1. Introduction
Hyperbranched polymers have emerged in the last few years as a new promising category of non-viral vehicles for drug and gene delivery applications [
1,
2,
3]. The advantages related to their use as complexation and delivery agents, such as their structural features, their multifunctionality and their favorable transport and thermodynamic properties, have surged the scientific as well as the industrial interest towards an effort for a better control of their physicochemical behavior [
4,
5,
6,
7,
8,
9,
10]. To this end, a large number of theoretical and experimental studies have been devoted to the elucidation of the structure/properties relation of complexes comprised by hyperbranched molecules (HBP) and linear polyelectrolytes (LPE), with complexes including nucleic acids being the most characteristic examples [
11,
12,
13,
14,
15].
Among other parameters, these studies have demonstrated the strong dependence of the physical properties of such complexes on the structural features of the hyperbranched hosts and on factors associated with the relevant thermodynamic environment [
16,
17,
18,
19,
20]. Due to the rather broad parameter space characterizing the actual systems, coarse-grained models have been widely utilized for the theoretical description and the computer modeling of such complexes [
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32]. These studies aimed at the elucidation of the generic behavior related to their conformational characteristics and to phenomena of key importance regarding their action as complexation agents and delivery vehicles, such as overcharging [
27,
31,
33,
34].
Results from stoichiometric complexes comprised by charged dendrimers and oppositely charged linear polyelectrolytes [
31] showed that the basic effects related to the overcharging phenomenon are in qualitative agreement with the correlation theory developed by Nguyen and Shklovskii [
21] for analogous complexes involving impenetrable macroions. The observed differences between computer simulations and the correlation theory were attributed to the deformable nature of the dendritic polymers. This attribute of the dendritic molecules resulted in a shift towards larger values of the predicted critical length (N
C) of the linear component at which a first order transition associated with the tail-release mechanism sets in [
31]. An amendment of the aforementioned theoretical description [
21] considering similar complexes but allowing an isotropic deformability of the spherical component, was found to provide a fair description for the behavior of stoichiometric complexes involving a charged linear chain and an oppositely charged dendritic component, the latter being characterized by a rather dense branching pattern close to the dendritic core [
27]. In the same study, it was also demonstrated that the anisotropic deformation of the hyperbranched component of the complexes with a preferential branching in the periphery, affected significantly the structural properties and the degree of overcharging of the formed complexes.
A step forward towards elucidating the specifics of the overcharging phenomena in linear LPE/dendrimer complexes was taken recently by considering non-stoichiometric complexes comprised by a linear polyelectrolyte and two oppositely charged dendrimers [
35]. In such complexes, a new LPE conformation is introduced, that of the “linker” between the two dendritic molecules [
35]. As in the stoichiometric systems [
27,
31,
33], the dendrimers were also found to be strongly overcharged upon increase of the length of the LPE, with the maximum degree of overcharging observed at a length of the LPE close to that corresponding to the single dendrimer/LPE systems. However, in contrast to the case of stoichiometric complexes, the degree of overcharging in the non-stoichiometric systems was not found to decrease significantly after the highest level of adsorption of the LPE was reached. Moreover, no first-order transition related to the tail-release mechanism was observed upon increase of the LPE, in sharp contrast to the behavior noted in the single dendrimer/LPE models. Instead, in the non-stoichiometric complexes increase of the LPE length resulted in the appearance of the “linker” conformation and the gradual separation of the two dendrimers, but this occurrence did not affect the length of the dendrimer-adsorbed LPE parts. An intriguing effect related to the “linker” conformation was the non-monotonic dependence of the “linker” size on the LPE length [
35]. Detailed conformational analysis of the LPE chain showed that the “linker” state of the LPE coexisted with a “tail” configuration which could not be accounted for by an approximate theoretical approach developed by the authors [
35].
In the present work, we extend our previous simulational study in non-stoichiometric complexes comprised by two terminally charged HBPs which are preferentially branched at the periphery [
36] and an oppositely charged LPE, as a function of the LPE length. Our goal is to compare the behavior of such systems to that observed in analogous models comprised by stoichiometric analogues and by two dendrimers and one LPE [
27,
31,
35], thus elucidating the effects of stoichiometry and topology in the conformational and the overcharging characteristics of these complexes. Understanding of the latter effects is expected to improve the perspectives towards a better control of the properties of such systems and consequently to facilitate a molecular-level engineering of their behavior.
2. Simulation Details
Following our previous works [
16,
27,
31,
35], Brownian dynamics simulations (Ermak-McCammon algorithm) [
37] with the explicit inclusion of hydrodynamic interactions, through Rotne-Prager-Yamakawa tensor [
38,
39], were employed in order to simulate coarse-grained models of LPE/HBP complexes in implicit solution, characterized by rigid bond spacers of length equal to
l (the SHAKE algorithm is employed for the implementation of rigid bonds [
40]). A Lennard-Jones potential is used for the description of excluded volume interactions and the Debye-Hückel potential is employed for the description of electrostatic interactions. Although the presence of explicit counterions is known to affect the electrostatic screening conditions and relevant thermodynamic aspects associated with the complexation process [
23,
26,
41], as in our previous studies [
31,
35,
36] the models are constructed to represent only the dilute solution limit where the degree of the counterion condensation remains low and thus does not affect significantly the structural properties of the polymeric components. It is therefore considered that the conditions of the screening of the electrostatic interactions are such that the Debye-Hückel approximation remains valid [
35,
42]. To verify that the Coulombic screening conditions adopted do not affect the complexation behavior of the studied models, we also performed additional simulations including explicit counterions employing the Particle Mesh Ewald method for the calculation of the electrostatic forces [
43,
44]. The results obtained compare favorably to those without the explicit presence of counterions (see later in the text), lending credence to the utilization of the Debye-Hückel approximation for the treatment of electrostatic interactions under the conditions realized in the current study.
All the information describing the specifics of the simulational algorithm and the form of the interaction potentials are identical to those quoted in our past studies [
28,
31,
36] where the interested reader can refer to for more details. We briefly repeat herein that the Lennard-Jones parameters were taken as
εLJ = 0.3
kT (
k is the Boltzmann constant and
T the temperature) and
σ = 0.8
l, with a cutoff distance of
rcut = 2.5
σ. For the electrostatic interactions, we considered the Bjerrum length
λB to be equal to
l without loss of generality (the value of
λB in water at room temperature is equal to 7.14 Å, that is close to the length of a segment of a common flexible linear polymer), while the Debye radius was taken as
rD = 8.96
l For comparison purposes, the aforementioned parameters were taken to be identical to those utilized in the previous study concerning the non-stoichiometric LPE/dendrimer complexes. The details regarding the explicit-counterion simulations are as they described in [
45]. All the dimensionless quantities used in the simulations (
i.e., bond length, thermal energy, translational friction coefficient and time) are set to unity. The dimensionless integration step is set to
Δt = 10
−4.
