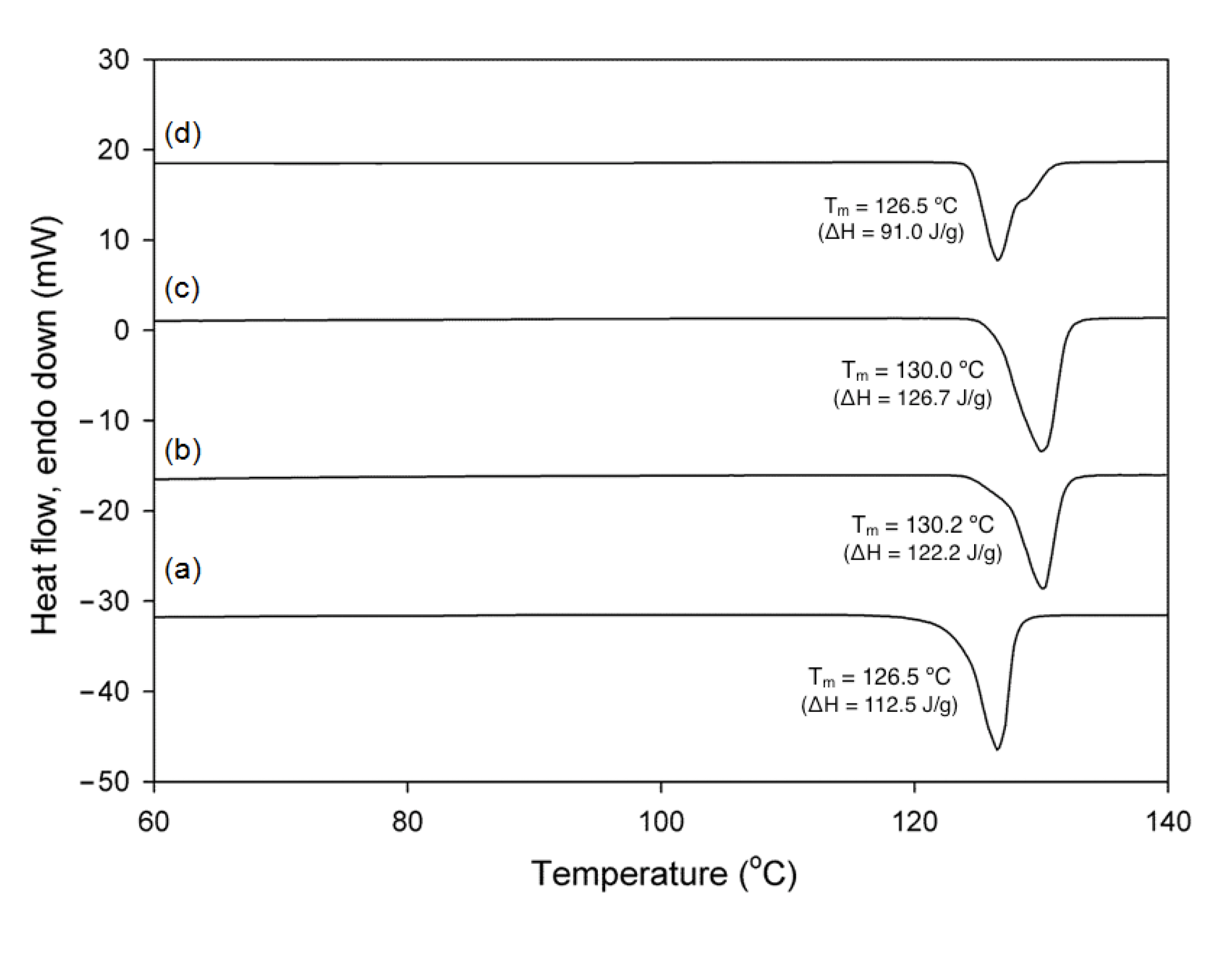
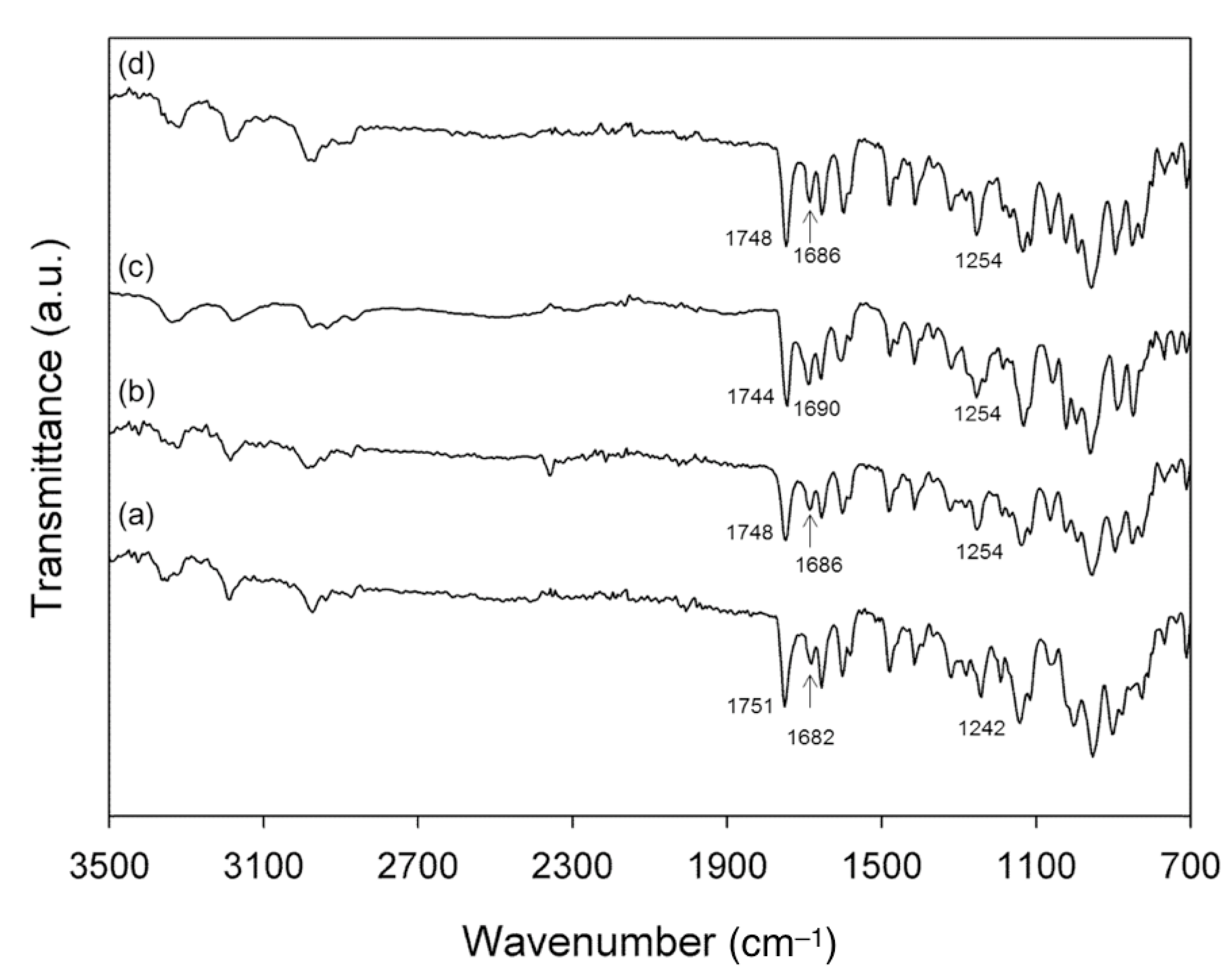
3.3. NMR, DSC and IR Analyses
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jones, W.; Motherwell, W.D.S.; Trask, A.V. Pharmaceutical cocrystals: An emerging approach to physical property enhancement. MRS Bull. 2006, 31, 875–879. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Physical stability enhancement of theophylline via cocrystallization. Int. J. Pharm. 2006, 320, 114–123. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, J.; Liu, Z.; Kan, H.; Zu, H.; Sun, W.; Zhang, J.; Qian, S. Coformer selection based on degradation pathway of drugs: A case study of adefovir dipivoxil-saccharin and adefovirdipivoxil-nicotinamide cocrystals. Int. J. Pharm. 2012, 438, 327–335. [Google Scholar] [CrossRef]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Jarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnel, E.; Park, A. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm. Res. 2012, 23, 1888–1897. [Google Scholar]
- Grobelny, P.; Mukherjee, A.; Desiraju, G.R. Drug-drug co-crystals: Temperature-dependent proton mobility in the molecular complex of isoniazid with 4-aminosalicylic acid. CrystEngComm 2011, 13, 4358–4364. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA, 2002; pp. 1–28. [Google Scholar]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989; pp. 25–49. [Google Scholar]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Solvent-drop grinding: Green polymorph control of cocrystallisation. Chem. Commun. 2004, 2004, 890–891. [Google Scholar]
- Babu, N.J.; Reddy, L.S.; Aitipamula, S.; Nangia, A. Polymorphs and polymorphic cocrystals of temozolomide. Chem. Asian J. 2008, 3, 1122–1133. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Sivachelvam, S.; Jones, W. Screening for polymorphs of cocrystals: A case study. CrystEngComm 2013, 15, 175–181. [Google Scholar] [CrossRef]
- Belcher, A.M.; Wu, X.H.; Christensen, R.J.; Hansma, P.K.; Stucky, G.D.; Morse, D.E. Control of crystal phase switching and orientation by soluble mollusc-shell proteins. Nature 1996, 381, 56–58. [Google Scholar] [CrossRef]
