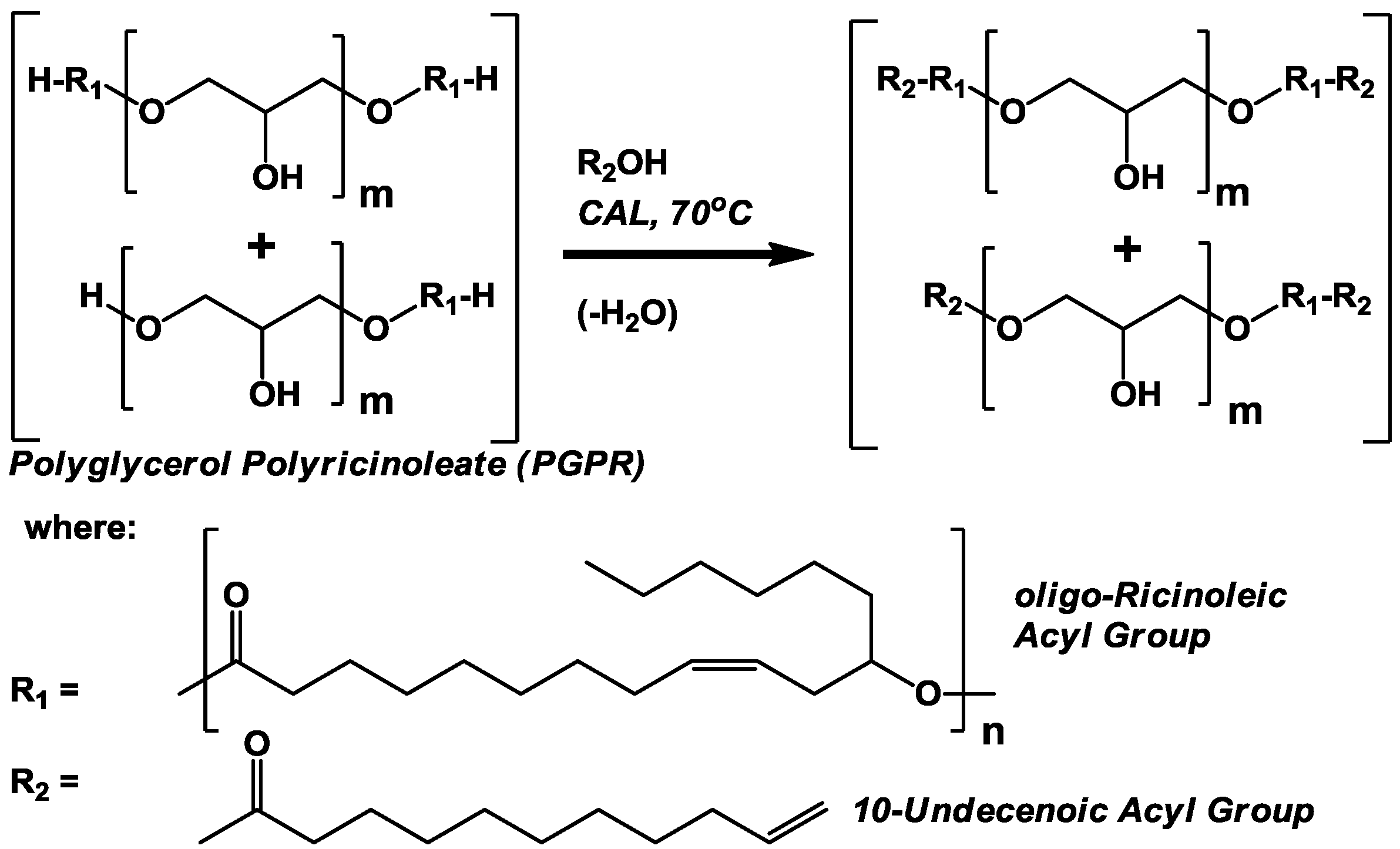
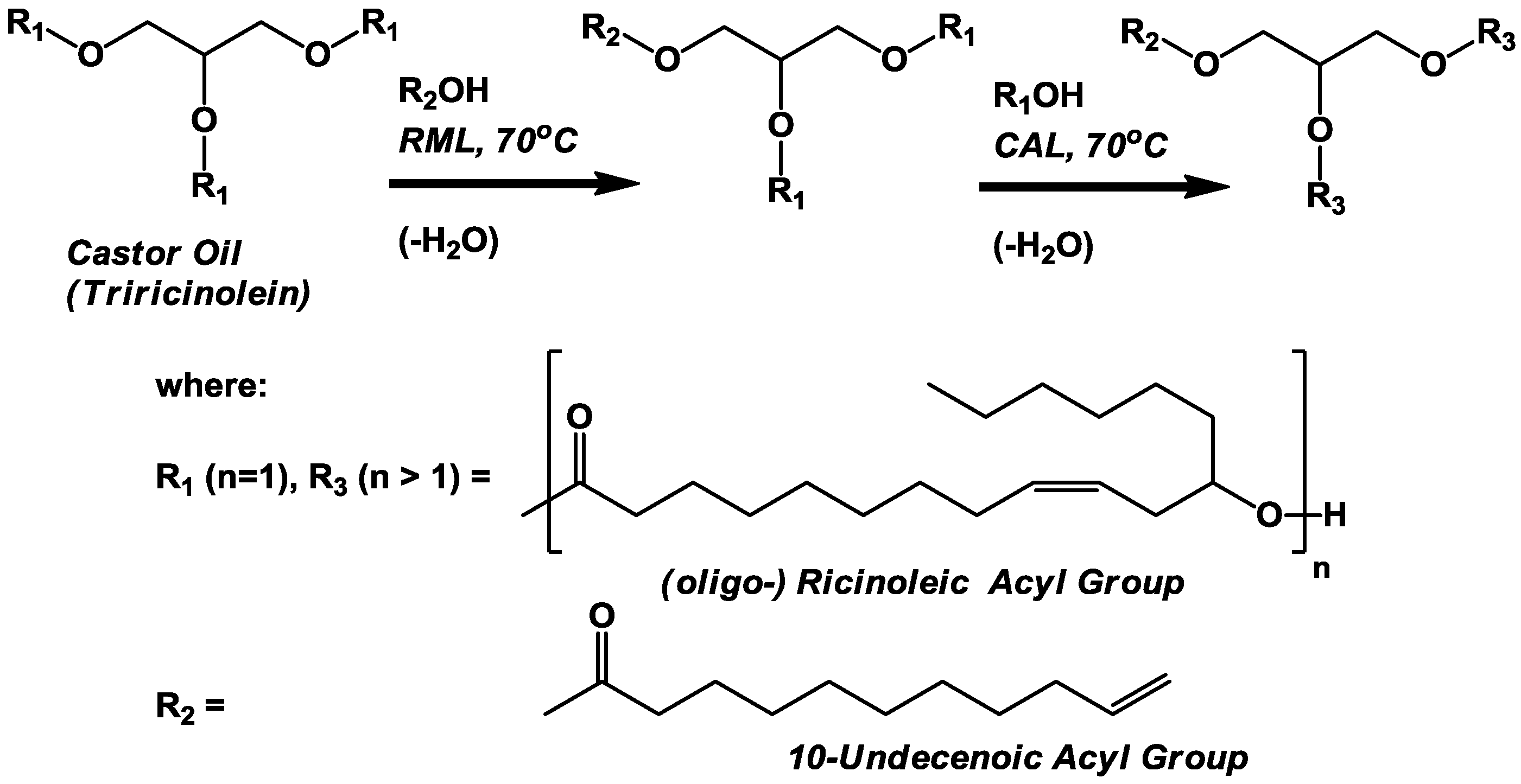
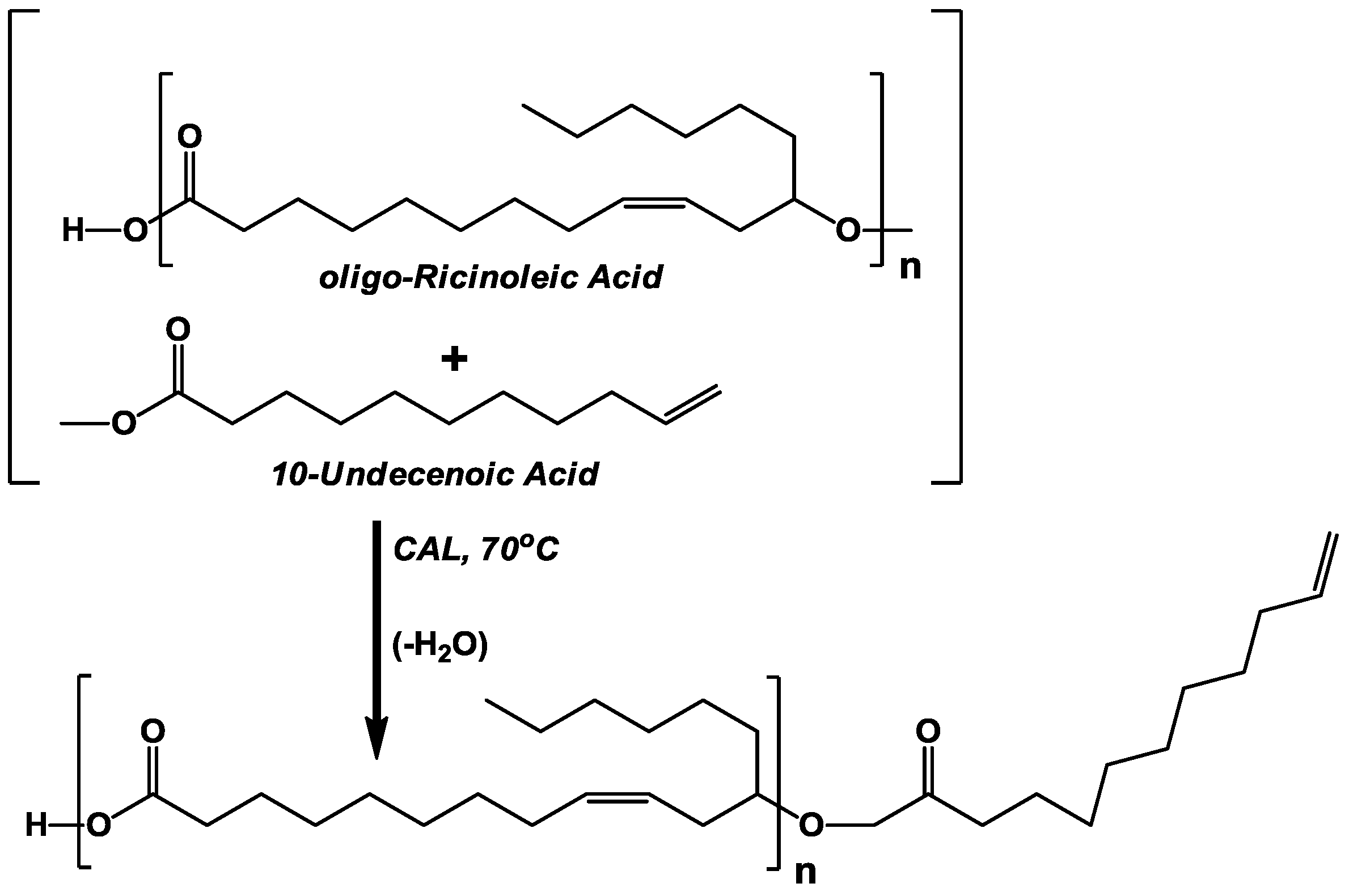
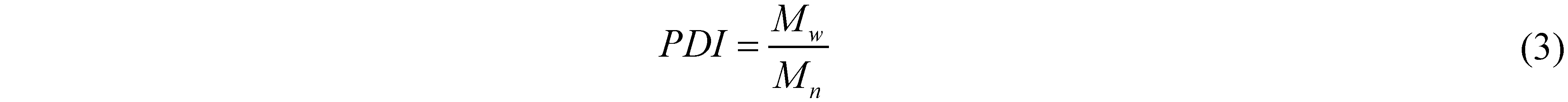The objective of this investigation is to incorporate 10-undecenoic acyl groups into
oligo-ricinoleic acid derivatives. Three approaches were employed. The first two, involving the esterification of undecenoic acid to the -OH end group of
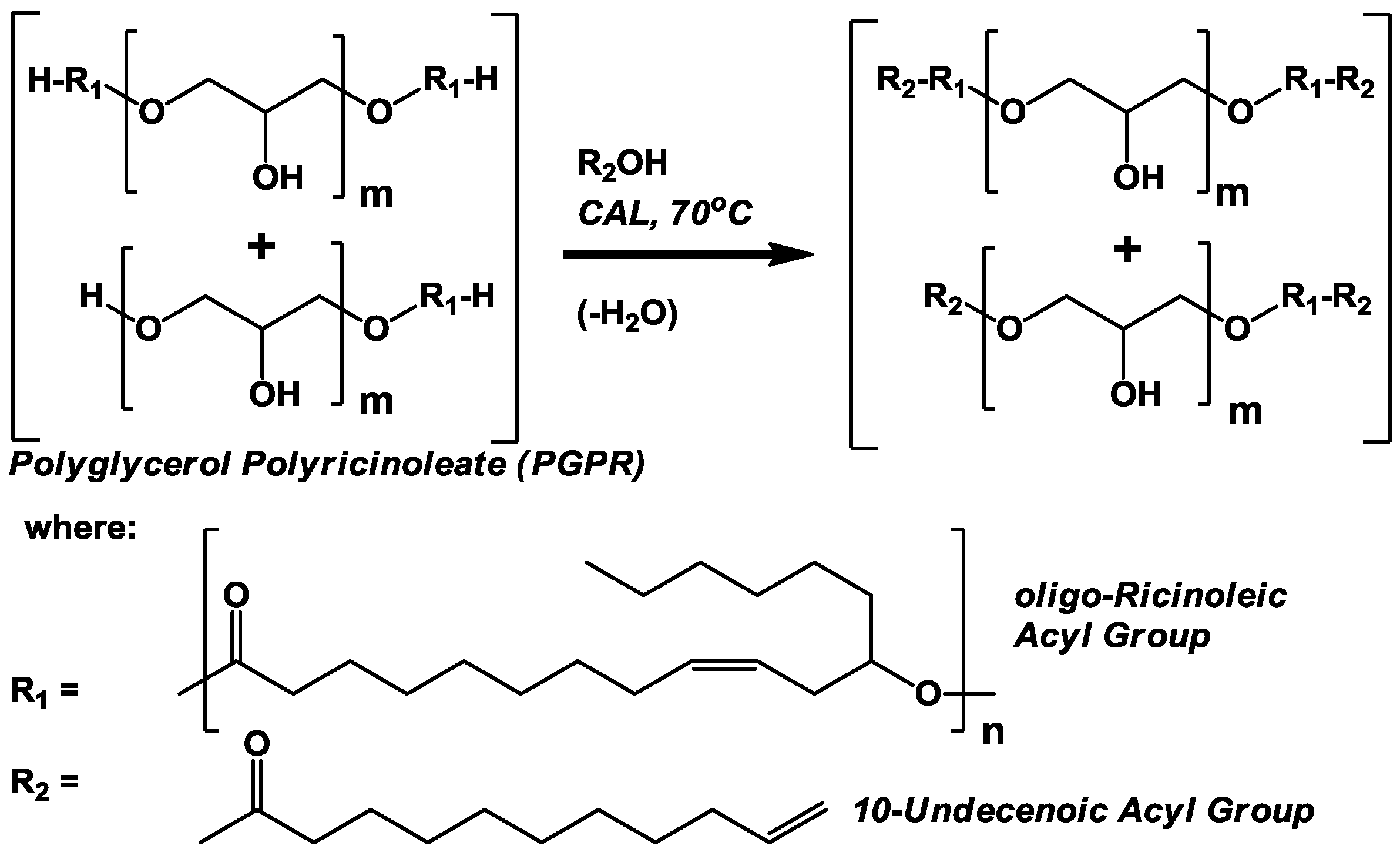
oligo-ricinoleic acyl chains, either as homopolymers or attached to polyglycerol (
i.e., PGPR),
Scheme Iand
Scheme II, respectively, were performed successfully. The second approach, the formation of