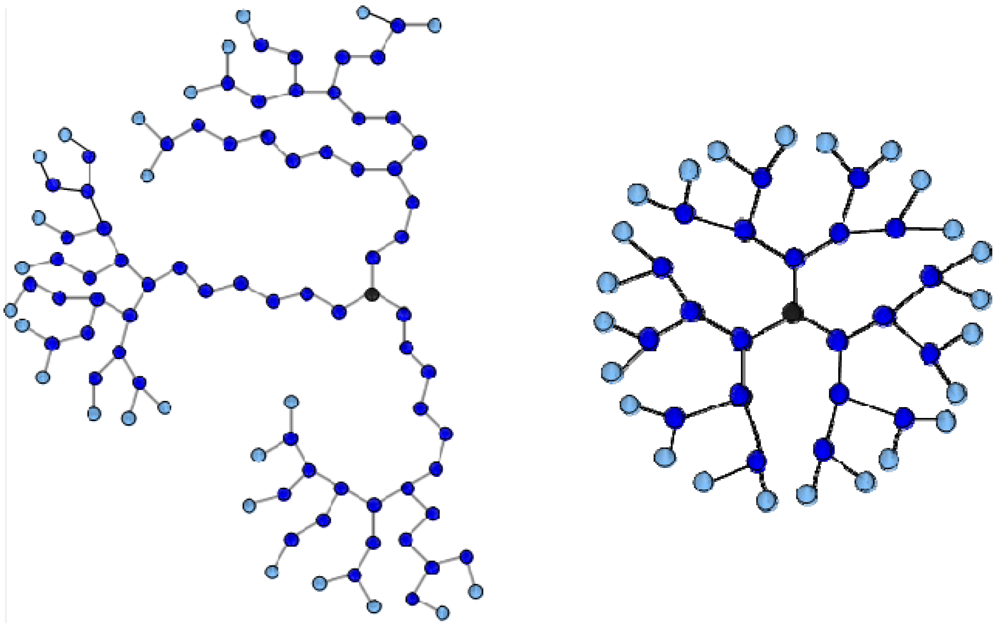
For the preparation of the systems a freely-jointed bead-rod model is chosen [
46]. The hyperbranched molecules were constructed using the topology builder described in [
47] where the structure emanates from a trifunctional core and is sequentially built by addition of bifunctional reactive monomers. Two terminally charged HBPs and one oppositely charged LPE form each one of the simulated systems. The HBP constituents studied herein, correspond to the so-called G4MAX HBP systems (see [
28]), which correspond to trifunctional randomly branched HBPs with an intermediate degree of branching,
DB equal to 0.5 [
48] and a high value of Wiener index,
W [
47,
49] representing an open-structured star-like macromolecular entity (referred to as MAX topology hereafter). Experimentally, there is always a degree of polydispersity in the synthesis of such hyperbranched systems, but with the recently developed topology-sensitive separation techniques [
50,
51] this topology-related polydispersity can be significantly narrowed. The structure of the aforementioned category of HBPs is characterized by rather long linear segments near the core and a higher degree of branching in the periphery (see
Figure 1). As was demonstrated by our previous studies [
27,
28,
36], substantial (and thus potentially experimentally observable) differences in the static and dynamic response between hyperbranched polymers of the same degree of branching (DB ~ 0.5) could mostly be observed at systems belonging to topological extremes characterized by a dense/homogeneous and a sparse/peripheral branching pattern. In these studies we have checked different variations of such peripherally branched and densely branched systems, as well as molecules of different sizes and we have found that the dynamic as well as the static/conformational properties of hyperbranched polymers belonging to these two categories, are determined not by the specific variations of the branching pattern within each family, but mainly by the distinctly different degree of deformability which arises from the generic differences between the two extreme branching patterns (
i.e., sparse/peripheral and dense/homogenous branching).
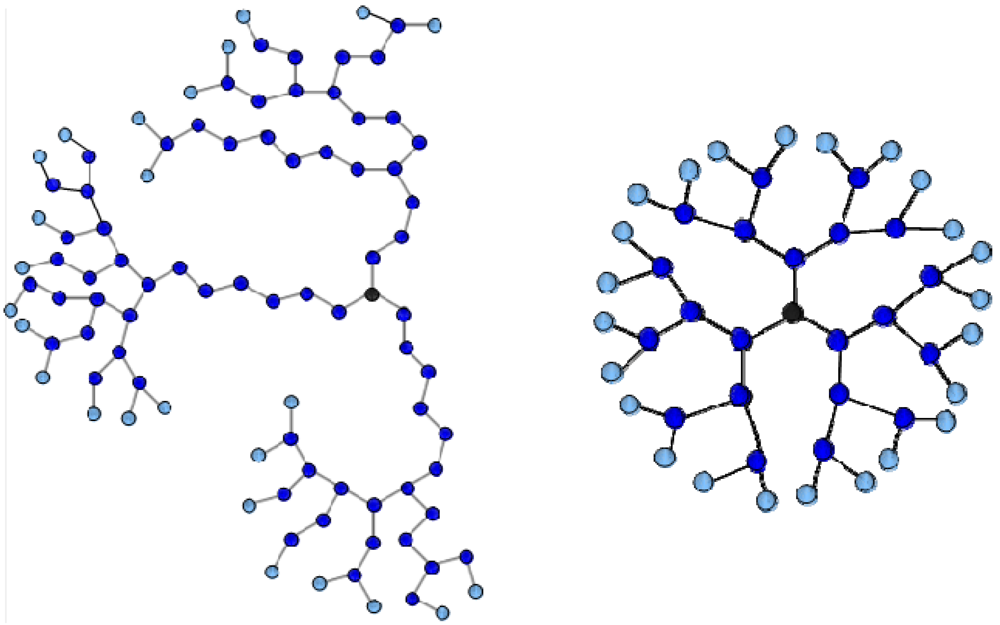
Figure 1.
Two-dimensional projections of (left) a G4MAX hyperbranched molecules (HBP) (N = 94, NT = 25) and (right) a G3 dendrimer (N = 46, NT = 24). Terminal charged beads are indicated in lighter colors.
Figure 1.
Two-dimensional projections of (left) a G4MAX hyperbranched molecules (HBP) (N = 94, NT = 25) and (right) a G3 dendrimer (N = 46, NT = 24). Terminal charged beads are indicated in lighter colors.
Therefore, without loss of generality (regarding the main characteristics of the targeted topology) we opted in simulating these systems on the basis of their similarities (in terms of the strength of electrostatic interactions) as well as on that of their distinct behavior (with respect to their conformation properties) compared to their regular-branched analogues [
28,
36]. In a more general approach where a generic behavior related to randomly branched systems (with no predominant branching pattern) was intended, an averaging over many different topologies should have been performed instead. This, however, is not within the scope of the present work.
Each of the studied complexes is comprised by the linear chain and by two G4MAX HBPs bearing a structure as depicted in
Figure 1, the latter possessing a total number of beads
N = 94 and a number of terminal charged monomers of
NT = 25. The number of charged beads is very close to the 24 terminal beads of a 3rd generation (G3) dendrimer (
Figure 1), so that a close comparison can be made with the behavior of the G3 complexes studied in our previous works [
27,
31,
35].
A wide range of LPE lengths complexed with two G4MAX HBPs were studied in the present work, starting from the overall electrical neutrality limit (
i.e., the total number of the charged LPE beads equal twice the number of the each of the oppositely charged G4MAX molecule), up to LPE lengths exceeding the double-charge limit:
Nch = 50, 55, 60, 70, 80, 90, 100, and 110 (
Nch stands for the number of monomers of the LPE chain). The protocol for the construction of the initial configurations was the same as that described in our previous work [
35] and thus not repeated here. All the systems were equilibrated for more than 2 × 10
7 simulation steps, followed by production runs of 7 × 10
6 steps. The average size of both components in the complex is stabilized already below 2 × 10
6 steps. A 10-fold longer equilibration period, results in a fair sampling of the configurational phase space, prior to the commencement of the production runs.
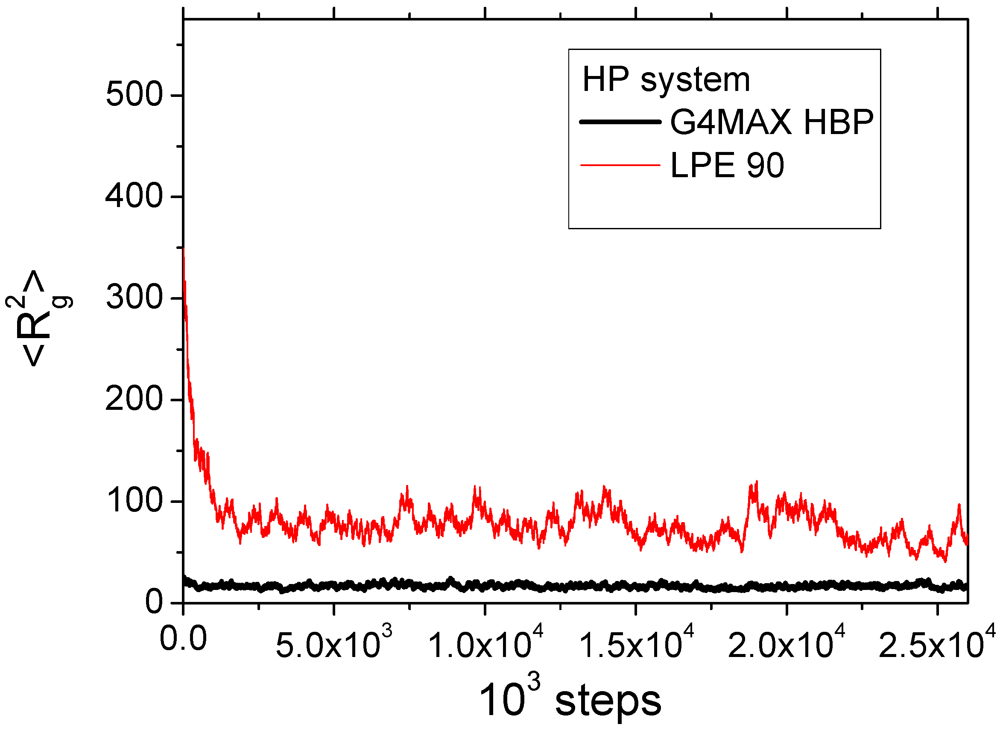
Figure 2 shows the variation of the squared radius of gyration of the two components in one of the studied complexes (the pictures for the rest of the systems are completely analogous).
