
Synthesis and Characterization of Fully Bio-Based Butylene Succinate Oligomers with Varying Molecular Weights for Sustainable Food Packaging Applications

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Oligomerization
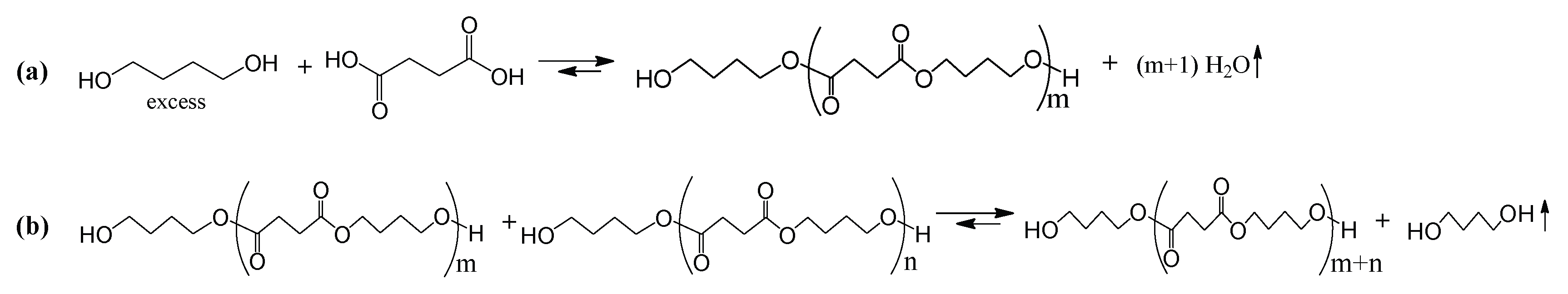
2.2.1. Esterification
2.2.2. Transesterification
2.3. Chemical Characterization
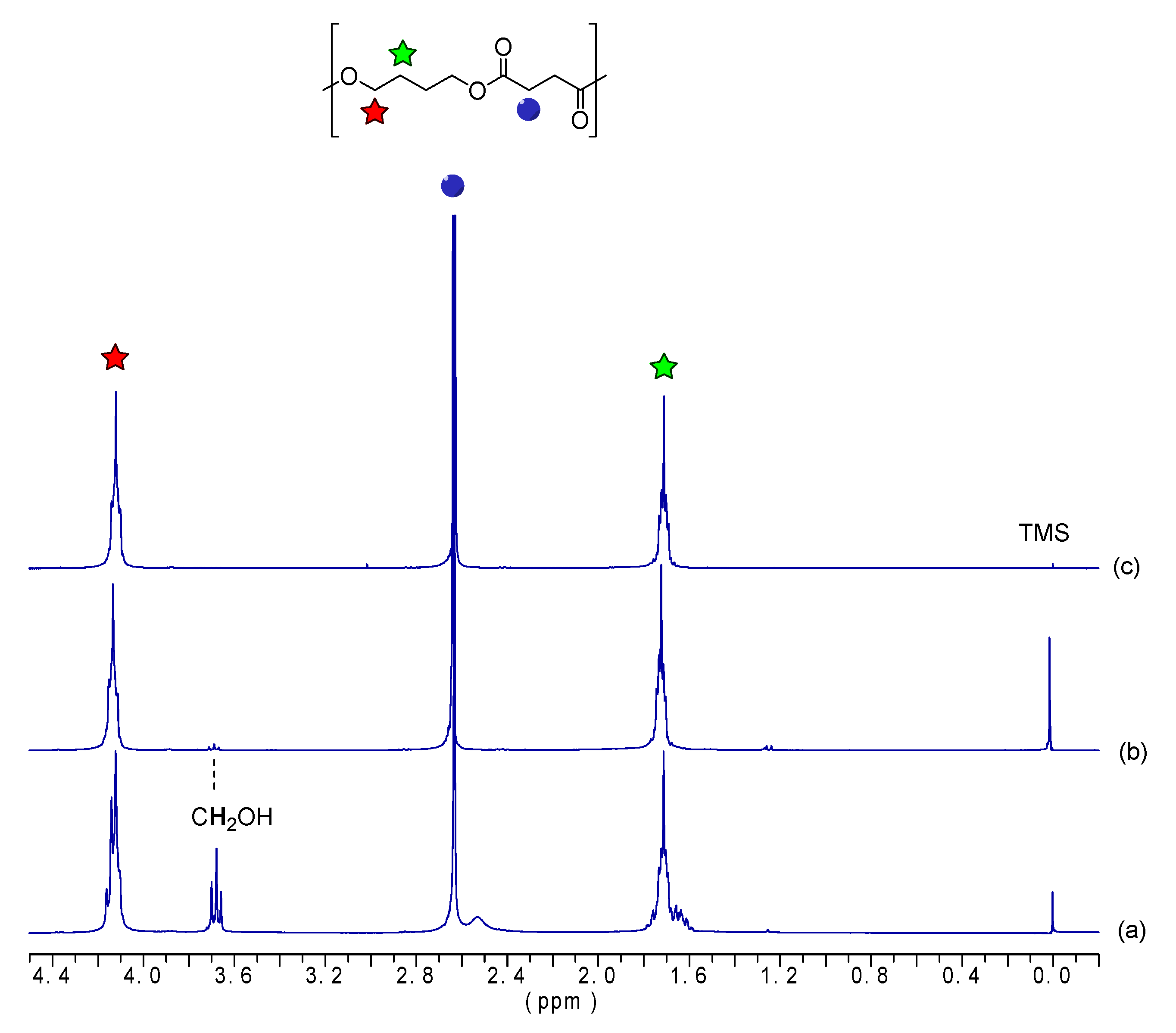
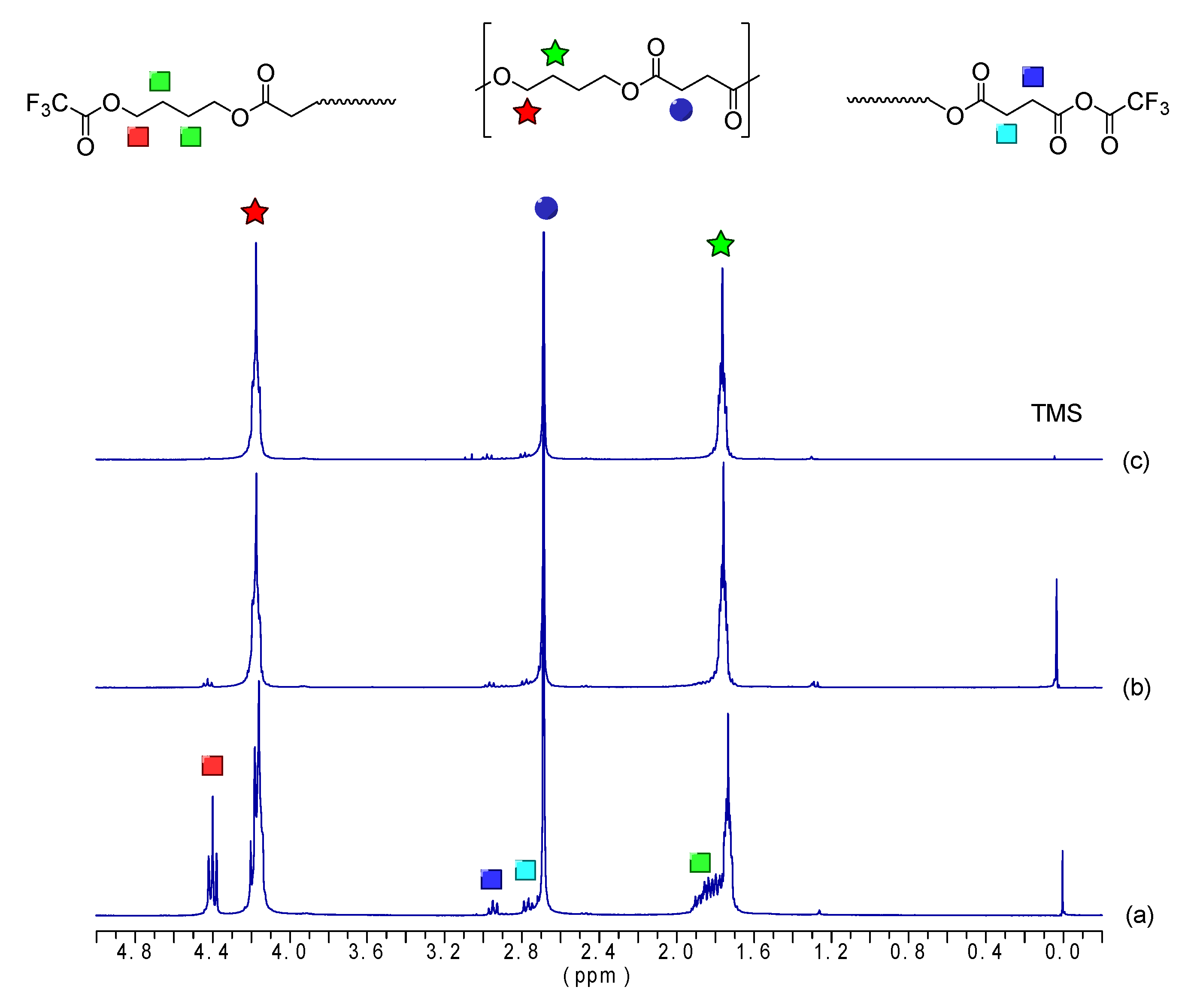
2.3.1. Nuclear Magnetic Resonance Spectroscopy
2.3.2. Gel Permeation Chromatography
2.4. Physical Characterization
2.4.1. Thermogravimetric Analysis
2.4.2. Differential Scanning Calorimetry
2.4.3. X-Ray Diffraction Analysis
2.5. Statistical Analysis
3. Results and Discussion
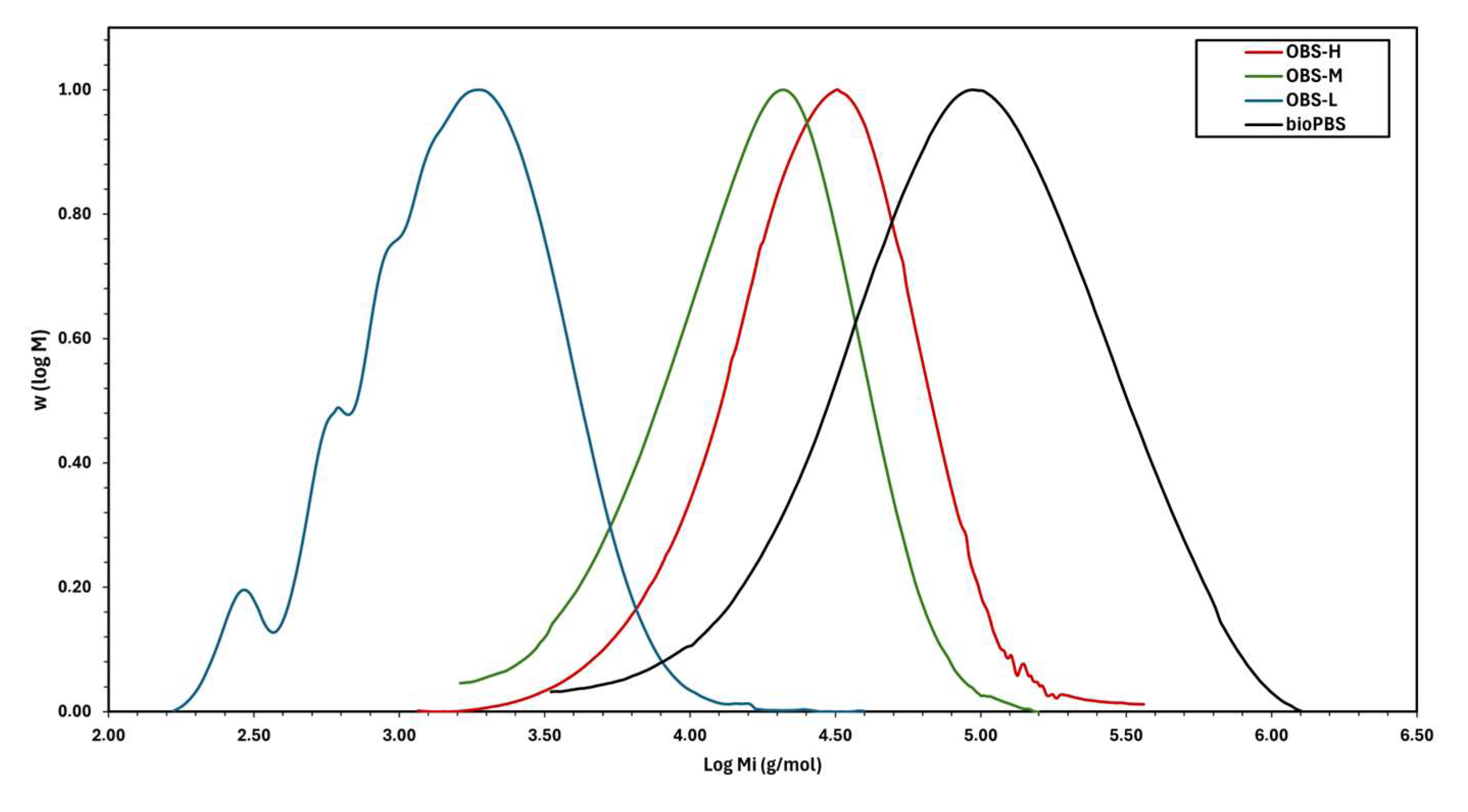
3.1. Molecular Weight of Oligomers
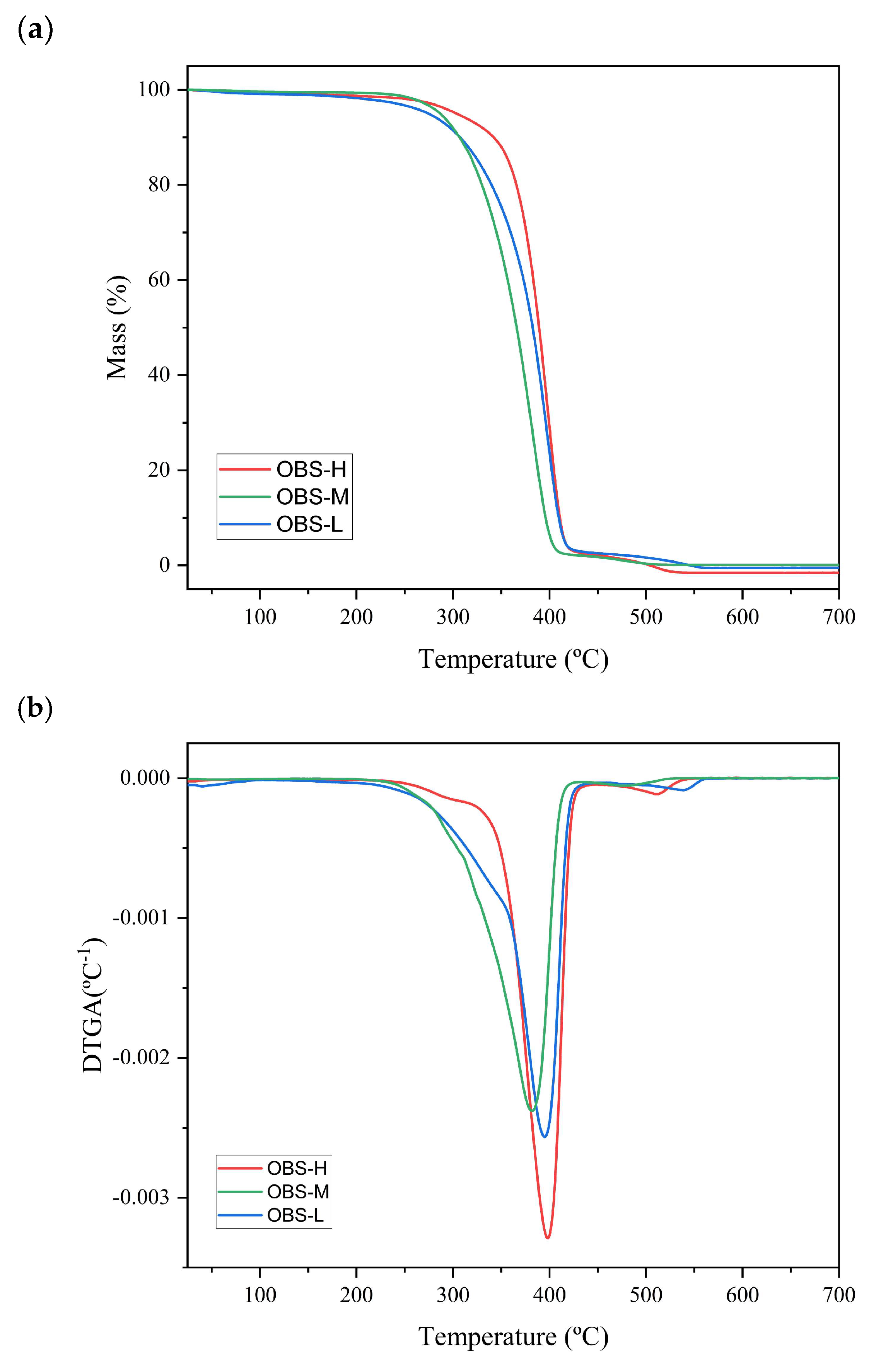
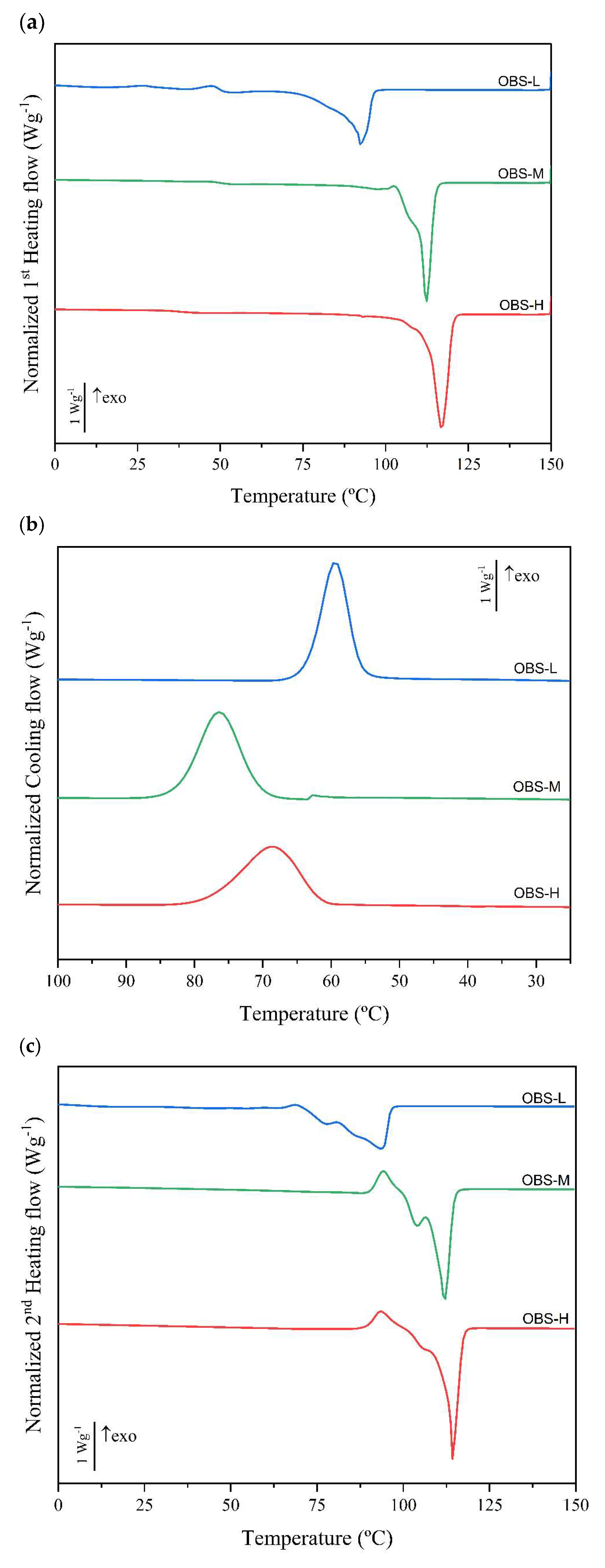
3.2. Thermal and Crystallization Behavior of Oligomers
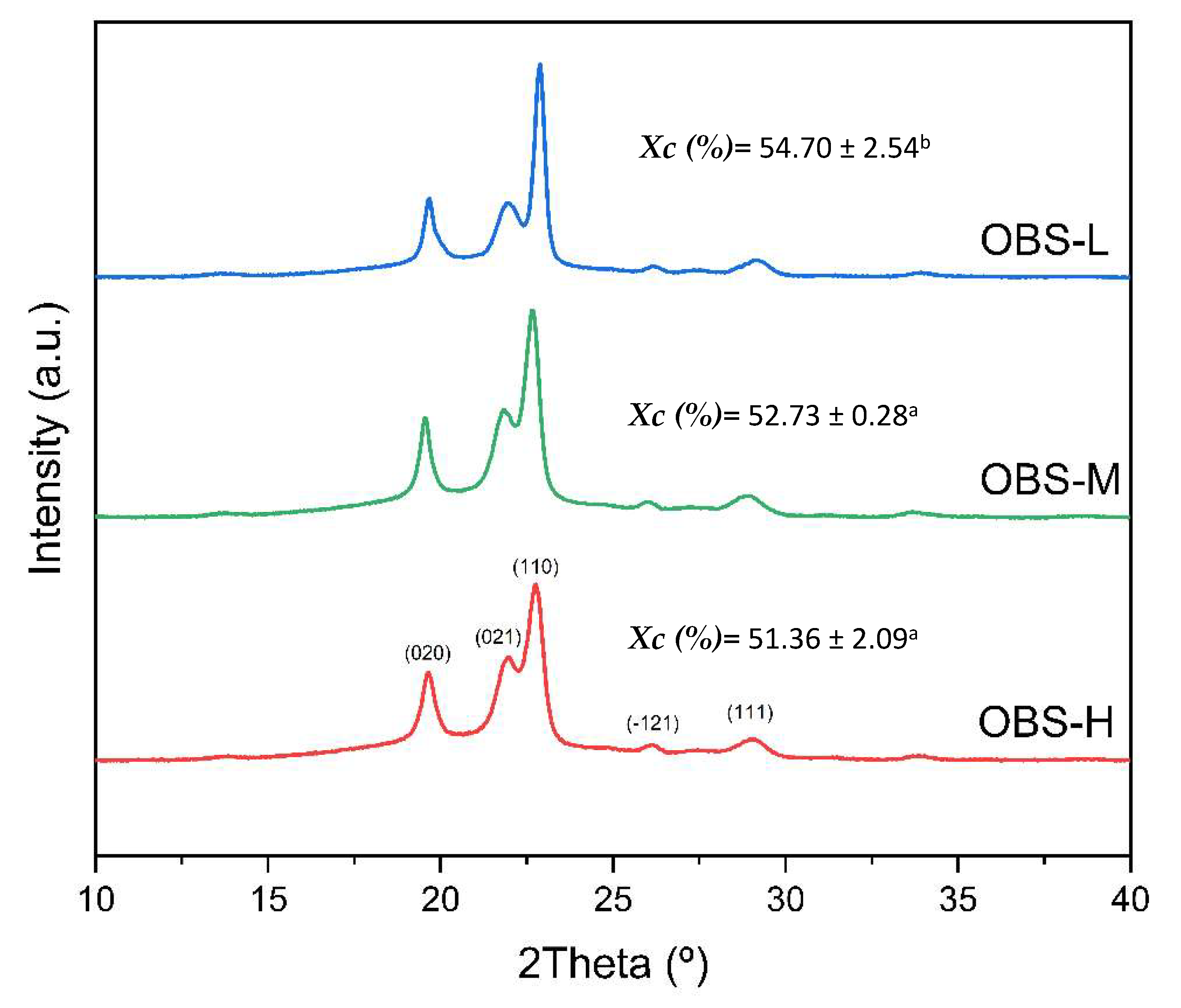
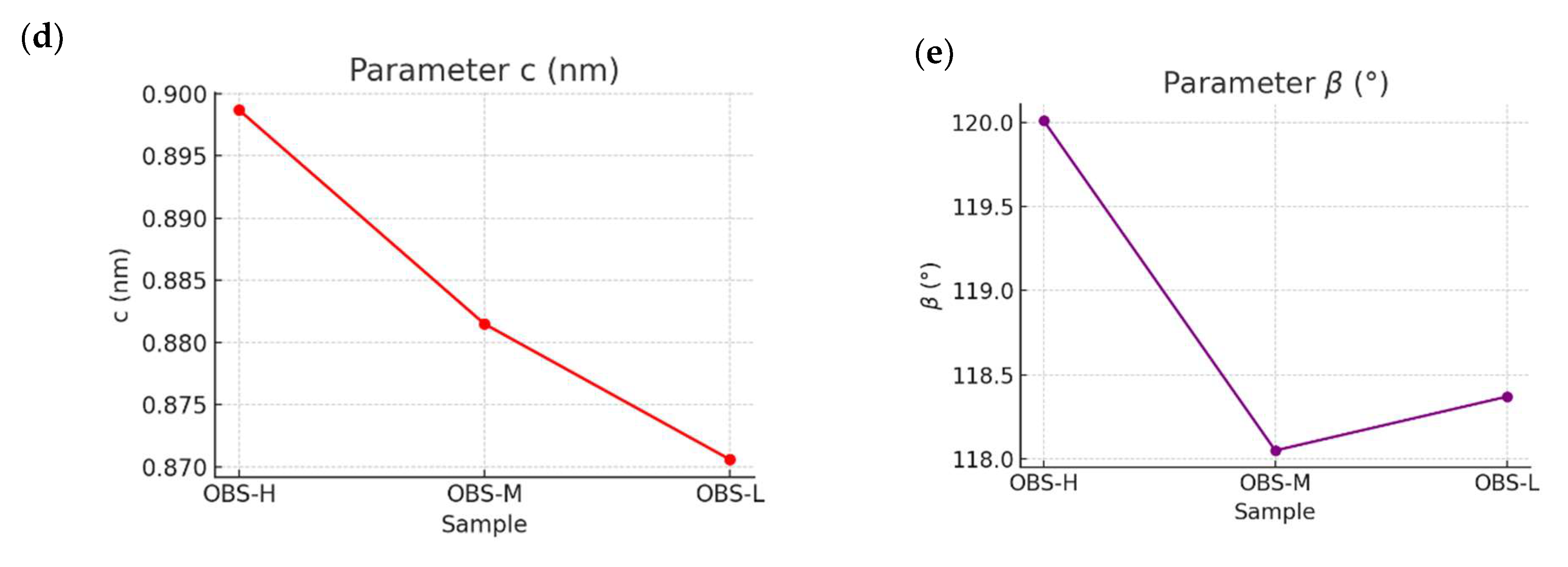
3.3. XRD Crystallinity Analysis of Oligomers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Adrah, K.; Ananey-Obiri, D.; Tahergorabi, R. Development of Bio-Based and Biodegradable Plastics. In Handbook of Nanomaterials and Nanocomposites for Energy and Environmental Applications; Springer International Publishing: Cham, Switzerland, 2020; pp. 3663–3687. [Google Scholar]
- Olivas-Alonso, C.; Freitas, P.A.V.; Torres-Giner, S.; Chiralt, A. Thermo-Compressed Films of Poly(butylene succinate) Reinforced with Cellulose Fibers Obtained from Rice Straw by Green Extraction Methods. Macromol. Mater. Eng. 2024, 309, 2400094. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Figueroa-Lopez, K.J.; Melendez-Rodriguez, B.; Prieto, C.; Pardo-Figuerez, M.; Lagaron, J.M. Emerging Trends in Biopolymers for Food Packaging. In Sustainable Food Packaging Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 1–33. [Google Scholar] [CrossRef]