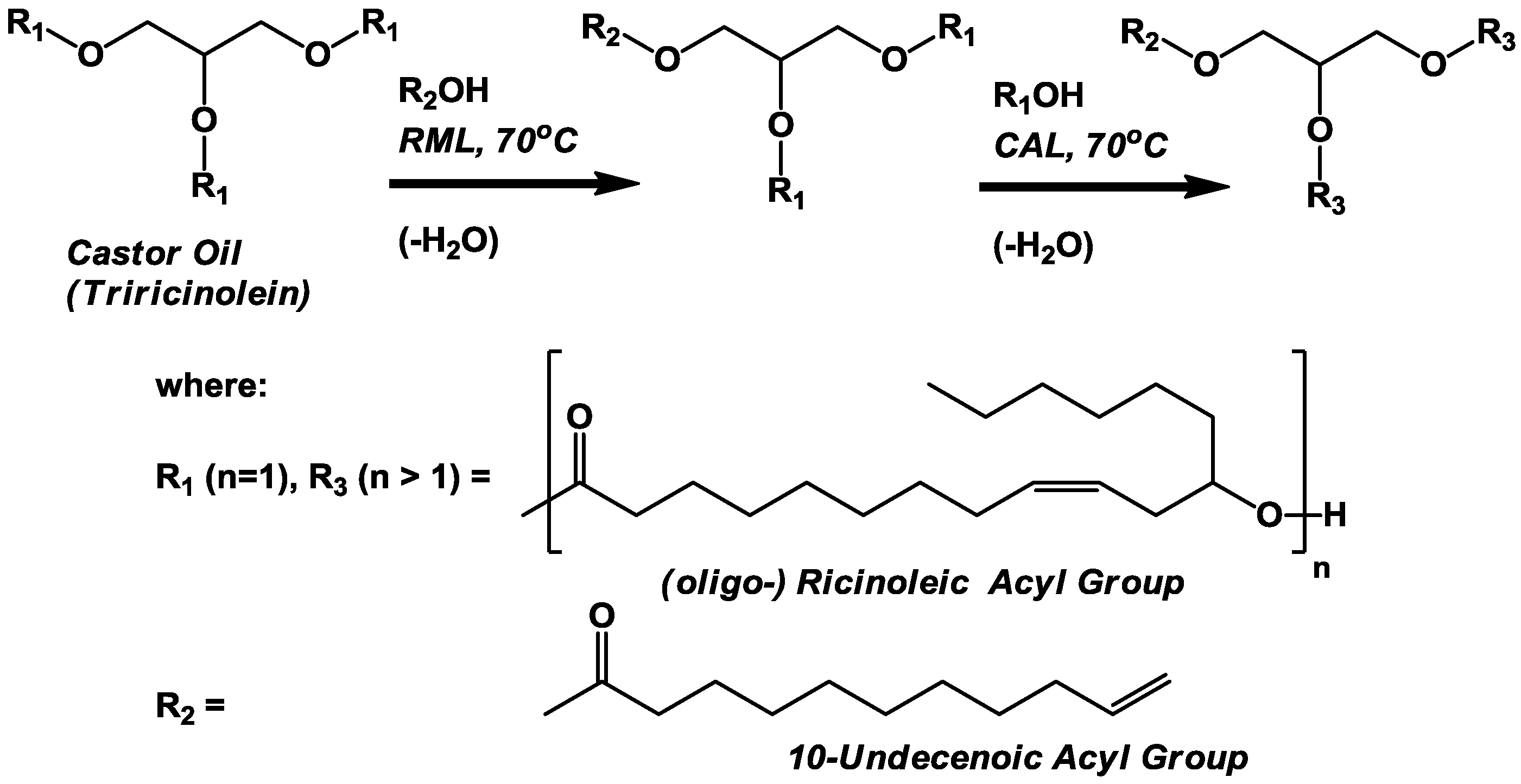
oligo-ricinoleyl, undecenoyl mixed TAG (
Scheme III) was successful to a lesser extent. The analysis of the reactions was challenging due to the several different co-products that could form, and weak detector signal exhibited by undecenoic acid when analyzed by GPC and HPLC. Moreover, undecenoic acid’s detector signal was approximately two orders of magnitude lower than signals for ricinoleic acid-containing analytes under most of the situations encountered in this investigation. Therefore, additional chemical analyses (AV and NMR) were required. The analysis of each reaction will now be discussed separately.