Figure 2.
Instant values of the square gyration radius of the components of one of the complexes during equilibration runs. The
![]()
for the HBP refers to the average of the two G4MAX molecules of the complex.
Figure 2.
Instant values of the square gyration radius of the components of one of the complexes during equilibration runs. The
![]()
for the HBP refers to the average of the two G4MAX molecules of the complex.
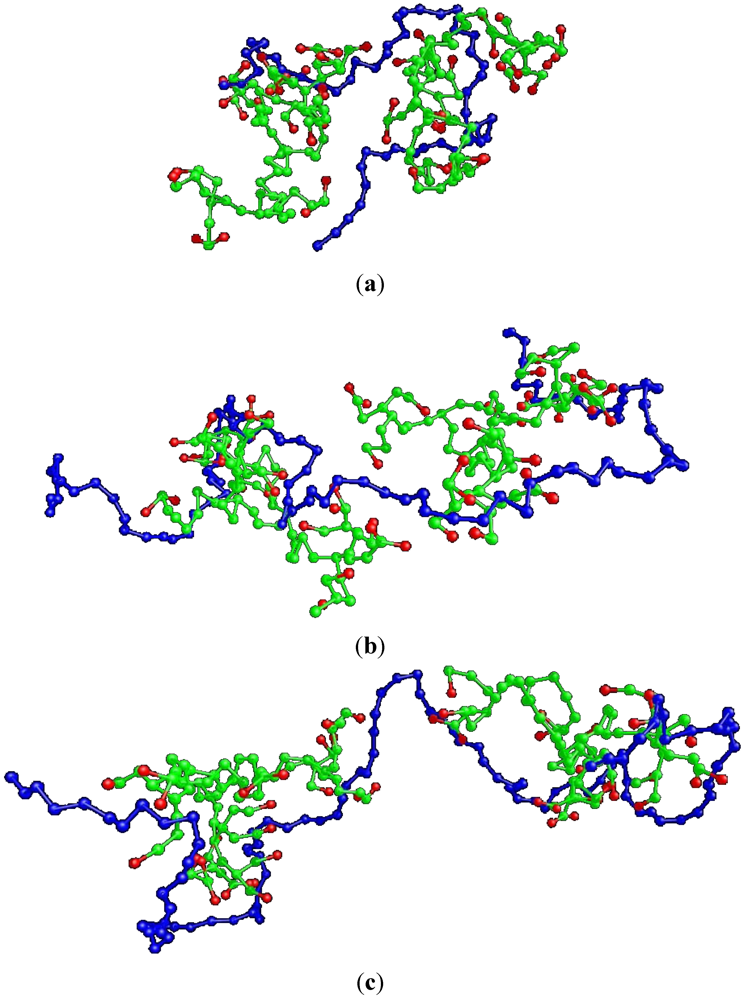
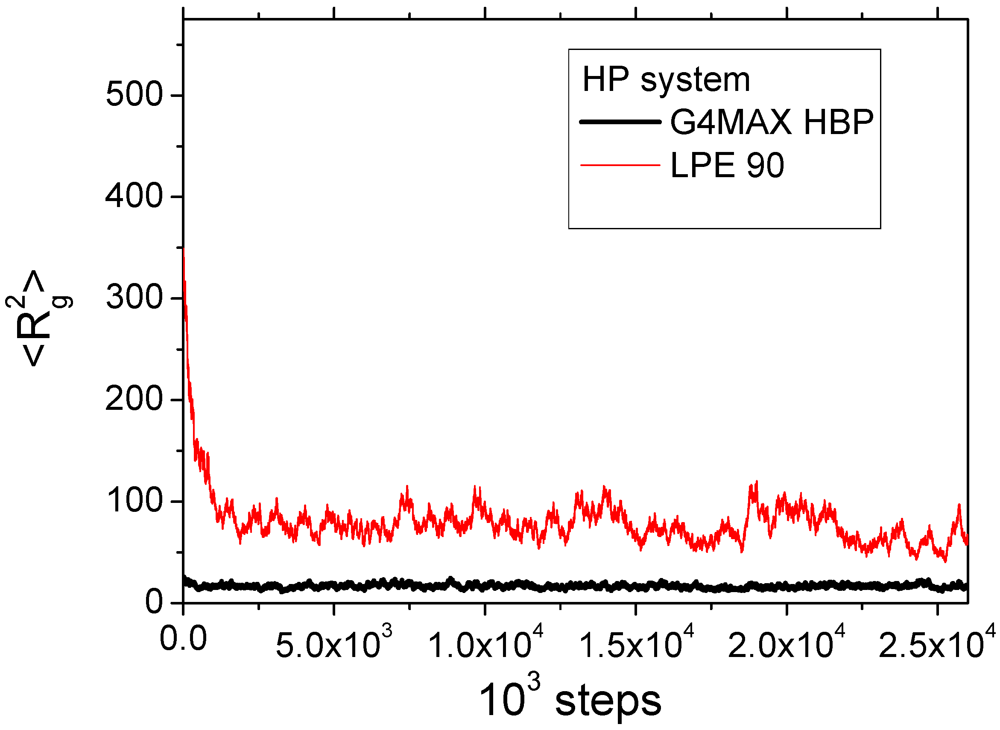
Figure 3 displays configurations of some characteristic studied systems after equilibration.
Figure 3.
Snapshots of some of the simulated non-stoichiometric complexes comprised by two G4MAX HBPs and a linear polyelectrolyte (LPE) of length: (a) 50 (electrically neutral complex) (b) 80 and (c) 100 (doubly charged complex). Red beads represent the charged monomers of the HBPs.
Figure 3.
Snapshots of some of the simulated non-stoichiometric complexes comprised by two G4MAX HBPs and a linear polyelectrolyte (LPE) of length: (a) 50 (electrically neutral complex) (b) 80 and (c) 100 (doubly charged complex). Red beads represent the charged monomers of the HBPs.
3. Results and Discussion
One of the key factors that can affect drastically the static, as well as the dynamic behavior, of the complexes formed by charged dendritic molecules and LPEs, is their ability to assume distinctly different conformations depending on the structural features of the two components. The energetic characteristics and the response of such complexes under external stimuli were found to be particularly sensitive to factors such as the topology of the dendrtitic molecule and the charge ratio between the branched and the linear components (the LPE charge usually taken proportional to its length) [
28,
29,
33,
36]. In stoichiometric complexes [
27], it was found that the finite (and in the case of the HBPs bearing the MAX topology, the anisotropic) deformability of the hyperbranched molecules may affect the characteristics of the first order transition related to the tail release mechanism observed when the length of the LPE exceeds a critical value. To check the effects triggered by the realization of the “linker” conformation (as described in [
35], see also
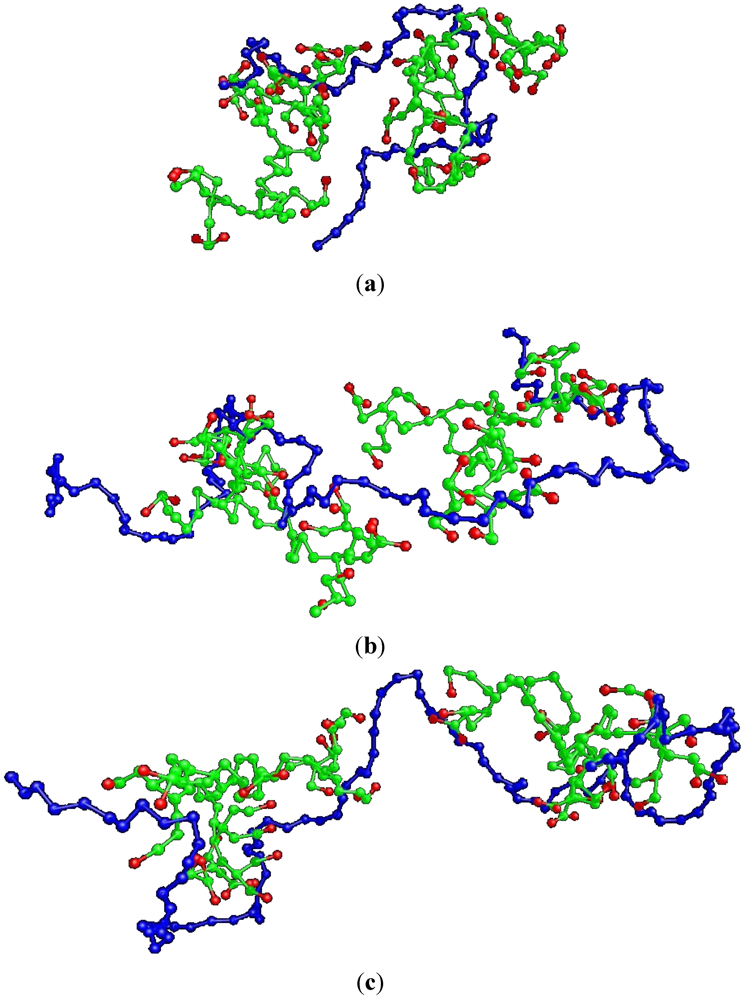
Figure 3) in non-stoichiometric complexes comprised by HBPs of the MAX topology, we have calculated the dependence of the semiaxes of the ellipsoid of inertia of the HBP components of the complexes as a function of the length of the LPE.