- Samaniego-Aguilar, K.; Sanchez-Safont, E.; Pisa-Ripoll, I.; Torres-Giner, S.; Flores, Y.; Lagaron, J.M.; Cabedo, L.; Gamez-Perez, J. Performance Enhancement of Biopolyester Blends by Reactive Compatibilization with Maleic Anhydride-Grafted Poly(butylene succinate-co-adipate). Polymers 2024, 16, 2325. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Baschnagel, J.; Cangialosi, D.; Fukao, K.; Glynos, E.; Janssen, L.M.C.; Müller, M.; Muthukumar, M.; Steiner, U.; Xu, J.; et al. Processing Pathways Decide Polymer Properties at the Molecular Level. Macromolecules 2019, 52, 7146–7156. [Google Scholar] [CrossRef]
- Jasso-Gastinel, C.F.; Soltero-Martínez, J.F.A.; Mendizábal, E. Introduction: Modifiable Characteristics and Applications. In Modification of Polymer Properties; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 1–21. ISBN 9780323443982. [Google Scholar]
- Viecelli, N.C.; Lovatel, E.R.; Cardoso, E.M.; Filho, I.N. Quantitative Analysis of Plasticizers in a Wastewater Treatment Plant: Influence of the Suspended Solids Parameter. J. Braz. Chem. Soc. 2011, 22, 1150–1155. [Google Scholar] [CrossRef]
- Poussard, L.; Mecheri, A.; Mariage, J.; Barakat, I.; Bonnaud, L.; Raquez, J.M.; Dubois, P. Synthesis of Oligo(butylene succinate)-Based Polyurethanes: Influence of the Chemical Structure on Thermal and Mechanical Properties. J. Renew. Mater. 2014, 2, 13–22. [Google Scholar] [CrossRef]
- Yin, G.Z.; Yang, X.M.; Zhou, Z.; Li, Q.F. A Green Pathway to Adjust the Mechanical Properties and Degradation Rate of PCL by Blending Bio-Sourced Poly(glycerol-succinate) Oligoesters. Mater. Chem. Front. 2018, 2, 544–553. [Google Scholar] [CrossRef]
- Bernabé, I.; Amarilla, E.; de la Orden, M.U.; Martínez Urreaga, J.; Beltrán, F.R. Effect of Oligomeric Lactic Acid Plasticizer on the Mechanical Recycling of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Environ. Sci. Pollut. Res. 2024. [Google Scholar] [CrossRef]
- Defelice, J.; Lipson, J.E.G. The Influence of Additives on Polymer Matrix Mobility and the Glass Transition. Soft Matter 2021, 17, 376–387. [Google Scholar] [CrossRef]
- Niiyama, E.; Uto, K.; Ebara, M. Electrospun PCL-PCL Polyblend Nanofibers with High- And Low-Molecular Weight for Controlled Degradation. Chem. Lett. 2019, 48, 623–626. [Google Scholar] [CrossRef]
- Burgos, N.; Tolaguera, D.; Fiori, S.; Jiménez, A. Synthesis and Characterization of Lactic Acid Oligomers: Evaluation of Performance as Poly(Lactic Acid) Plasticizers. J. Polym. Environ. 2014, 22, 227–235. [Google Scholar] [CrossRef]