3.1. CAL-Catalyzed Esterification of oligo-Ricinoleic Acid and 10-Undecenoic Acid (Scheme I)
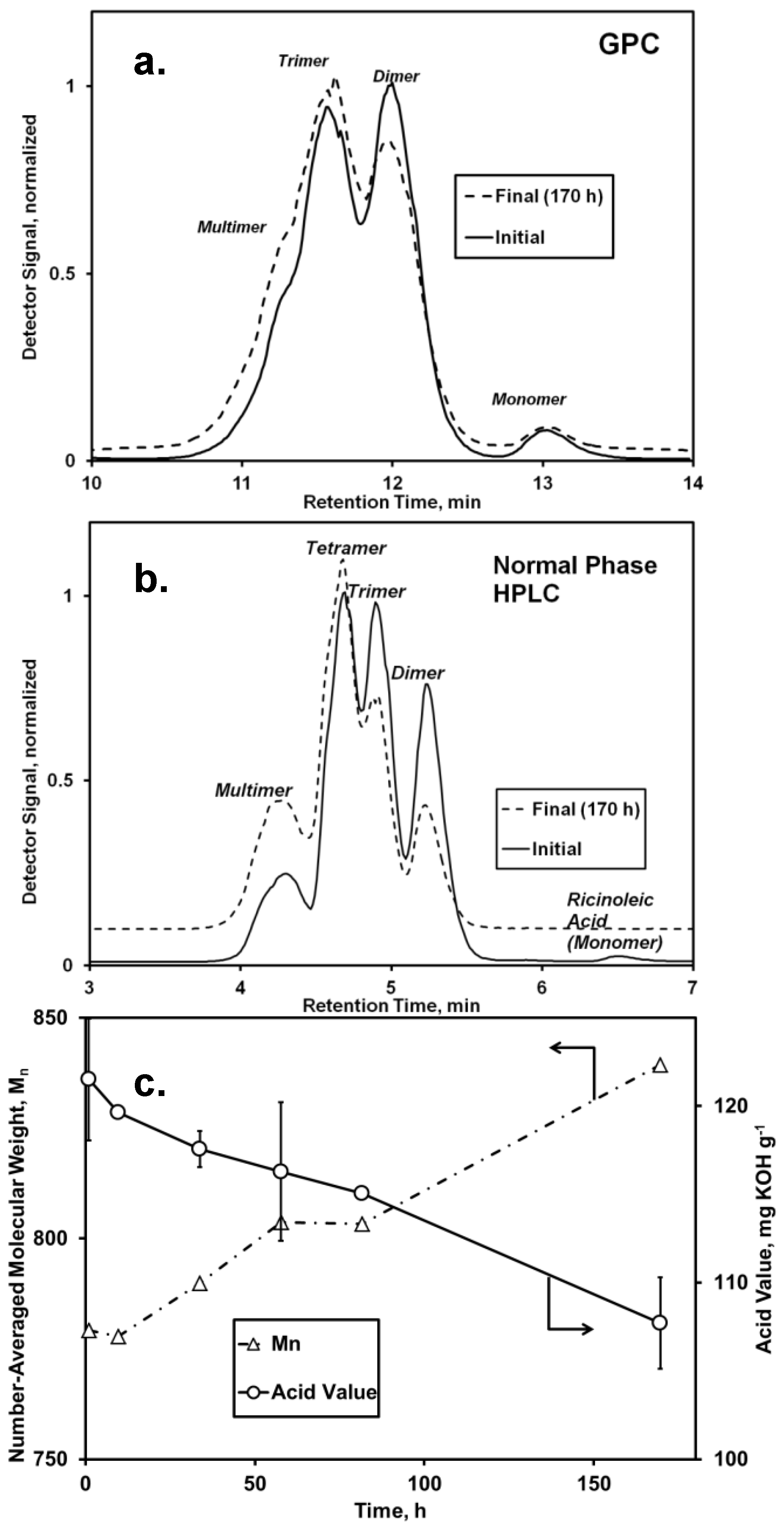
Based on the modestly successful esterification of lauric acid to
oligo-ricinoleic acid in our previous work [
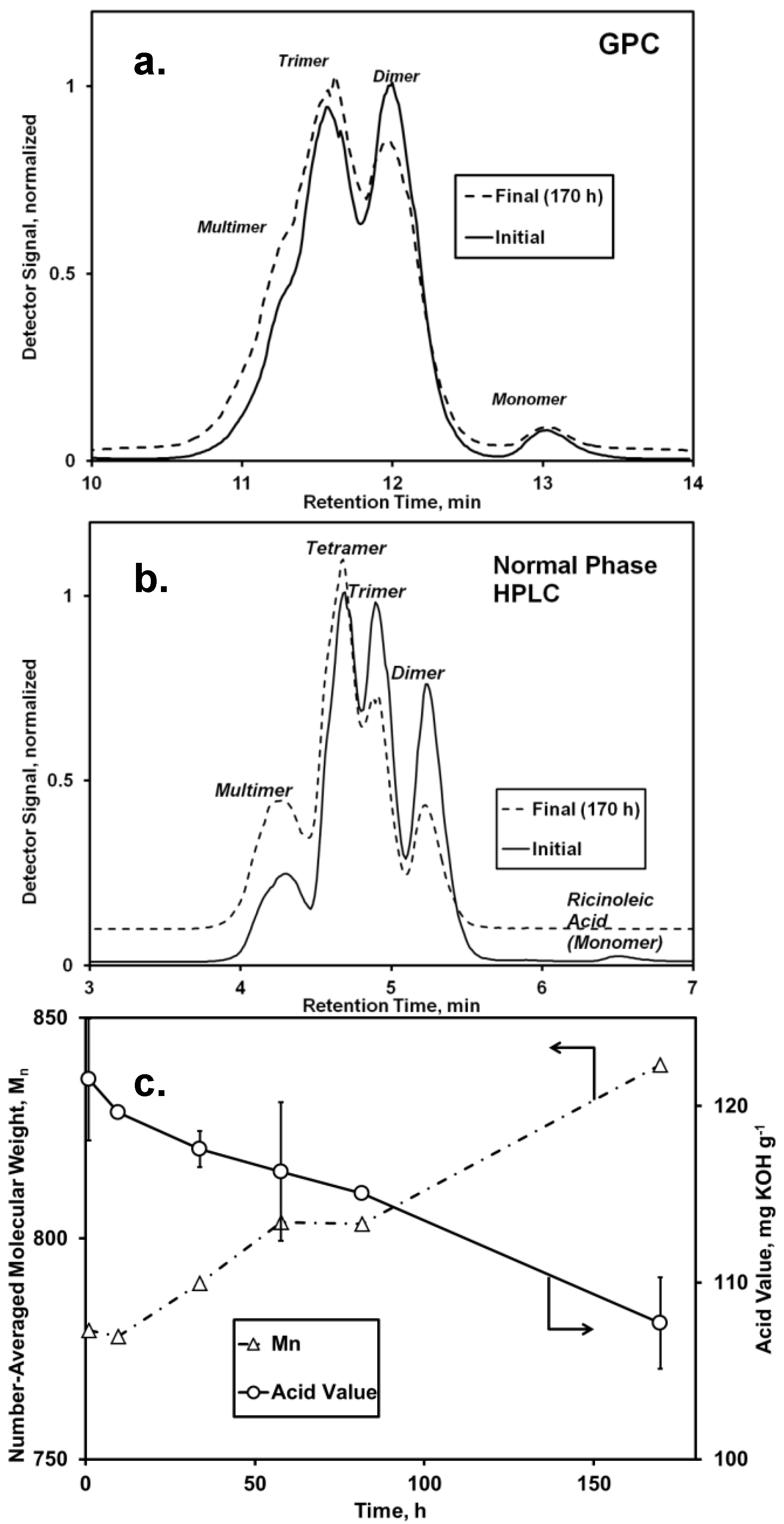
17], it was anticipated that the attachment of 10-undecenoic acid to -OH end groups would also occur readily. Both GPC and normal phase HPLC analysis demonstrated a slight increase of molecular weight, shown by the growth of peaks representing tetramers and multimers and a reduction in the peak area corresponding to monomers and dimers (
Figure 1). Concurrently, the AV decreased from 121 to 107 mg KOH g
−1 (
Figure 1c), with the decrease attributed mainly to consumption of undecenoic acid, since the amount of free ricinoleic acid initially was <3 wt %. Formation of free ricinoleic acid via hydrolysis or chain scission probably did not occur. Moreover, the fraction of ricinoleic acyl groups that participated in intermolecular ester bonds did not change during the time course of reaction, as determined from
1H-NMR analysis using bands at 4.80 and 5.25–5.57 ppm to represent the fraction esterified and total number of ricinoleic acyl groups, respectively. The absence of chain scission for
oligo-ricinoleic acid during its biocatalytic modification was previously reported by us [
9]. (DP could not be calculated accurately by
1H-NMR due to the high degree of uncertainty in integrating the band at 3.6 ppm attributable to free OH end groups.) Therefore, under this assumption, the decrease in AV of 13.8 mg KOH g
−1 represents a 28% conversion of undecenoic acid. In agreement, the increase of M
n for the oligomer, 61 g·mol
−1 (
Figure 1c), equates to 33% of undecenoic acid’s molecular weight,
i.e., a 33% conversion of the latter. This level of conversion strongly agrees with the incorporation of lauric acid onto
oligo-ricinoleic acid reported previously by us, 34% [
17]. Both the reactions to incorporate undecenoic acid and lauric acid into
oligo-ricinoleic acid contained 12–13 wt% of unreacted medium-chain free fatty acid (FFA) in the final product. PDI values remained low throughout the time course of reaction, residing between 1.1 and 1.2. Further research is needed to determine if the percent incorporation of undecenoic acid can be increased without increasing the final FFA concentration, perhaps by adding undecenoic acid into the reaction mixture in small batchwise increments throughout the time course of reaction, and/or by applying a more effective means of water removal than free evaporation, such as high vacuum pressure [
7,
8]. (It is known that the small amount of non-hydroxy fatty acid present in technical grade ricinoleic acid is readily incorporated as end groups for
oligo-ricinoleic acid [
9], suggesting the former approach may be successful.)