- Weiner, S.; Addadi, L. Design strategies in mineralized biological materials. J. Mater. Chem. 1997, 7, 689–702. [Google Scholar] [CrossRef]
- Kim, I.W.; Robertson, R.E.; Zand, R. Effects of some nonionic polymeric additives on the crystallization of calcium carbonate. Cryst. Growth Des. 2005, 5, 513–522. [Google Scholar] [CrossRef]
- Lang, M.; Grzesiak, A.L.; Matzger, A.J. The use of polymer heteronuclei for crystalline polymorph selection. J. Am. Chem. Soc. 2002, 124, 14834–14835. [Google Scholar] [CrossRef]
- Yin, Y.; Alivisatos, A.P. Colloidal nanocrystal synthesis and the organic-inorganic interface. Nature 2005, 437, 664–669. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.H.; Pouton, C.W. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef]
- Lee, M.K.; Lee, H.; Kim, I.W.; Lee, J. Novel polymorphic form of adefovir dipivoxil derived from polymer-directed crystallization. Pharmazie 2011, 66, 766–770. [Google Scholar]
- Jung, S.; Kim, I.W. Effects of polymers on the cocrystallization of adefovir dipivoxil and suberic acid. Polymer (Korea) 2013, 37, 663–668. [Google Scholar]
- Porter, W.W., III; Elie, S.C.; Matzger, A.J. Polymorphism in carbamazepine cocrystals. Cryst. Growth Des. 2008, 8, 14–16. [Google Scholar] [CrossRef]
- Jung, S.; Ha, J.-M.; Kim, I.W. Bis[(2,2-dimethylpropanoyloxy)methyl] {[2-(6-amino-9H-purin-9-yl)ethoxy]methyl}phosphonate–succinic acid (2/1). Acta Crystallogr. E 2012, 68, o809–o810. [Google Scholar] [CrossRef]
- Chung, J.; Kim, I.W. Effects of some polymeric additives on the cocrystallization of caffeine. J. Cryst. Growth 2011, 335, 106–109. [Google Scholar] [CrossRef]
- Cullity, B.D.; Stock, S.R. Elements of X-ray Diffraction, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2001; pp. 347–361. [Google Scholar]
- Mullin, J.W. Crystallization, 4th ed.; Butterworth-Heinemann: Oxford, UK, 2001; pp. 181–314. [Google Scholar]
- Sangwal, K. Additives and Crystallization Processes: From Fundamentals to Applications; John Wiley & Sons: Chichester, UK, 2007; pp. 1–176. [Google Scholar]
- Curran, D.P.; Hadida, S. Tris(2-(perfluorohexyl)ethyl)tin hydride: A new fluorous reagent for use in traditional organic synthesis and liquid phase combinatorial synthesis. J. Am. Chem. Soc. 1996, 118, 2531–2532. [Google Scholar] [CrossRef]