- Ambrosio-Martín, J.; Fabra, M.J.; Lopez-Rubio, A.; Lagaron, J.M. An Effect of Lactic Acid Oligomers on the Barrier Properties of Polylactide. J. Mater. Sci. 2014, 49, 2975–2986. [Google Scholar] [CrossRef]
- Terroba-Delicado, E.; Fiori, S.; Gomez-Caturla, J.; Montanes, N.; Sanchez-Nacher, L.; Torres-Giner, S. Valorization of Liquor Waste Derived Spent Coffee Grains for the Development of Injection-Molded Polylactide Pieces of Interest as Disposable Food Packaging and Serving Materials. Foods 2022, 11, 1162. [Google Scholar] [CrossRef] [PubMed]
- Flores, Y.; Pacheco-Romeralo, M.; Mboumba, E.; Alla, A.; Lagaron, J.M.; Lopez Granados, M.; Martínez de Ilarduya, A.; Perret, N.; Torres-Giner, S. High-Pressure Catalytic Hydrogenation of Renewable Succinic Acid to 1,4-Butanediol for the Synthesis of Fully Bio-Based Poly(butylene succinate). Am. Chem. Soc. 2025, in press. [CrossRef]
- Peñas, M.I.; Pérez-Camargo, R.A.; Hernández, R.; Müller, A.J. A Review on Current Strategies for the Modulation of Thermomechanical, Barrier, and Biodegradation Properties of Poly(butylene succinate) (PBS) and Its Random Copolymers. Polymers 2022, 14, 1025. [Google Scholar] [CrossRef] [PubMed]
- Savitha, K.S.; Ravji Paghadar, B.; Senthil Kumar, M.; Jagadish, R.L. Polybutylene Succinate, a Potential Bio-Degradable Polymer: Synthesis, Copolymerization and Bio-Degradation. Polym. Chem. 2022, 13, 3562–3612. [Google Scholar] [CrossRef]
- Yang, R.; Xu, G.; Dong, B.; Guo, X.; Wang, Q. Selective, Sequential, and “One-Pot” Depolymerization Strategies for Chemical Recycling of Commercial Plastics and Mixed Plastics. ACS Sustain. Chem. Eng. 2022, 10, 9860–9871. [Google Scholar] [CrossRef]
- Rajgond, V.; Mohite, A.; More, N.; More, A. Biodegradable Polyester-Polybutylene Succinate (PBS): A Review. Polym. Bull. 2024, 81, 5703–5752. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X.W.; Ye, H.M. Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight. Crystals 2021, 11, 1530. [Google Scholar] [CrossRef]
- Hernández-García, E.; Pacheco-Romeralo, M.; Pascual-Ramírez, L.; Vargas, M.; Torres-Giner, S. Synthesis and Characterization of Polyamide 1010 and Evaluation of Its Cast-Extruded Films for Meat Preservation. Food Packag. Shelf Life 2023, 36, 101058. [Google Scholar] [CrossRef]