Figure 1.
Analysis of the
Candida antarctica lipase (CAL)-catalyzed esterification of
oligo-ricinoleic acid and 10-undecenoic acid (
Scheme I). Chromatograms: (
a) gel permeation chromatography (GPC) and (
b) normal phase HPLC of the reaction mixture and initial and final conditions (170 h); (
c) Number-averaged molecular weight (
Mn), obtained from GPC and acid value (AV)
vs. time. Reaction conditions: 10-undecenoic acid/
oligo-ricinoleic acid 0.20 g·g
−1 (0.32 mol·mol
−1); 7.1% w/w CAL, 69 °C.
Figure 1.
Analysis of the
Candida antarctica lipase (CAL)-catalyzed esterification of
oligo-ricinoleic acid and 10-undecenoic acid (
Scheme I). Chromatograms: (
a) gel permeation chromatography (GPC) and (
b) normal phase HPLC of the reaction mixture and initial and final conditions (170 h); (
c) Number-averaged molecular weight (
Mn), obtained from GPC and acid value (AV)
vs. time. Reaction conditions: 10-undecenoic acid/
oligo-ricinoleic acid 0.20 g·g
−1 (0.32 mol·mol
−1); 7.1% w/w CAL, 69 °C.
3.2. CAL-Catalyzed Esterification of Polyglyerol Polyricinoleate (PGPR) and 10-Undecenoic Acid (Scheme II)
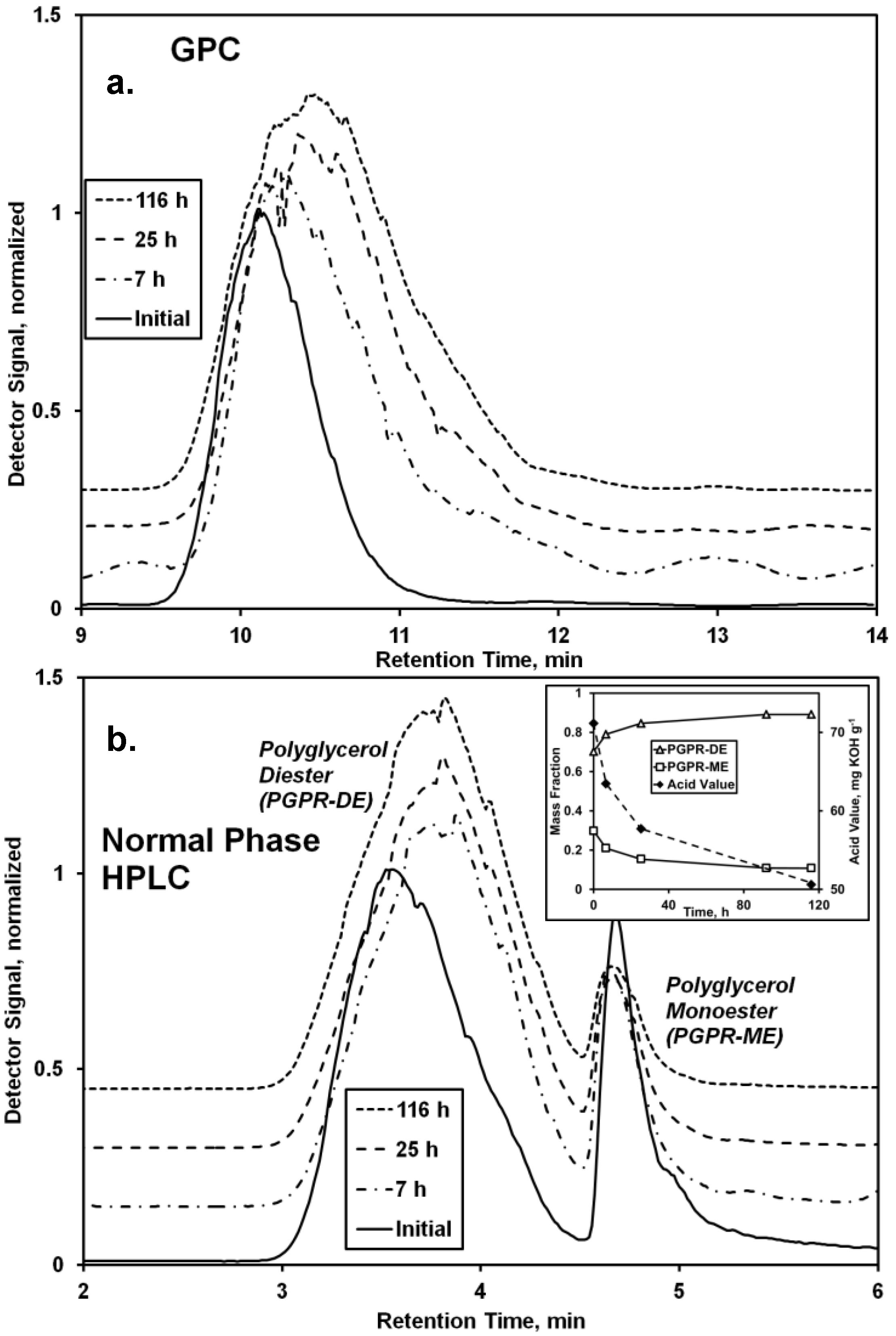
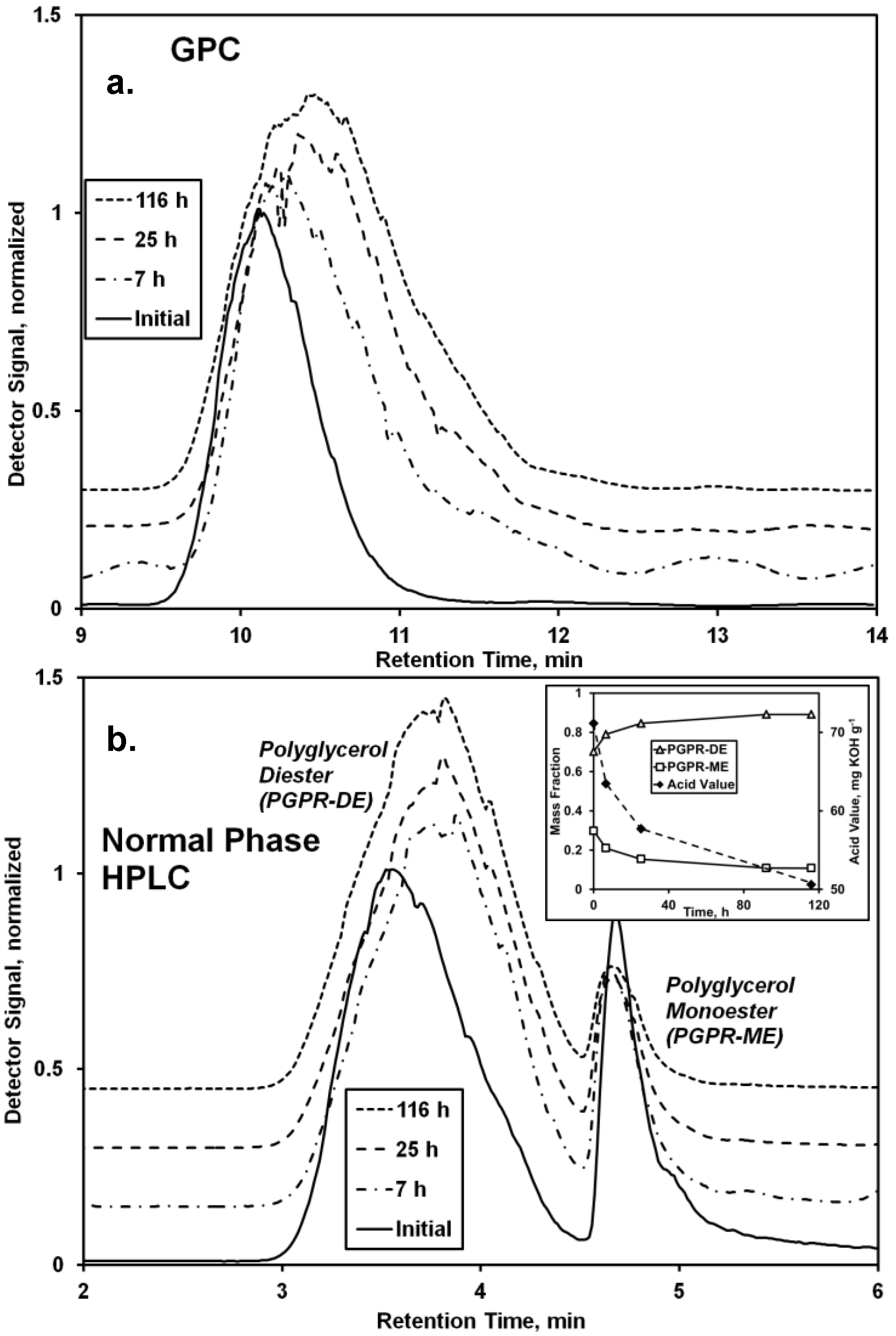
Chromatographic analyses demonstrate the reaction between PGPR (prepared using lipases under solvent-free conditions [
7,
8]) and undecenoic acid greatly affected the chemical nature of the polymer (
Figure 2). Normal phase HPLC provided the more useful data for interpretation, demonstrating the existence of two main bands for the starting material, a main and broad band between 3.0 and 4.5 min retention time and a second sharper band at 4.5–5.0 min. It is believed the former and latter represent di- and mono-esters of polyglycerol and polyricinoleic acid, respectively. The larger number of free hydroxyls of the monoester promote its increased polarity, hence its longer retention on the polar silica HPLC column. From a previous series of experiments performed by the authors examining the covalent attachment of
oligo-ricinoleic acid to mono- di-, and tri-glycerol, it appears that the primary, or endgroup, hydroxyls of polyglycerol are the principal acyl acceptors [
20]. Moreover, our research group has observed that secondary hydroxyls of polyols are poor acyl acceptors for
oligo-hydroxy fatty acids. Therefore, it is probable that ester bonds between polyglycerol and
oligo-ricinoleic acid form only at the former’s primary hydroxyl end groups.