Figure 4 illustrates the comparison of the latter with those corresponding to the HBPs participating in the corresponding stoichiometric complexes, as described in our previous study [
27]. Apparently, no significant change is observed in the elongation of the HBP in the non-stoichiometric complexes, in contrast to the case of their stoichiometric analogues. Apart from a weak trend for a monotonic increase of the longer semiaxis, the shape of the HBP components in the present systems remains rather insensitive to the LPE length, up to the longer length examined.
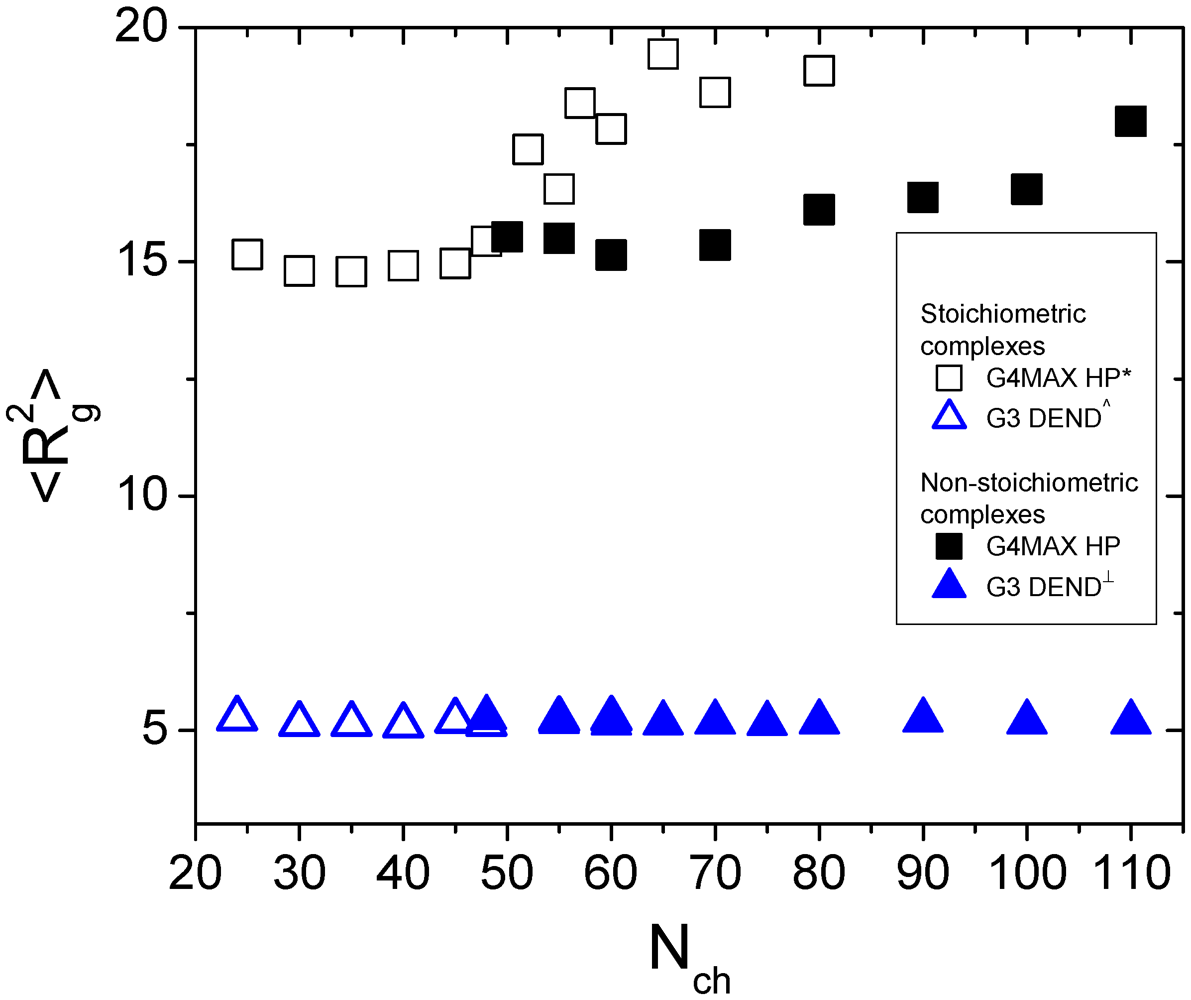
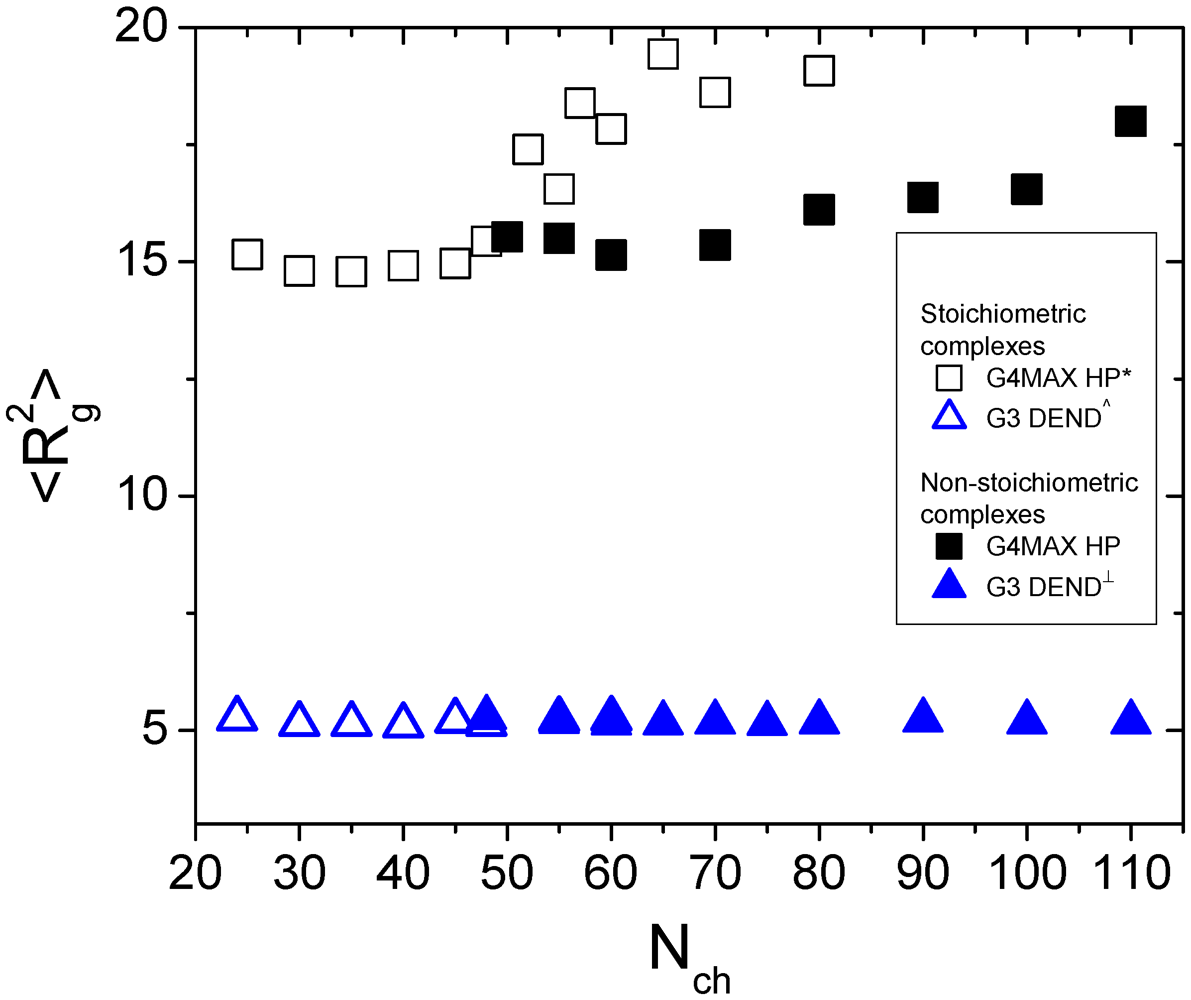
This occurrence is reflected to the average size of the HBPs as is shown in
Figure 5, where the square radius of gyration of the G4MAX HBPs of the present systems is compared to that of the analogous stoichiometric complexes from [
27]. For comparison purposes, the square radii of gyration of both the stoichiometric and non-stoichiometric complexes, comprised by G3 dendrimers from our previous works, are included in the same plot as well. While for the G3 systems the average size of the dendrimer components appears to be independent of the kind of complex (
i.e., stoichiometric or not) they participate in, a clear differentiation is observed between the stoichiometric and non-stoichiometric G4MAX HBP systems.