- Chun Na Cui, J.T.H. Synthesis and Characterization of Poly(butylenes succinate) with Control Different End-Group. Adv. Mater. Res. 2012, 430–432, 497–500. [Google Scholar]
- Vandermeulen, G.W.M.; Boarino, A.; Klok, H.A. Biodegradation of Water-Soluble and Water-Dispersible Polymers for Agricultural, Consumer, and Industrial Applications—Challenges and Opportunities for Sustainable Materials Solutions. J. Polym. Sci. 2022, 60, 1797–1813. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Kausch, H.H. GPC Data Interpretation in Mechanochemical Polymer Degradation. Int. J. Polym. Anal. Charact. 1998, 4, 447–470. [Google Scholar] [CrossRef]
- Held, D.; Kilz, P. Size-Exclusion Chromatography as a Useful Tool for the Assessment of Polymer Quality and Determination of Macromolecular Properties. Chem. Teach. Int. 2021, 3, 77–103. [Google Scholar] [CrossRef]
- Gkountela, C.I.; Markoulakis, D.; Mathioudaki, M.; Pitterou, I.; Detsi, A.; Vouyiouka, S.N. Scalable Enzymatic Polymerization and Low-Temperature Post-Polymerization of Poly(butylene succinate): Process Parameters and Application. Eur. Polym. J. 2023, 198, 112423. [Google Scholar] [CrossRef]
- Liu, G.C.; Zhang, W.Q.; Zhou, S.L.; Wang, X.L.; Wang, Y.Z. Improving Crystallization and Processability of PBS: Via Slight Cross-Linking. RSC Adv. 2016, 6, 68942–68951. [Google Scholar] [CrossRef]
- Wu, S.S.; Lu, H.J.; Li, Y.D.; Zhang, S.D.; Zeng, J.B. Unlocking the Potential of Poly(butylene succinate) through Incorporation of Vitrimeric Network Based on Dynamic Imine Bonds. Chin. J. Polym. Sci. 2024, 42, 1414–1424. [Google Scholar] [CrossRef]
- Gowman, A.; Wang, T.; Rodriguez-Uribe, A.; Mohanty, A.K.; Misra, M. Bio-Poly(butylene succinate) and Its Composites with Grape Pomace: Mechanical Performance and Thermal Properties. ACS Omega 2018, 3, 15205–15216. [Google Scholar] [CrossRef]
- Chen, K.; Li, P.; Li, X.; Liao, C.; Li, X.; Zuo, Y. Effect of Silane Coupling Agent on Compatibility Interface and Properties of Wheat Straw/Polylactic Acid Composites. Int. J. Biol. Macromol. 2021, 182, 2108–2116. [Google Scholar] [CrossRef]