The normal phase HPLC chromatogram reflects a decrease of the percentage of the monoester from 30% to 11% (
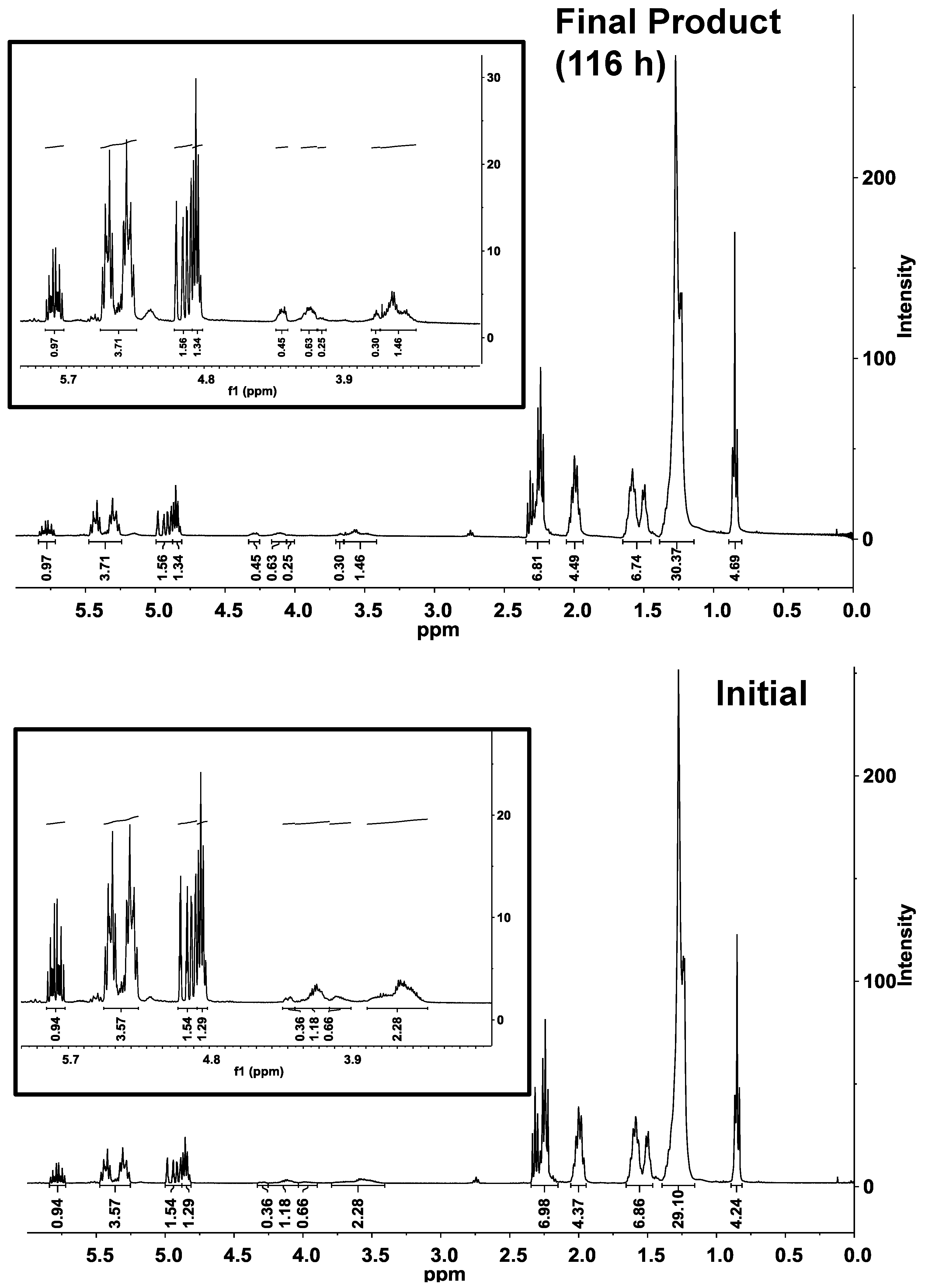
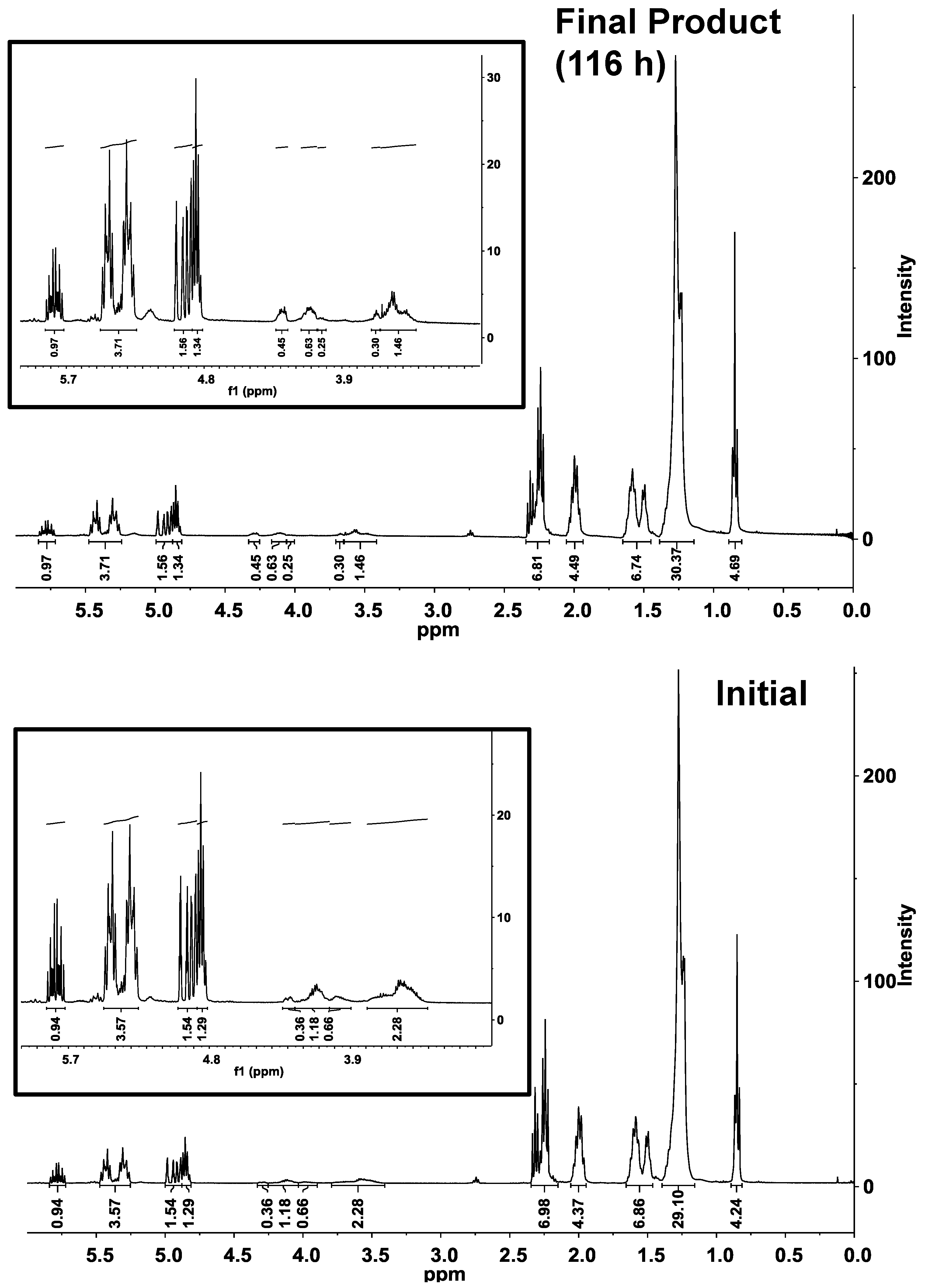
Figure 2b, inset), and a concurrent increase of the diester percentage. Consistent with the conversion of monoester to diester is an increase of intensity for bands between 4.0 and 4.3 ppm, consistent with ester formation by the -OH groups of the polyglycerol moiety (
Figure 3). The normal-phase HPLC diester peak shifted to slightly higher retention times as the reaction progressed, indicating increased polarity. This is attributed to the formation of
oligo-ricinoleic acid-polyglycerol-undecenoic acid diester (
Scheme II), anticipated to be more polar than PGPR diester.
During the time course of reaction, the AV decreased from 71 to 51 mg KOH g
−1, reflecting the consumption of undecenoic acid (
Figure 2b, inset). The measured AV for the initial reaction medium (71 mg KOH g
−1) is reasonably close to a calculated value based on the initial amount of undecenoic acid (20 wt %; 60.9 mg KOH g
−1), plus the AV reported for PGPR (5.3 mg KOH g
−1), or, 65.2 mg KOH g
−1. The decrease of 20 mg KOH g
−1 is equivalent to a 31% consumption of undecenoic acid.