Figure 4.
HBP complex constituents’ shape anisotropy, in terms of their inertia ellipsoid semi-axes. The results of the present simulations are collated to those describing the HBPs participating in the analogous stoichiometric complexes [
27]. The description of each semiaxes is shown in the schematic of the ellipsoid of inertia.
Figure 4.
HBP complex constituents’ shape anisotropy, in terms of their inertia ellipsoid semi-axes. The results of the present simulations are collated to those describing the HBPs participating in the analogous stoichiometric complexes [
27]. The description of each semiaxes is shown in the schematic of the ellipsoid of inertia.
Figure 5.
Comparison of the square radii of gyration for the HBP and the analogous dendrimer components of stoichiometric and non-stoichiometric complexes, as a function of the length of the LPE, from the present and from previous works (* [
27], ^ [
31] and
┴ [
35]).
Figure 5.
Comparison of the square radii of gyration for the HBP and the analogous dendrimer components of stoichiometric and non-stoichiometric complexes, as a function of the length of the LPE, from the present and from previous works (* [
27], ^ [
31] and
┴ [
35]).
The picture emerging from
Figure 4 and
Figure 5 already indicates that the conformational properties of the open-structure G4MAX polymers are strongly dependent on the presence of the second hyperbranched polymer. The simpler rationalization of this finding is to correlate it to the conformational characteristics of the linear component of the complexes. This is corroborated by our past findings regarding the conformational behavior of the linear components when comparing the systems comprised by one LPE and a one [
31] or two [
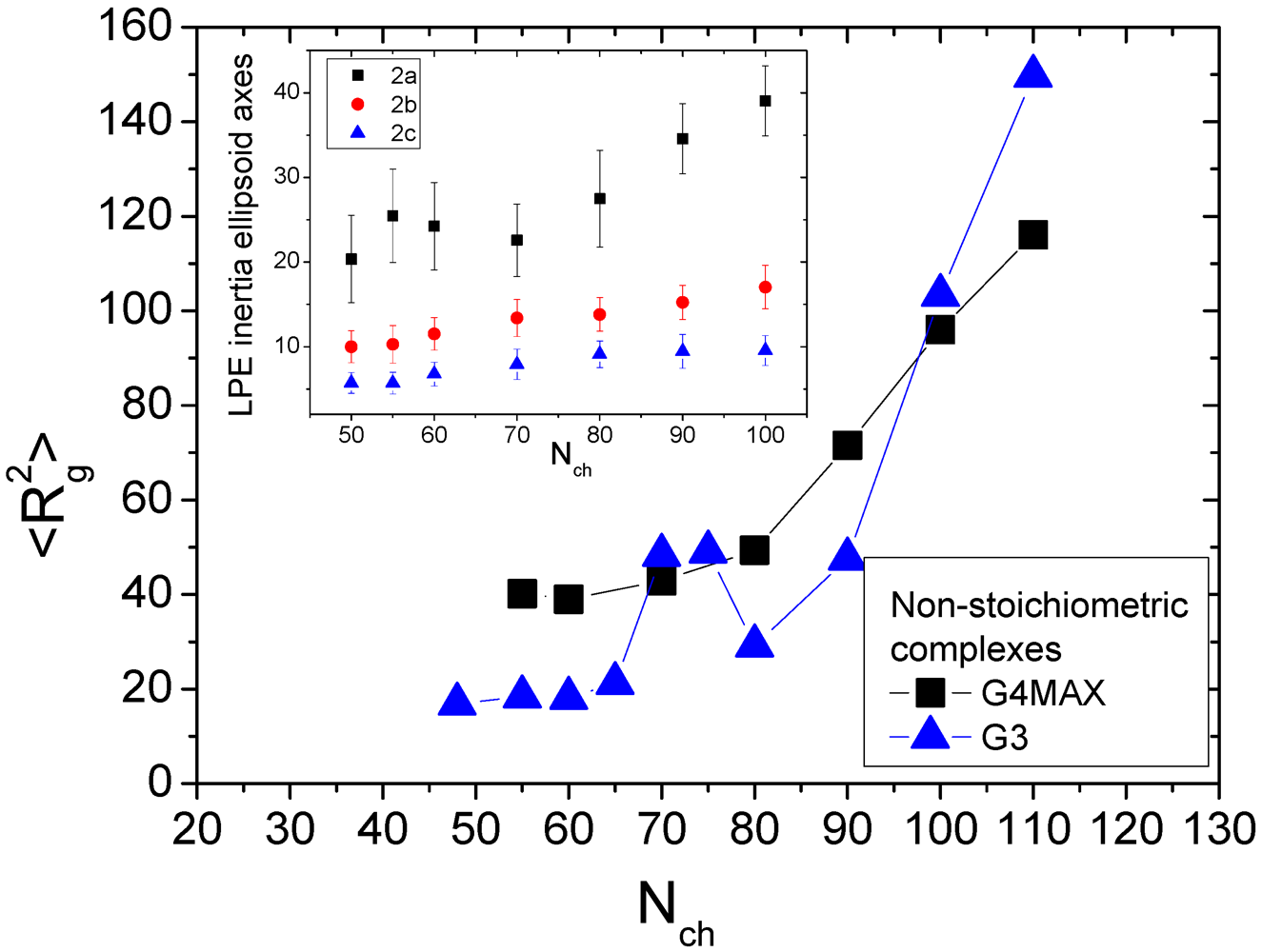
35] dendrimers. In the latter complexes, which are analogous to those examined in the present work, a non-monotonic behavior of the average size of LPE with its length was observed. To check whether this feature is present in the non-stoichiometric G4MAX systems, we compare in
Figure 6 the square radii of gyration of the LPEs to that of the corresponding LPEs in the non-stoichiometric G3 complexes from our previous work [
35].
Figure 6.
Main panel: average square radius of gyration of the LPE component of the present systems (
![]()
), compared to that from the analogous non-stoichiometric G3 systems (
![]()
) from [
35]. Inset: dependence of the axes of the ellipsoid of inertia for the LPE participating in the G4MAX non-stoichiometric systems. The definition of the axes is identical to that of
Figure 4.
Figure 6.