- Feijoo, P.; Samaniego-Aguilar, K.; Sánchez-Safont, E.; Torres-Giner, S.; Lagaron, J.M.; Gamez-Perez, J.; Cabedo, L. Development and Characterization of Fully Renewable and Biodegradable Polyhydroxyalkanoate Blends with Improved Thermoformability. Polymers 2022, 14, 2527. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, J.; Li, L. Multiple Melting Behavior of Poly(butylene succinate). Eur. Polym. J. 2007, 43, 3163–3170. [Google Scholar] [CrossRef]
- Kelly, A.; Knowles, K.M. Appendix 3: Interplanar Spacings and Interplanar Angles. In Crystallography and Crystal Defects; Wiley: Hoboken, NJ, USA, 2012; pp. 469–472. [Google Scholar]
- Lee, C.W.; Masutani, K.; Kimura, Y. Ring-Opening Polymerization of a Macrocyclic Lactone Monomer Isolated from Oligomeric Byproducts of Poly(butylene succinate) (PBS): An Efficient Route to High-Molecular-Weight PBS and Block Copolymers of PBS. Polymer 2014, 55, 5673–5679. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, C.; Ouyang, C.; Zeng, X.; Guo, Z.; Lai, F.; Li, J. Analysis of Oligomers in Poly (Butylene Succinate) and Poly (Butylene Adipate-co-Terephthalate). Polym. Bull. 2023, 80, 4487–4502. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, X.; Wei, B.; Wang, X.; Lu, W.; Dai, J.; Xu, S.; Zha, Q. Biodegradable Polybutylene Succinate: Purified Method and Oligomers Investigation. Mater. Today Commun. 2024, 39, 109157. [Google Scholar] [CrossRef]
- Charlier, Q.; Girard, E.; Freyermouth, F.; Vandesteene, M.; Jacquel, N.; Ladavière, C.; Rousseau, A.; Fenouillot, F. Solution Viscosity -Molar Mass Relationships for Poly(butylene succinate) and Discussion on Molar Mass Analysis. Express Polym. Lett. 2015, 9, 424–434. [Google Scholar] [CrossRef]
- Gomoll, A.; Hallstein, J.; Lieske, A.; Metzsch-Zilligen, E.; Pfaendner, R.; Zehm, D. Unraveling the Cause for the Unusual Processing Behavior of Poly(butylene succinate). Part 2. The Effect of Synthesis on the Processing Stability of Poly(Butylene Succinate). J. Appl. Polym. Sci. 2025, 142, e56897. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Vouyiouka, S.; Georgousopoulou, I.-N.; Marinkovic, S.; Estrine, B.; Joly, C.; Dole, P. Feasibility of Solid-State Postpolymerization on Fossil- and Bio-Based Poly(butylene succinate) Including Polymer Upcycling Routes. Ind. Eng. Chem. Res. 2016, 55, 5832–5842. [Google Scholar] [CrossRef]