GPC analysis reveals a single peak for PGPR which shifts to higher retention times during the time course of reaction (
Figure 2a). Also, the peak broadens slightly, shown by an increase of the PDI from 1.05 to 1.12. Typically, an increase of retention time would reflect a decrease of molecular weight. However, events that would lead to a decrease of molecular weight, such as chain scission or hydrolysis, appear to be absent. First, peaks representing ricinoleic acid and its dimer are absent in the chromatogram obtained by normal phase HPLC,
Figure 2b (
cf.
Figure 1b). Second, the area underneath
1H-NMR bands at 4.8 ppm and 5.25–5.57 ppm, representing the fraction esterified and total number of ricinoleic acyl groups, respectively, do not change appreciably (
Figure 3), similar to that discussed in the previous subsection. Third, the absence of major GPC chromatographic peaks associated with ricinoleic acid and its oligomers, 10.6–13.5 min (
Figure 1a), further supports this hypothesis (
Figure 2a). The shift in retention time may result from major differences in conformation of the biopolymer in solution resulting from the conjugation of undecenoic acyl groups to the polyglycerol moiety’s -OH groups.
The results are inconclusive whether the desired reaction, attachment of undecenoic acid to the hydroxyl endgroups of oligo-ricinoleic acyl chains occurred or not. However, the unintended result, the apparent attachment of FFA onto free hydroxyls of the polyglycerol moiety, may lead to improved homogeneity of the material’s hydrophilic-lipophilic balance, hence to improved performance, of PGPR prepared enzymatically for its employment as a food or pharmaceutical emulsifier.
Figure 2.
Analysis of the CAL-catalyzed modification of polyglycerol polyricinoleate (PGPR) with 10-undecenoic acid. Chromatograms from (a) GPC and (b) normal phase HPLC analysis of the reaction mixture and initial and final conditions (170 h) are depicted. Figure b inset shows the change of oligo-fatty acid-polyglycerol mono- and di-esters and acid value (AV) vs. time. Reaction conditions: PGPR (2.48 g) was mixed with 10-undecenoic acid (0.62 g, 3.4 mmol) and CAL (11 wt %). The solvent-free mixture was magnetically stirred at 68 °C.
Figure 2.
Analysis of the CAL-catalyzed modification of polyglycerol polyricinoleate (PGPR) with 10-undecenoic acid. Chromatograms from (a) GPC and (b) normal phase HPLC analysis of the reaction mixture and initial and final conditions (170 h) are depicted. Figure b inset shows the change of oligo-fatty acid-polyglycerol mono- and di-esters and acid value (AV) vs. time. Reaction conditions: PGPR (2.48 g) was mixed with 10-undecenoic acid (0.62 g, 3.4 mmol) and CAL (11 wt %). The solvent-free mixture was magnetically stirred at 68 °C.
Figure 3.
1H-NMR analysis of the CAL-catalyzed esterification of polyglycerol polyricinoleate (
PGPR) and 10-undecenoic acid under initial and final conditions. Inset shows expanded view of 3–6 ppm region. Reaction conditions are given in
Figure 2.
Figure 3.
1H-NMR analysis of the CAL-catalyzed esterification of polyglycerol polyricinoleate (
PGPR) and 10-undecenoic acid under initial and final conditions. Inset shows expanded view of 3–6 ppm region. Reaction conditions are given in
Figure 2.
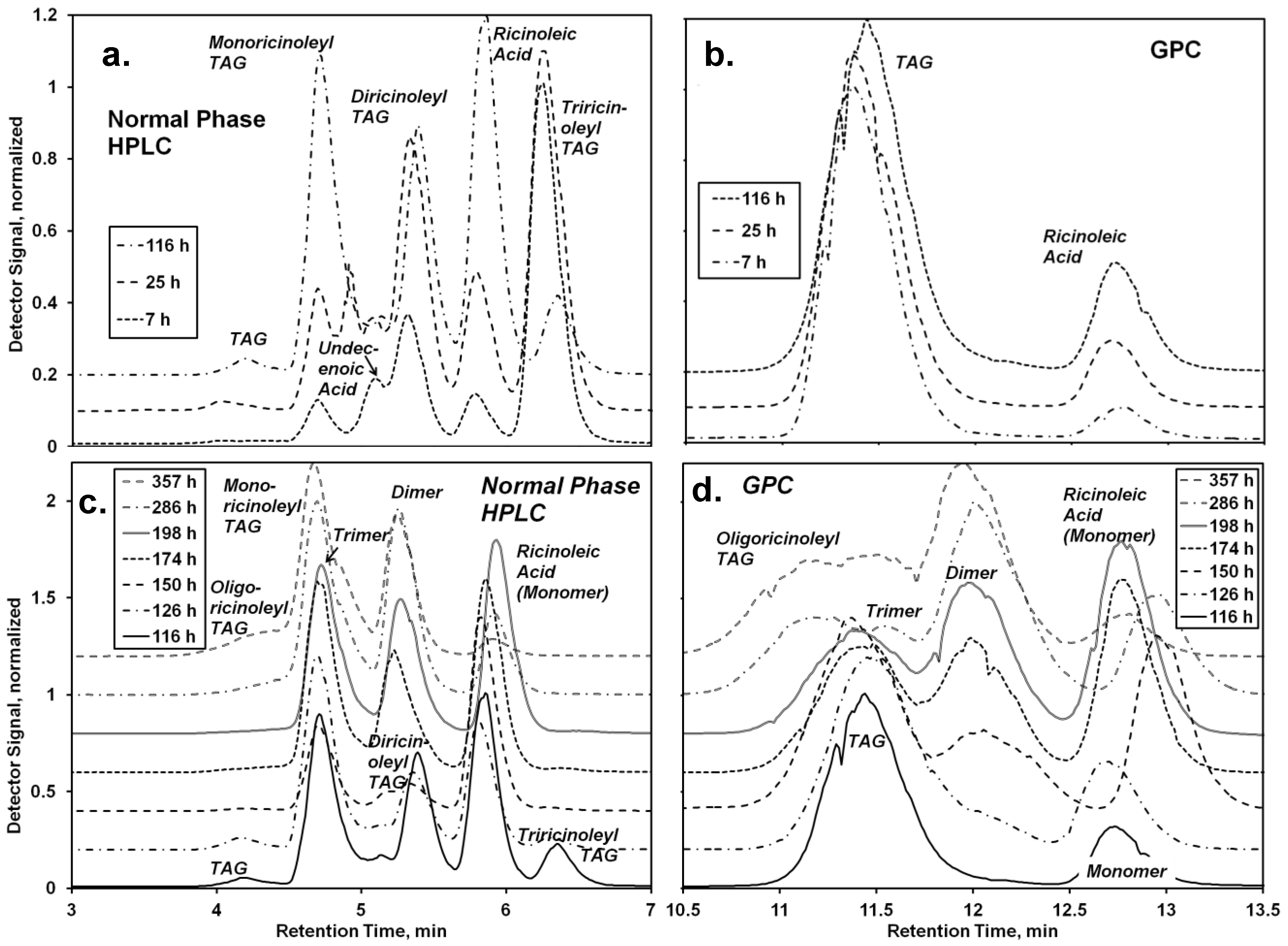
3.3. Lipase-Catalyzed Synthesis of oligo-Ricinoleyl, Undecenoyl TAG (Scheme III)
The solvent-free synthesis of
oligo-ricinoleyl, undecenoyl TAG entailed two steps, the 1,3-selective lipase (RML)-catalyzed interesterification of castor oil (consisting of >80% triricinolein and the remainder primarily as diricinoleyl TAG) with undecenoic acid, followed by the CAL-catalyzed formation and elongation of
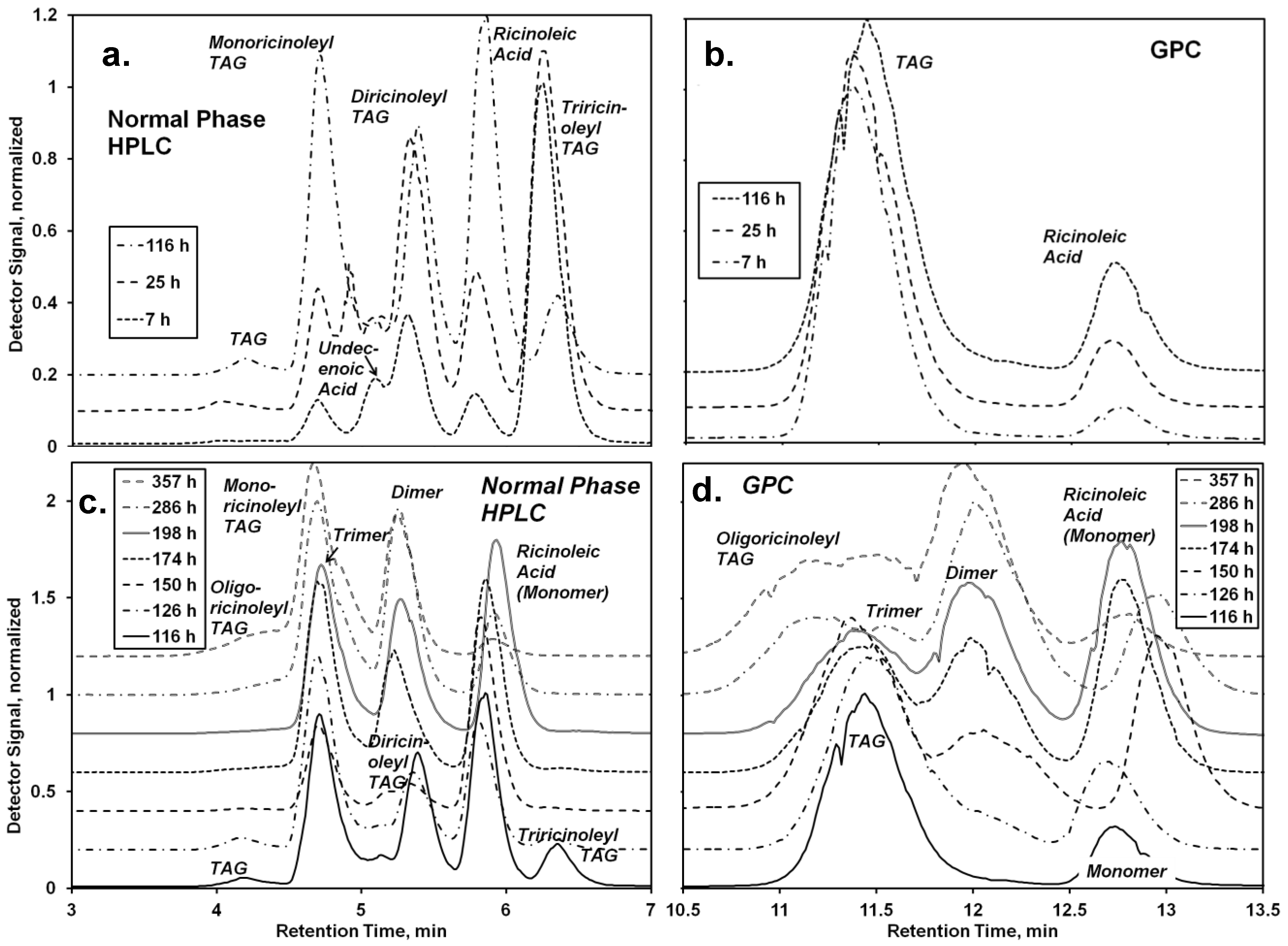
oligo-ricinoleyl acyl groups in TAG molecules. For the first step, a mole ratio of 1:1 for undecenoic acid to TAG acyl groups was employed initially; moreover a 50 mol% excess of undecenoic acid per acyl groups in the 1- and 3-TAG position was employed. These conditions would drive acidolysis in the forward position, to produce a mixture of monoundecenoyl and diundecenoyl TAGs. Normal-phase HPLC and GPC chromatograms for the first step are contained in
Figure 4a,b, respectively. The former method provided very good resolution of non-, mono-, di- and tri-hydroxylacyl (
i.e., -ricinoleyl) TAG, and ricinoleic acid. Peak positions for the former three were confirmed using
Lesquerella fendleri oil as a standard, due to its prominence of mono- and di-hydoxyacyl TAG [
21]. Undecenoic acid appears as a shoulder at 5.0 min, but disappears during the time course of reaction due in part to its consumption, and to the incommensurately large decrease of its detector response with a small decrease of concentration (detector signal
α concentration
5.5). The undecenoic acid concentration was calculated from the AV after subtracting the contribution attributable to ricinoleic acid, obtained chromatographically (
Figure 4a,b). Of note, the absence of
oligo-ricinoleic acyl groups in the TAG or in free acyl form was confirmed from chromatographic and NMR analyses, consistent with a previously study demonstrating the inability of RML to catalyze
oligo-ricinoleic acid, and to utilize secondary hydroxyl acyl acceptors [
9].