Main panel: average square radius of gyration of the LPE component of the present systems (
![]()
), compared to that from the analogous non-stoichiometric G3 systems (
![]()
) from [
35]. Inset: dependence of the axes of the ellipsoid of inertia for the LPE participating in the G4MAX non-stoichiometric systems. The definition of the axes is identical to that of
Figure 4.
Evidently, the average size of the LPEs in the present systems increases monotonously with its length, in contrast to the behavior observed in the G3 complexes.
This increase of the radius of gyration of the LPE in the present systems appears to be originating mainly from an analogous monotonic elongation of the G4MAX component (inset of
Figure 6) as the LPE length grows.
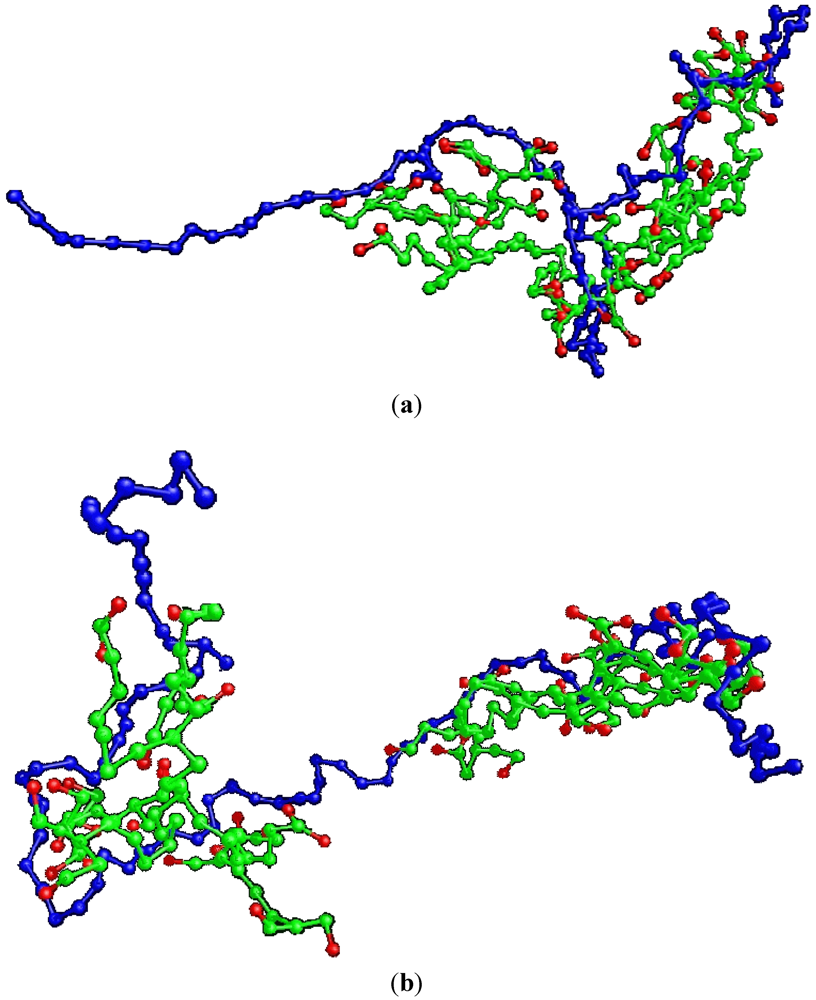
Figure 7 shows two snapshots of the N
CH = 90 and N
CH = 100 G4MAX complexes studied in the present work, where HBPs assuming such elongated shapes are shown. In the case of the non-stoichiometric G3 complexes, the behavior shown in
Figure 6 was attributed to the nonmonotonic dependence of the size of the linkers (
i.e., parts of the LPEs belonging to the “linker” conformation) on the LPE length. It is therefore straightforward to assume that the differentiation between the G4MAX and the G3 complexes, regarding the dependence of the size of the LPE on its length, may arise from differences in the conformations assumed by the linear components in the two cases.
Figure 7.
Snapshots of the presently examined G4MAX systems corresponding to lengths of the LPE (a) NCH = 90 and (b) NCH = 100.
Figure 7.
Snapshots of the presently examined G4MAX systems corresponding to lengths of the LPE (a) NCH = 90 and (b) NCH = 100.
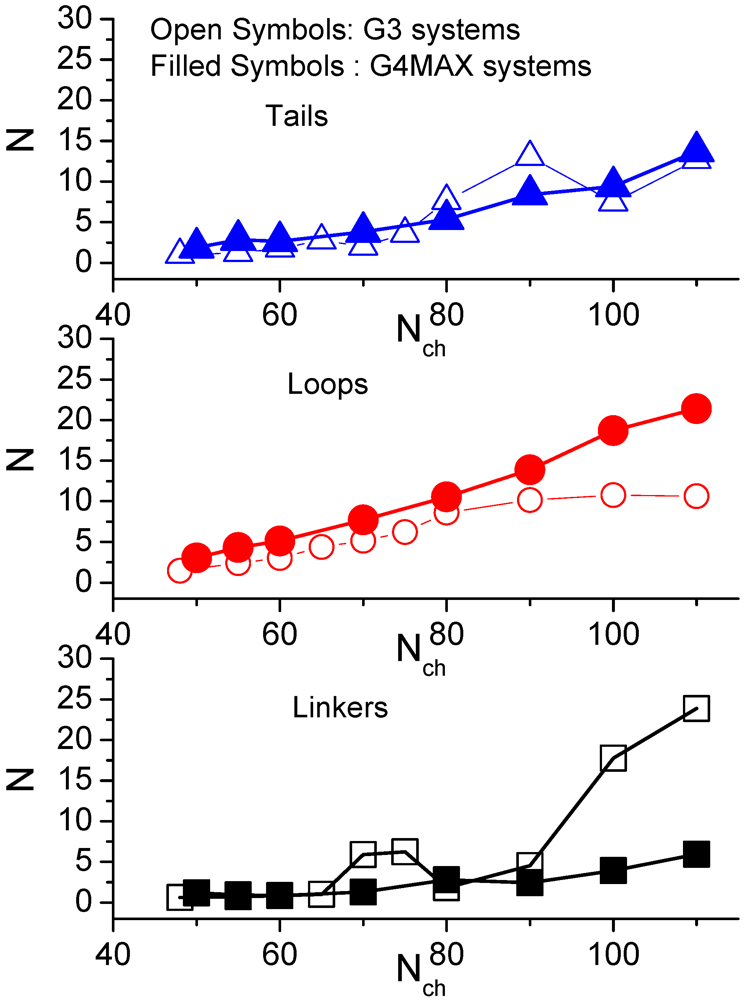
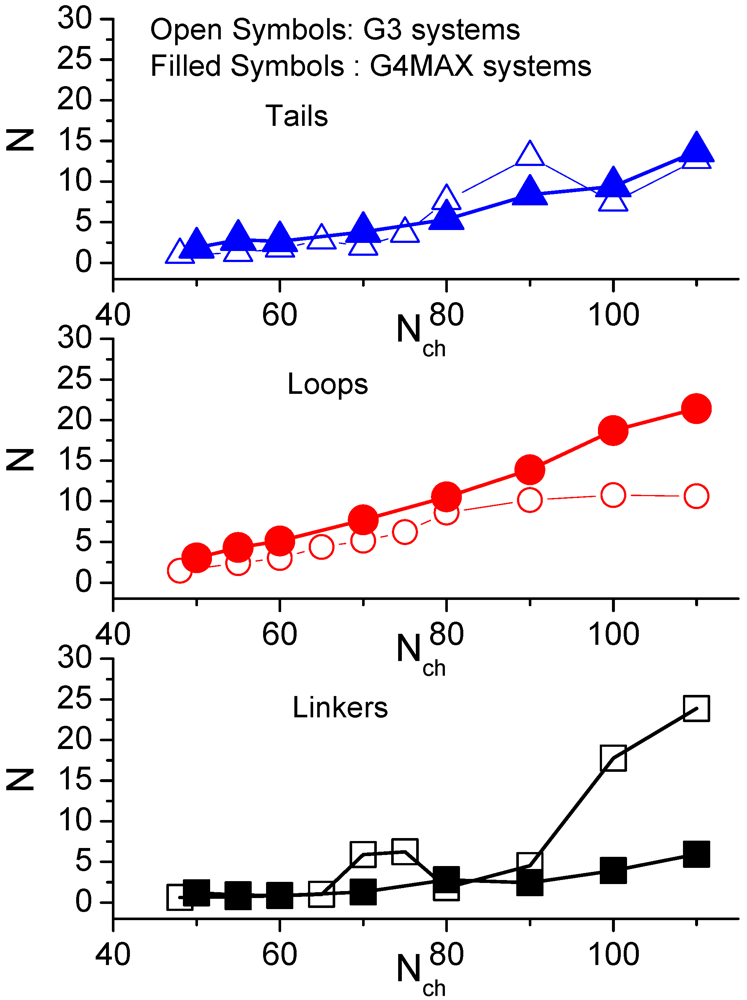
Figure 8 compares the average number of monomers of the LPEs in the non-stoichiometric G3 complexes [
35] and in the G4MAX systems.