- Khopade, K.V.; Chikkali, S.H.; Barsu, N. Metal-Catalyzed Plastic Depolymerization. Cell Rep. Phys. Sci. 2023, 4, 101341. [Google Scholar] [CrossRef]
- Chiarcos, R.; Sparnacci, K.; Antonioli, D.; Ivaldi, C.; Gianotti, V.; Po, R.; Biagini, P.; Losio, S.; Laus, M. Catalyst Residues Severely Impact the Thermal Stability and Degradation Mechanism of Polycarbonates: How to Turn a Flaw into an Opportunity. Eur. Polym. J. 2024, 214, 113148. [Google Scholar] [CrossRef]
- Ndiaye, M.; Myler, P.; Kandola, B.K. Thermoplastic Composites: Modelling Melting, Decomposition and Combustion of Matrix Polymers. J. Compos. Sci. 2022, 6, 27. [Google Scholar] [CrossRef]
- Hernández-García, E.; Pacheco-Romeralo, M.; Zomeño, P.; Viscusi, G.; Malvano, F.; Gorrasi, G.; Torres-Giner, S. Development and Characterization of Thermoformed Bilayer Trays of Paper and Renewable Succinic Acid Derived Biopolyester Blends and Their Application to Preserve Fresh Pasta. Materials 2023, 16, 3872. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L.; Cinelli, P.; Gazzano, M. Temperature Dependence of the Rigid Amorphous Fraction of Poly(butylene succinate). RSC Adv. 2021, 11, 25731–25737. [Google Scholar] [CrossRef]
- Lv, Z.y.; Zhang, M.C.; Zhang, Y.; Guo, B.h.; Xu, J. Study on Melting and Recrystallization of Poly(butylene succinate) Lamellar Crystals via Step Heating Differential Scanning Calorimetry. Chin. J. Polym. Sci. 2017, 35, 1552–1560. [Google Scholar] [CrossRef]
- Yoo, E.S.; Im, S.S. Melting Behavior of Poly(butylene succinate) during Heating Scan by DSC. J. Polym. Sci. B Polym. Phys. 1999, 37, 1357–1366. [Google Scholar] [CrossRef]
- Ferreira, L.P.; Moreira, A.N.; Pinto, J.C.; De Souza, F.G. Synthesis of Poly(butylene succinate) Using Metal Catalysts. Polym. Eng. Sci. 2015, 55, 1889–1896. [Google Scholar] [CrossRef]
- Garin, M.; Tighzert, L.; Vroman, I.; Marinkovic, S.; Estrine, B. The Kinetics of Poly(butylene succinate) Synthesis and the Influence of Molar Mass on Its Thermal Properties. J. Appl. Polym. Sci. 2014, 131, 40639. [Google Scholar] [CrossRef]