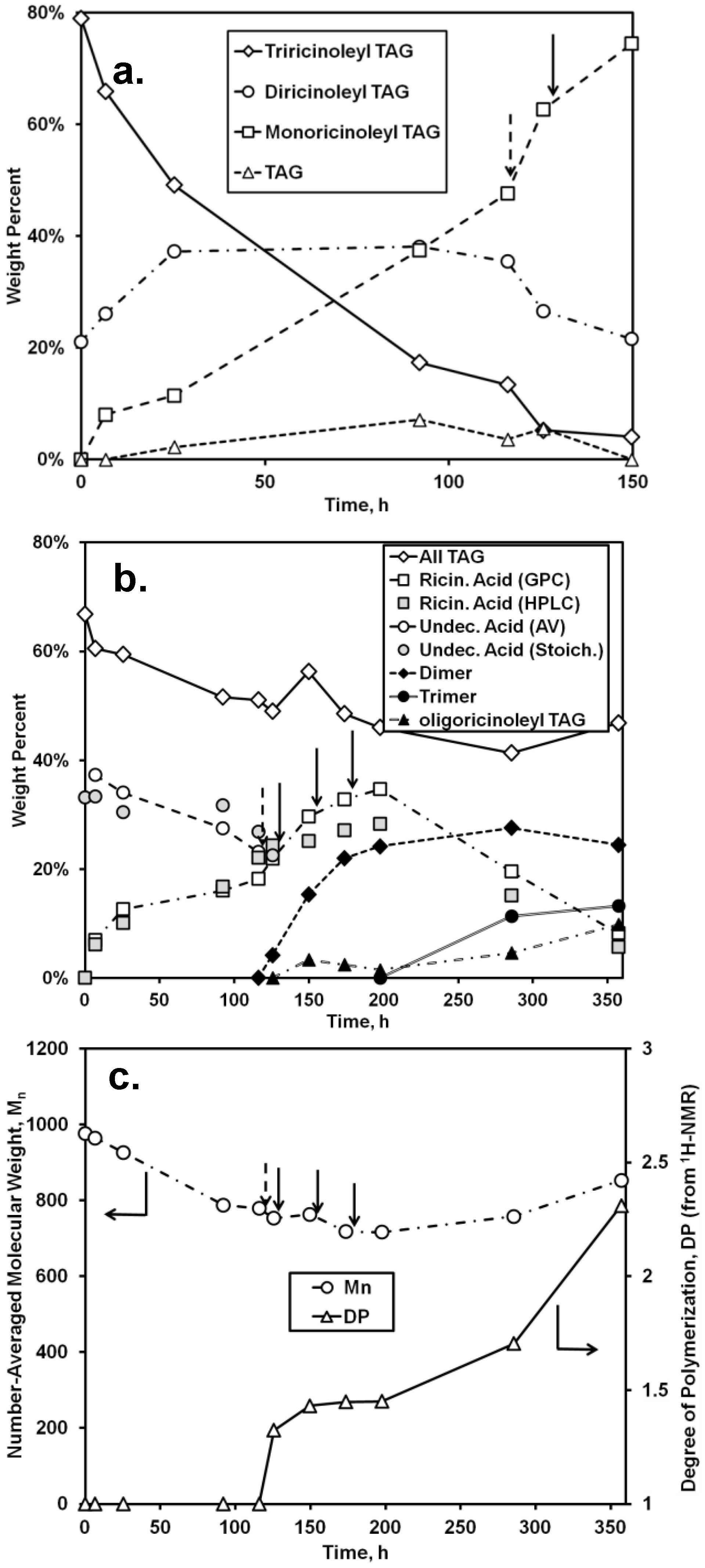
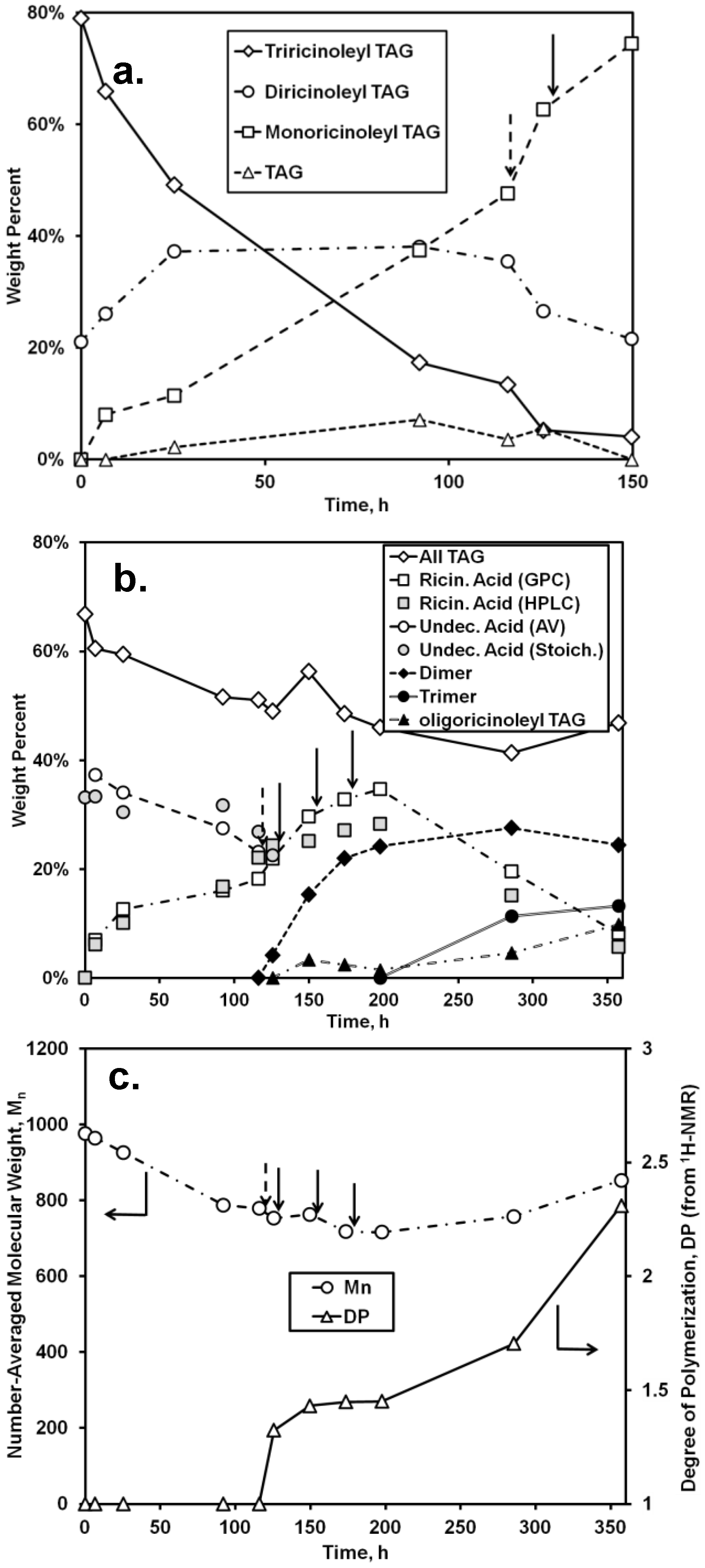
Quantitative analysis of the chromatographic and AV data for the first stage of the two-step synthesis (0–116 h) and the first portion of the second stage (up to 150 h) is given in
Figure 5. As shown in
Figure 5a, the relative amount of triricinoleyl TAG decreased, accompanied by a successive growth of diricinoleyl TAG, in agreement with the measured decrease of the undecenoic acid and increase of ricinoleic acid that would occur during of acidolysis. However, the relative amount of diricinoleyl TAG reached a plateau and then decreased as the reaction time increased past 100 h, due to its conversion into monoricinoleyl TAG. In addition, the amount of TAG containing no ricinoleyl groups slowly increased, probably reflecting the occurrence of acyl migration, where neutral lipids undergo isomerization, with acyl exchange occurring between the 2- and 1- + 3-positions on the glycerol backbone (thereby allowing for undecenoic acyl groups at the 1- and/or 3-glycerol positions to migrate to the 2-positon). The shift to higher retention times for the TAG peak of the GPC chromatogram (
Figure 4b), representing a decrease of molecular weight, is consistent with the replacement of the higher molecular weight ricinoleic acyl groups with lower molecular weight undecenoic acyl groups attached to glycerol. There is good agreement between HPLC and GPC-derived estimates of the released ricinoleic acid, and between AV-derived concentration of undecenoic acid and the latter’s concentration calculated based on the moles of released ricinoleic acid, equated to moles of undecenoic acid incorporated into TAG. The approximate conversion of undecenoic acid for the first stage of reaction is therefore 38%. The latter calculation assumes that hydrolysis of TAG did not occur at appreciable levels, an assumption supported by chromatographic analysis which showed the absence of partial glycerides. In addition,
1H-NMR spectra did not contain bands in the 3.7–4.0 range, attributable to free -OH groups for glycerol. In sum, RML-catalyzed acidolysis of castor oil on a 30 g scale occurred successfully, consistent with the previously obtained successful interesterification of castor oil with vinyl undecenoin on a 0.1 gram scale [
18].