A “tail” is defined as an unadsorbed part of LPE which has only one adsorbed end, a “loop” when both ends are adsorbed on the same hyperbranched molecule while a “linker” when the ends are adsorbed on different ones. While the behavior describing the “loop” and the “tail” conformations is comparable between the two categories of complexes, distinct behaviors are noted for the “linker” parts of the LPEs. The parts of the LPE belonging to different configurations of the linear chains appear to increase in a monotonic fashion with the LPE length, with the “linker” parts assuming the shorter length compared to the “tails” and the “loops”.
Figure 8.
Comparison of the average number of the LPE monomers belonging to different conformations, between the non-stoichiometric G3 (from [
35]) and the G4MAX complexes.
Figure 8.
Comparison of the average number of the LPE monomers belonging to different conformations, between the non-stoichiometric G3 (from [
35]) and the G4MAX complexes.
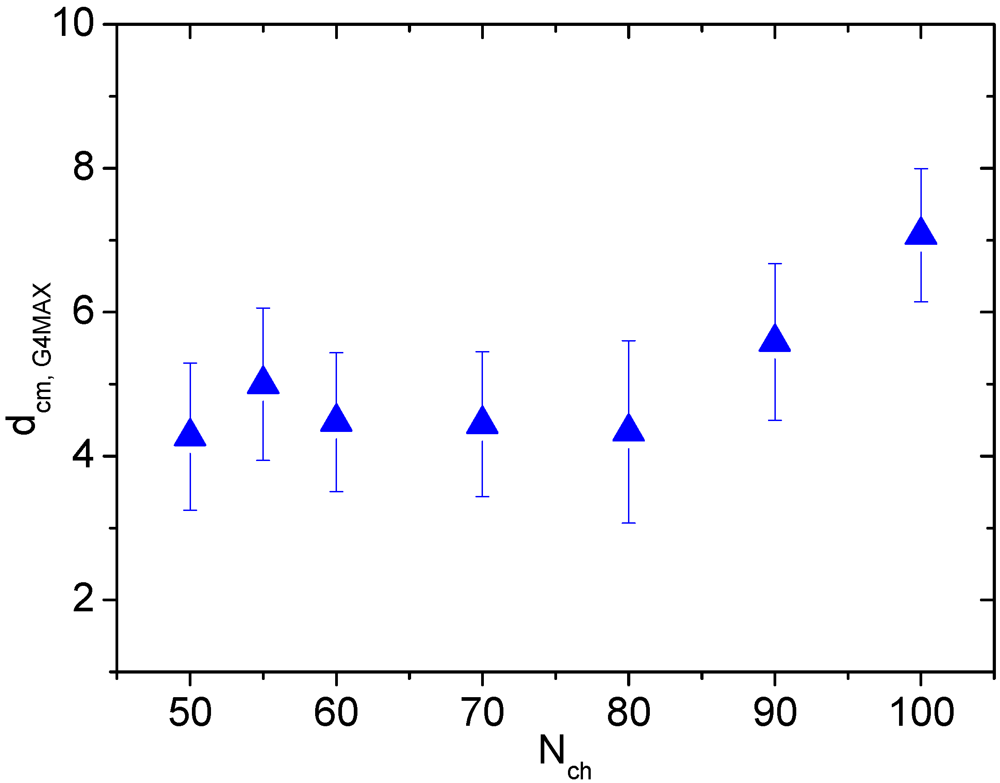
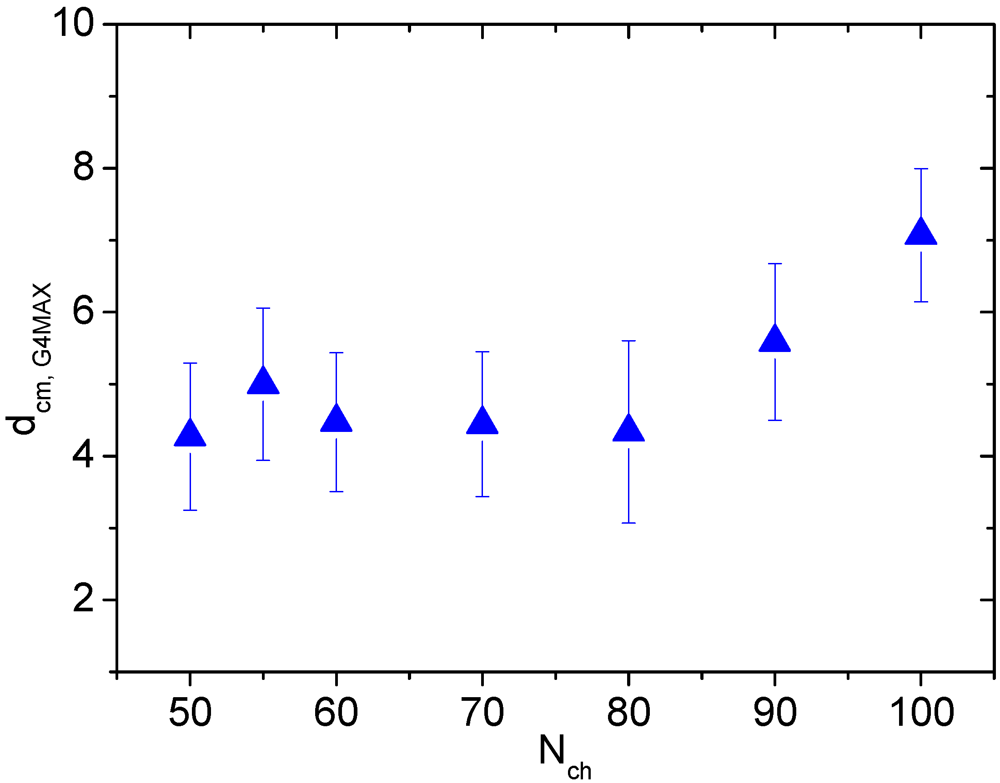
Such a weak dependence of the number of the “linker” monomers on the LPE length in the G4MAX complexes, is consistent with a similar weak dependence of the average separation between the two HBPs as shown in
Figure 9.
Figure 9.
Average distance between the centers of mass of the two hyperbranched polymers in the G4MAX non-stoichiometric complexes.
Figure 9.
Average distance between the centers of mass of the two hyperbranched polymers in the G4MAX non-stoichiometric complexes.