- Zeng, J.B.; Huang, C.L.; Jiao, L.; Lu, X.; Wang, Y.Z.; Wang, X.L. Synthesis and Properties of Biodegradable Poly(butylene succinate-co-diethylene glycol succinate) Copolymers. Ind. Eng. Chem. Res. 2012, 51, 12258–12265. [Google Scholar] [CrossRef]
- Hariraksapitak, P.; Suwantong, O.; Pavasant, P.; Supaphol, P. Effectual Drug-Releasing Porous Scaffolds from 1,6-Diisocyanatohexane-Extended Poly(1,4-butylene succinate) for Bone Tissue Regeneration. Polymer 2008, 49, 2678–2685. [Google Scholar] [CrossRef]
- Palamidi, A.; Kapourani, A.; Christodoulou, E.; Klonos, P.A.; Kontogiannopoulos, K.N.; Kyritsis, A.; Bikiaris, D.N.; Barmpalexis, P. Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications. Polymers 2021, 13, 146. [Google Scholar] [CrossRef]
- Peng, S.; Bu, Z.; Wu, L.; Li, B.G.; Dubois, P. High Molecular Weight Poly(butylene succinate-co-furandicarboxylate) with 10 Mol% of BF Unit: Synthesis, Crystallization-Melting Behavior and Mechanical Properties. Eur. Polym. J. 2017, 96, 248–255. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.; Han, J.; Xie, Z.; Xu, J.; Guo, B. Copolymerization with Polyether Segments Improves the Mechanical Properties of Biodegradable Polyesters. ACS Omega 2017, 2, 2639–2648. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L.; Cavallo, D.; Müller, A.J.; Gazzano, M. Structural Evolution of Poly(butylene succinate) Crystals on Heating with the Formation of a Dual Lamellar Population, as Monitored by Temperature-Dependent WAXS/SAXS Analysis. Polymer 2023, 268, 125711. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CH2OOC | CH2OH | CH2OTFA | CH2COO | CH2COTFA | CH2CH2O |
| Chemical Shifts | ||||||
| δ = 4.12 | δ = 3.67 | δ = 4.40 | δ = 2.63 | δ = 2.95 | δ = 1.70 | |
| Integrals before Derivatization | ||||||
| OBS-H | 572.2 | 2.0 | - | 576.0 | - | 574.0 |
| OBS-M | 110.7 | 2.0 | - | 110.9 | - | 112.5 |
| OBS-L | 8.1 | 2.0 | - | 6.6 | - | 10.1 |
| Integrals after derivatization | ||||||
| OBS-H | 573.0 | - | 2.0 | 570.0 | 5.6 | 574.5 |
| OBS-M | 110.5 | - | 2.0 | 109.3 | 1.3 | 112.0 |
| OBS-L | 8.1 | - | 2.0 | 6.4 | 0.3 | 10.1 |
| Sample | Mn (g·mol−1) a | Mn (g·mol−1) b | Mw (g·mol−1) b | Ð |
|---|---|---|---|---|
| OBS-H | 12,900 | 18,650 | 33,147 | 1.7 |
| OBS-M | 5950 | 8700 | 16,150 | 1.9 |
| OBS-L | 900 | 1100 | 2050 | 1.8 |