The second stage of the reaction, the formation of oligo-ricinoleyl groups within the TAG formed from the first stage, was conducted in a manner that would minimize formation of free oligo-ricinoleic acid. Moreover, the ricinoleic acid concentration was kept intentionally low to allow the hydroxyls of ricinoleic acyl groups contained in the TAG to be as high as possible relative to free ricinoleic acid’s hydroxyl groups, thereby permitting the former to serve as the principal acyl acceptor. To begin the second stage of reaction, at 116 h, RML was replaced by CAL to enable oligomer formation. Additional ricinoleic acid was added in small batchwise increments (32 mmol) at 126, 150, and 174 h during the time course of reaction.
During the first 10 h of the second stage (116–126 h) the most major change was the depletion of tri- and diricinoleyl-TAG and increase of monoricinoleyl TAG (
Figure 5b), reflecting the ability of CAL to catalyze acyl exchange at the 2-glycerol position. The amount of TAG containing no ricinoleic acyl groups also decreased, suggesting the acidolysis of the TAG by ricinoleic acid, resulting in additional monoricinoleyl TAG formation. In addition, the dimer of ricinoleic acid (diricinoleic acid) clearly forms by 126 h, as evidenced by the HPLC and GPC chromatograms (
Figure 4c and d); however, monoricinoleyl TAG may contribute slightly to the GPC peak. Moreover, the retention times of the newly formed peak are identical with those of dimer present in
oligo-ricinoleic acid (
cf. Figure 1). As additional ricinoleic acid is added to the reaction mixture, additional dimer and trimer is formed, with little or no
oligo-ricinoleyl TAG formed (
Figure 4 and
Figure 5).
oligo-Ricinoleyl TAG did not form until the latter stages of the reaction, reaching approximately 10% when the reaction was stopped. The formation of this product by 285 h is evidenced by the occurrence of new peaks at lower retention times for both HPLC and GPC suggesting the new product is relatively hydrophobic and higher in molecular weight, respectively (
Figure 4). The increase of Mn during the latter stage of the time course of reaction reflects the increased levels of
oligo-ricinoleyl TAG. However, a small peak representing tetramer or multimer hidden within the
oligo-ricinoleyl TAG band (GPC analysis) cannot be discounted (
cf. Figure 1a), with its fraction being small, based on the DP value obtained for the final reaction mixture (2.3 according to 1H-NMR analysis). Concurrently, the amount of free ricinoleic acid decreased greatly; and, the concentration of dimer and trimer reach plateaus, with the amount of both being significantly higher than free ricinoleic acid. Therefore, it is believed the incorporation of
oligo-ricinoleic acyl groups into TAG results from acidolysis of TAG by dimer and trimer.
The AV of the reaction mixture after the reaction was terminated via removal of CAL was 82.4 ± 1.0 mg KOH g
−1. Using approximate concentrations for ricinoleic acid and its dimers and trimers derived via normal phase HPLC (
Figure 5b), and subtracting their contributions to the AV, the remaining concentration of COOH groups, if attributed solely to free undecenoic acid, would be 13.0 wt %.,
i.e., a 65% conversion for undecenoic acid for the entire two-stage reaction.
In sum, the formation of
oligo-ricinoleyl TAG appears to have been moderately successful. Perhaps the direct acidolysis of undecenoyl TAG by dimer, trimer, and multimer of ricinoleic acid may produce the desired higher yield. In addition, the extent of incorporation of undecenoic acid into
oligo-ricinoleic TAG is not known. Due to the prominence of monoricinoleyl (diundecenoyl) TAG during the latter stages of reaction, it is believe that the majority of
oligo-ricinoleyl TAG contain undecenoic acyl groups. The amount of undecenoic acid conjugated to dimers and trimers is also not clear. However, the HPLC chromatogram,
Figure 4c, suggests column chromatography using a silica gel-based stationary phase can be used to isolate the
oligo-ricinoleyl TAG, dimer, and trimer fractions, to allow for their more thorough analysis, and to obtain the former in purified form.
Figure 4.
Analysis of the lipase-catalyzed synthesis of TAG containing oligo-ricinoleic acid and 10-undecenoic acid. (a) and (b) refer to the first step, the Rhizomucor miehei lipase (RML) -catalyzed acidolysis of castor oil with 10-undecenoic acid; (c) and (d) to the second step of the reaction, the CAL-catalyzed reaction between ricinoleic acid and the product from the first step. Chromatograms are from (a,c) normal phase HPLC and (b,d) GPC analyses. Reaction conditions: For the first step, castor oil (18.7 mg, 20.0 mmol) was mixed with 10-undecenoic acid (11.1 g, 60 mmol) and RML (9.5% w/w). The reaction mixture was held at 67 °C. After 116 h, the reaction was stopped; RML was removed by centrifugation, and CAL (5.8 wt %) was added. Additional ricinoleic acid (9.5 g, 32 mmol) was added 10 h, 34 h, and 58 h after the addition of CAL to the reaction mixture. The reaction was conducted at 72 °C.
Figure 4.
Analysis of the lipase-catalyzed synthesis of TAG containing oligo-ricinoleic acid and 10-undecenoic acid. (a) and (b) refer to the first step, the Rhizomucor miehei lipase (RML) -catalyzed acidolysis of castor oil with 10-undecenoic acid; (c) and (d) to the second step of the reaction, the CAL-catalyzed reaction between ricinoleic acid and the product from the first step. Chromatograms are from (a,c) normal phase HPLC and (b,d) GPC analyses. Reaction conditions: For the first step, castor oil (18.7 mg, 20.0 mmol) was mixed with 10-undecenoic acid (11.1 g, 60 mmol) and RML (9.5% w/w). The reaction mixture was held at 67 °C. After 116 h, the reaction was stopped; RML was removed by centrifugation, and CAL (5.8 wt %) was added. Additional ricinoleic acid (9.5 g, 32 mmol) was added 10 h, 34 h, and 58 h after the addition of CAL to the reaction mixture. The reaction was conducted at 72 °C.
Figure 5.
Time course of reaction for the lipase-catalyzed synthesis of TAG containing
oligo-ricinoleic acid and 10-undecenoic acid. (
a) change in distribution of TAG species (determined from normal phase HPLC analysis; “TAG” refers to TAG not containing ricinoleic acyl groups); (
b) change in concentration of TAG, fatty acid, and oligomer (determined from normal phase HPLC, unless noted otherwise; “AV” and “Stoich” refer to the wt % of 10-undecenoic acid determined from acid value measurements and from stoichiometry: equating the amount of released ricinoleic acid to the amount of undecenoic acid consumed, respectively); (
c) change of number-averaged molecular weight and degree of polymerization for fatty acids (determined from, GPC and
1H-NMR, respectively). Dashed arrow refers to the replacement of RML with CAL; solid arrows refer to the addition of (9.5 g, 32 mmol) of ricinoleic acid. Reaction conditions are given in
Figure 4.
Figure 5.
Time course of reaction for the lipase-catalyzed synthesis of TAG containing
oligo-ricinoleic acid and 10-undecenoic acid. (
a) change in distribution of TAG species (determined from normal phase HPLC analysis; “TAG” refers to TAG not containing ricinoleic acyl groups); (
b) change in concentration of TAG, fatty acid, and oligomer (determined from normal phase HPLC, unless noted otherwise; “AV” and “Stoich” refer to the wt % of 10-undecenoic acid determined from acid value measurements and from stoichiometry: equating the amount of released ricinoleic acid to the amount of undecenoic acid consumed, respectively); (
c) change of number-averaged molecular weight and degree of polymerization for fatty acids (determined from, GPC and
1H-NMR, respectively). Dashed arrow refers to the replacement of RML with CAL; solid arrows refer to the addition of (9.5 g, 32 mmol) of ricinoleic acid. Reaction conditions are given in
Figure 4.


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}