- Robins, M.J.; Sarker, S.; Wnuk, S.F. What are the practical limits for detection of minor nucleoside reaction products with HPLC (UV detection), H NMR, and TLC (UV detection)? Nucleo. Nucleot. 1998, 17, 785–790. [Google Scholar] [CrossRef]
- Cölfen, H.; Antonietti, M. Mesocrystals: Inorganic superstructures made by highly parallel crystallization and controlled alignment. Angew. Chem. Int. Ed. 2005, 44, 5576–5591. [Google Scholar] [CrossRef]
- Kim, R.; Kim, C.; Lee, S.; Kim, J.; Kim, I.W. In situ atomic force microscopy study on the crystallization of calcium carbonate modulated by poly(vinyl alcohol)s. Cryst. Growth Des. 2009, 9, 4584–4587. [Google Scholar] [CrossRef]
- Suzuki, M.; Shimanouchi, T. Infrared and Raman spectra of succinic acid crystal. J. Mol. Spectrosc. 1968, 28, 394–410. [Google Scholar] [CrossRef]
- Bullen, H.A.; Oehrle, S.A.; Bennett, A.F.; Taylor, N.M.; Barton, H.A. Use of attenuated total reflectance Fourier transform infrared spectroscopy to identify microbial metabolic products on carbonate mineral surfaces. Appl. Environ. Microbiol. 2008, 74, 4553–4559. [Google Scholar] [CrossRef]
- Sigma-Aldrich Catalog. Available online: http://www.sigmaaldrich.com/spectra/rair/RAIR001162.PDF (accessed on 10 November 2013).
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons: Chichester, UK, 2000; pp. 10815–10837. [Google Scholar]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.R. Introduction to Spectroscopy, 4th ed.; Brooks/Cole: Belmont, CA, USA, 2008; p. 56. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, S.; Ha, J.-M.; Kim, I.W. Phase Transformation of Adefovir Dipivoxil/Succinic Acid Cocrystals Regulated by Polymeric Additives. Polymers 2014, 6, 1-11. https://doi.org/10.3390/polym6010001
Jung S, Ha J-M, Kim IW. Phase Transformation of Adefovir Dipivoxil/Succinic Acid Cocrystals Regulated by Polymeric Additives. Polymers. 2014; 6(1):1-11. https://doi.org/10.3390/polym6010001
Chicago/Turabian StyleJung, Sungyup, Jeong-Myeong Ha, and Il Won Kim. 2014. "Phase Transformation of Adefovir Dipivoxil/Succinic Acid Cocrystals Regulated by Polymeric Additives" Polymers 6, no. 1: 1-11. https://doi.org/10.3390/polym6010001