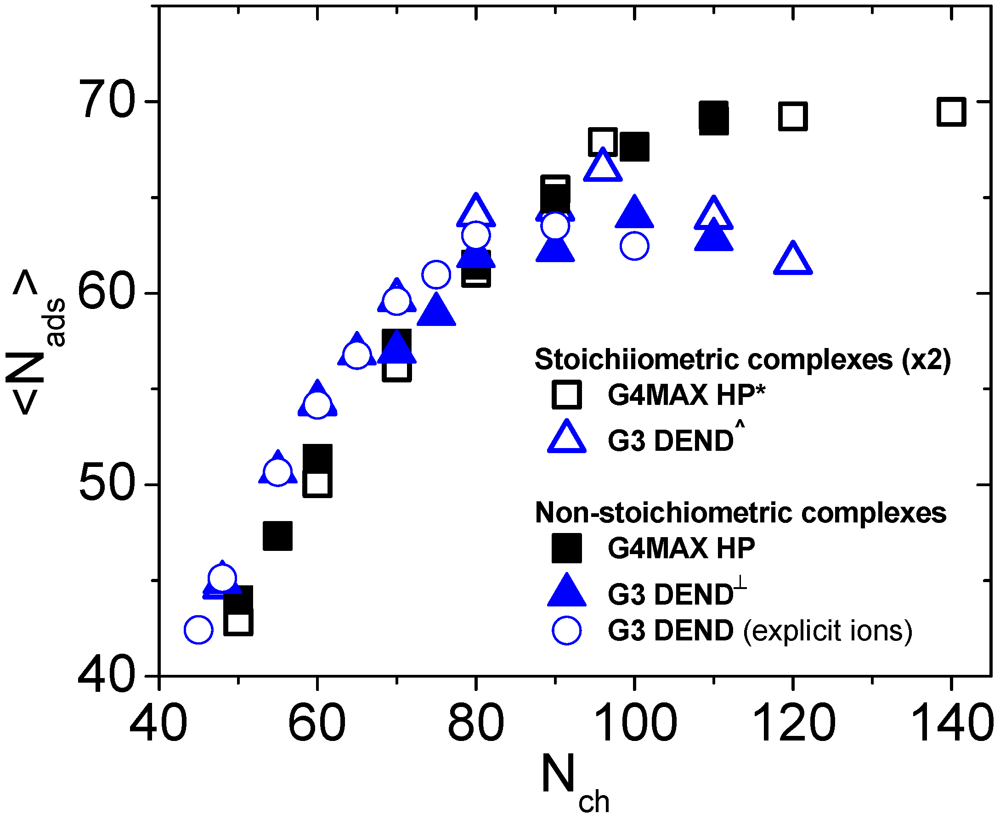
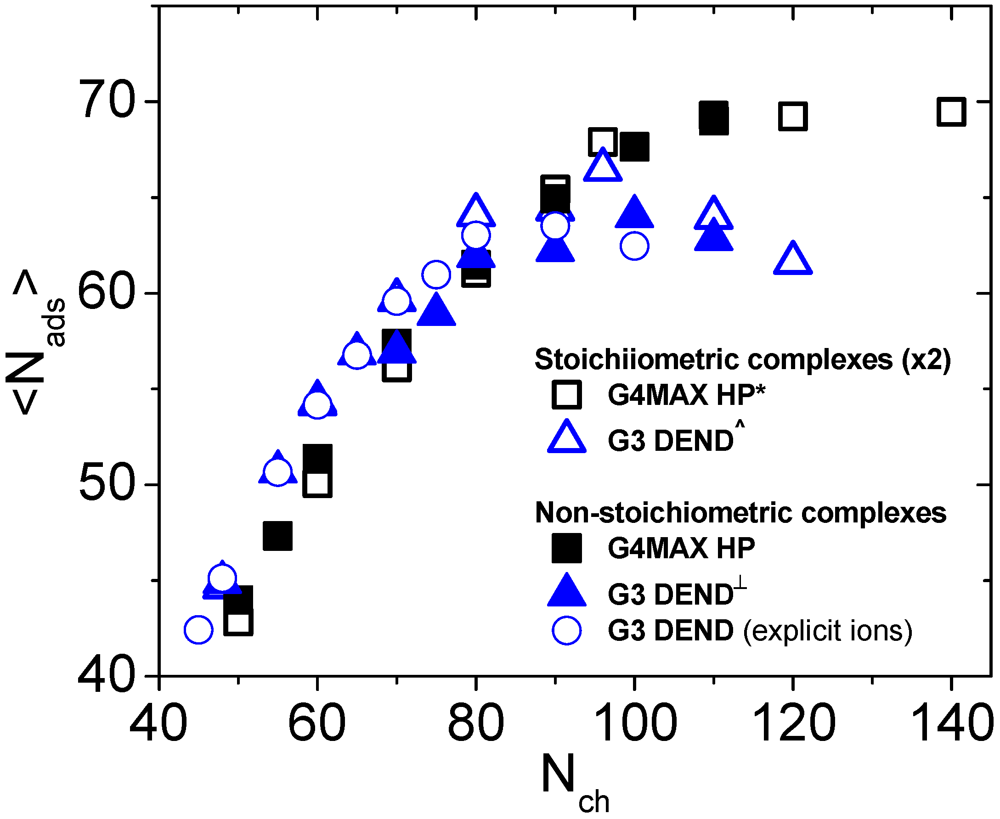
In view of the similarities regarding the “tails” and “loops” and the differences in the “linker” conformations of the LPE in the two kinds of complexes, it is interesting to examine whether the number of adsorbed beads of the LPE on the branched molecules depends on the topology and the stoichiometry of the complexes. The relevant results are presented in
Figure 10. The estimation of the adsorbed monomers was performed following the “local” criterion as in our previous works [
27,
31,
35].
Figure 10.
Dependence of the number of the adsorbed LPE beads onto the branched polymers, on the linear chain length. Results of the non-stoichiometric G4MAX complexes of the present study, are compared to those from previous works (* [
27], ^ [
31] and
┴ [
35]). G3 DEND refers to complexes comprised by 3rd generation dendrimers while G4MAX for systems comprised by G4MAX hyperbranched molecules. Values corresponding of the stoichiometric complexes are doubled, for comparison purposes. Results from the explicit-ion simulations performed for the G3 non-stoichiometric systems are shown as well (
![]()
).
Figure 10.
Dependence of the number of the adsorbed LPE beads onto the branched polymers, on the linear chain length. Results of the non-stoichiometric G4MAX complexes of the present study, are compared to those from previous works (* [
27], ^ [
31] and
┴ [
35]). G3 DEND refers to complexes comprised by 3rd generation dendrimers while G4MAX for systems comprised by G4MAX hyperbranched molecules. Values corresponding of the stoichiometric complexes are doubled, for comparison purposes. Results from the explicit-ion simulations performed for the G3 non-stoichiometric systems are shown as well (
![]()
).
As was noted previously for the G3 systems [
35], no dependence of the number of the adsorbed beads on the stoichiometry is present for the G4MAX systems, as well. It appears, therefore, that for systems bearing two HBPs (either with a sparse or with a dense branching pattern) the degree of adsorption of the linear chain on the LPE is not affected by the conformational changes of the linear chain imparted by the presence of the second branched molecule.
4. Conclusions
In this work we have examined the effects of the HBP topology in non-stoichiometric complexes consisting of two terminally charged G4MAX hyperbranched polymers and an oppositely charged linear chain, by comparing relevant conformational properties to those characterizing similar stoichiometric systems or analogous models comprised by dendrimer components with the same number of charged monomers. It was found that the higher degree of deformability of the peripherally branched HBPs (see
Figure 1), drastically affected the conformational properties of both components of the complex, with respect to those of the stoichiometric counterparts or those in complexes formed by the densely branched dendrimer molecules. The step-wise changes to properties, like the shape and the average size which characterized the HBP component of the G4MAX stoichiometric models, have been smeared out (
Figure 4 and
Figure 5), while LPE conformational changes that have been observed in non-stoichiometric G3 complexes, were not present (
Figure 6). The latter effect could be accounted for by the absence of the corresponding non-monotonic dependence of the “linker” conformation of the LPE on its length in the examined G4MAX complexes (
Figure 8). This observation is consistent with a weak dependence of the average separation between the two G4MAX HBPs on the LPE length (
Figure 9). Such a behavior can be rationalized by the higher degree of deformation of the hyperbranched components so that the number of the energetically favorable contacts between the charged LPE and HBP monomers remains at the same levels as in their stoichiometric analogues (
Figure 10).
The above findings, which differentiate the behavior of the examined systems with respect to their stoichiometric counterparts and their dendrimer-based analogues, are expected to arise mainly by entropic factors which should be taken into account in future amendments of the correlation theory [
21,
27,
35,
52].
We believe that the results from the present study offer new insight regarding the structural features of hyperbranched/linear polyelectrolyte complexes and can be of particular importance in the efforts towards a molecular-level design of hyperbranched-based nanovectors with optimized properties.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
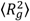 for the HBP refers to the average of the two G4MAX molecules of the complex.
for the HBP refers to the average of the two G4MAX molecules of the complex.
 ), compared to that from the analogous non-stoichiometric G3 systems (
), compared to that from the analogous non-stoichiometric G3 systems (  ) from [35]. Inset: dependence of the axes of the ellipsoid of inertia for the LPE participating in the G4MAX non-stoichiometric systems. The definition of the axes is identical to that of Figure 4.
) from [35]. Inset: dependence of the axes of the ellipsoid of inertia for the LPE participating in the G4MAX non-stoichiometric systems. The definition of the axes is identical to that of Figure 4.

 ).
).