| Oligomer | 25–450 °C | ||
|---|---|---|---|
| Tonset (°C) | Tpeak (°C) | Tend (°C) | |
| OBS-H | 249 ± 4 c | 398 ± 0 b | 423 ± 3 b |
| OBS-M | 223 ± 1 b | 379± 2 a | 410 ± 1 a |
| OBS-L | 183 ± 3 a | 396 ± 1 b | 420 ± 1 b |
| Oligomer | Cooling | Second Heating | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Tc (°C) | ∆Hc (J/g) | Tcc (°C) | Tm1 (°C) | Tm2 (°C) | ∆Hm (J/g) | Xc (%) (ΔH°m = 110.3 J/g) | Xc (%) (ΔH°m = 102 J/g) | Xc (%) (ΔH°m = 200 J/g) | |
| OBS-H | 69.4 ± 1.2 b | 73.0 ± 2.6 a | 93.8 ± 0.4 c | 106.62 ± 0.3 c | 114.3 ± 0.1 c | 73.5 ± 0.7 a | 66.6 ± 0.6 a | 72.0 ± 0.6 a | 36.7 ± 0.3 a |
| OBS-M | 76.7 ± 0.4 c | 83.4 ± 2.9 a | 94.3 ± 0.1 b | 103.77 ± 0.4 b | 111.7 ± 0.2 b | 70.8 ± 3.3 a | 64.2 ± 2.9 a | 69.4 ± 3.2 a | 35.4 ± 1.6 a |
| OBS-L | 59.0 ± 0.5 a | 68.6 ± 8.7 a | 71.1 ± 0.9 a | 79.81 ± 0.8 a | 95.6 ± 0.7 a | 78.9 ± 4.5 a | 71.6 ± 4.1 a | 77.4 ± 4.4 a | 39.4 ± 2.2 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Alonso, C.; Flores, Y.; Martínez de Ilarduya, A.; Chiralt, A.; Torres-Giner, S. Synthesis and Characterization of Fully Bio-Based Butylene Succinate Oligomers with Varying Molecular Weights for Sustainable Food Packaging Applications. Polymers 2025, 17, 1276. https://doi.org/10.3390/polym17091276
Olivas-Alonso C, Flores Y, Martínez de Ilarduya A, Chiralt A, Torres-Giner S. Synthesis and Characterization of Fully Bio-Based Butylene Succinate Oligomers with Varying Molecular Weights for Sustainable Food Packaging Applications. Polymers. 2025; 17(9):1276. https://doi.org/10.3390/polym17091276
Chicago/Turabian StyleOlivas-Alonso, Carmen, Yaiza Flores, Antxon Martínez de Ilarduya, Amparo Chiralt, and Sergio Torres-Giner. 2025. "Synthesis and Characterization of Fully Bio-Based Butylene Succinate Oligomers with Varying Molecular Weights for Sustainable Food Packaging Applications" Polymers 17, no. 9: 1276. https://doi.org/10.3390/polym17091276
APA StyleOlivas-Alonso, C., Flores, Y., Martínez de Ilarduya, A., Chiralt, A., & Torres-Giner, S. (2025). Synthesis and Characterization of Fully Bio-Based Butylene Succinate Oligomers with Varying Molecular Weights for Sustainable Food Packaging Applications. Polymers, 17(9), 1276. https://doi.org/10.3390/polym17091276