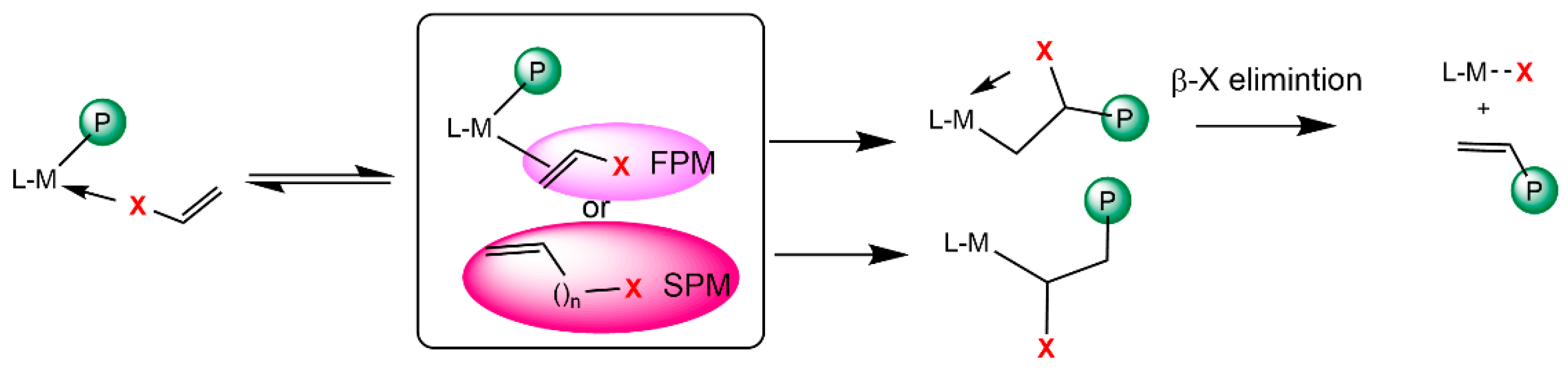
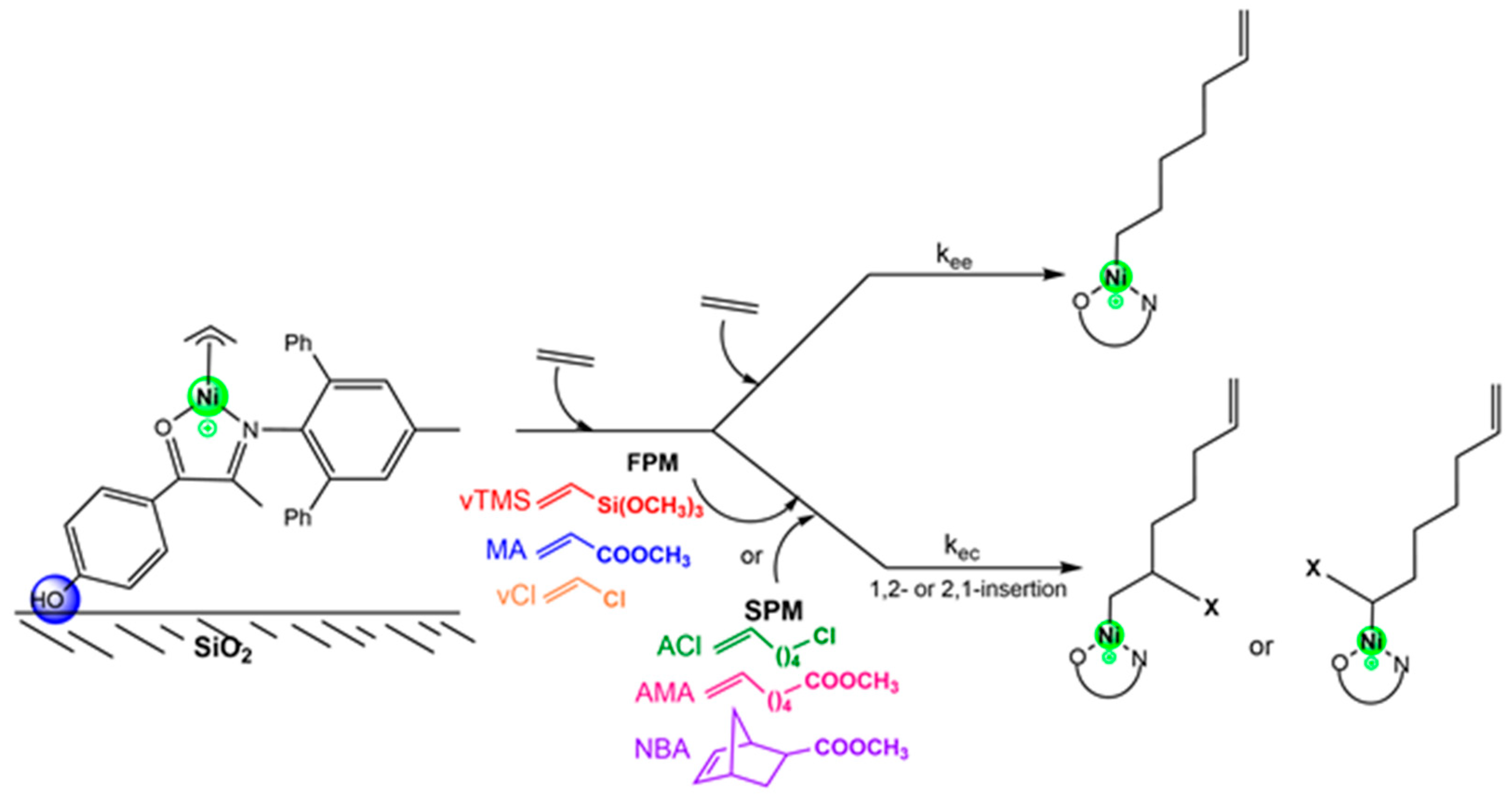
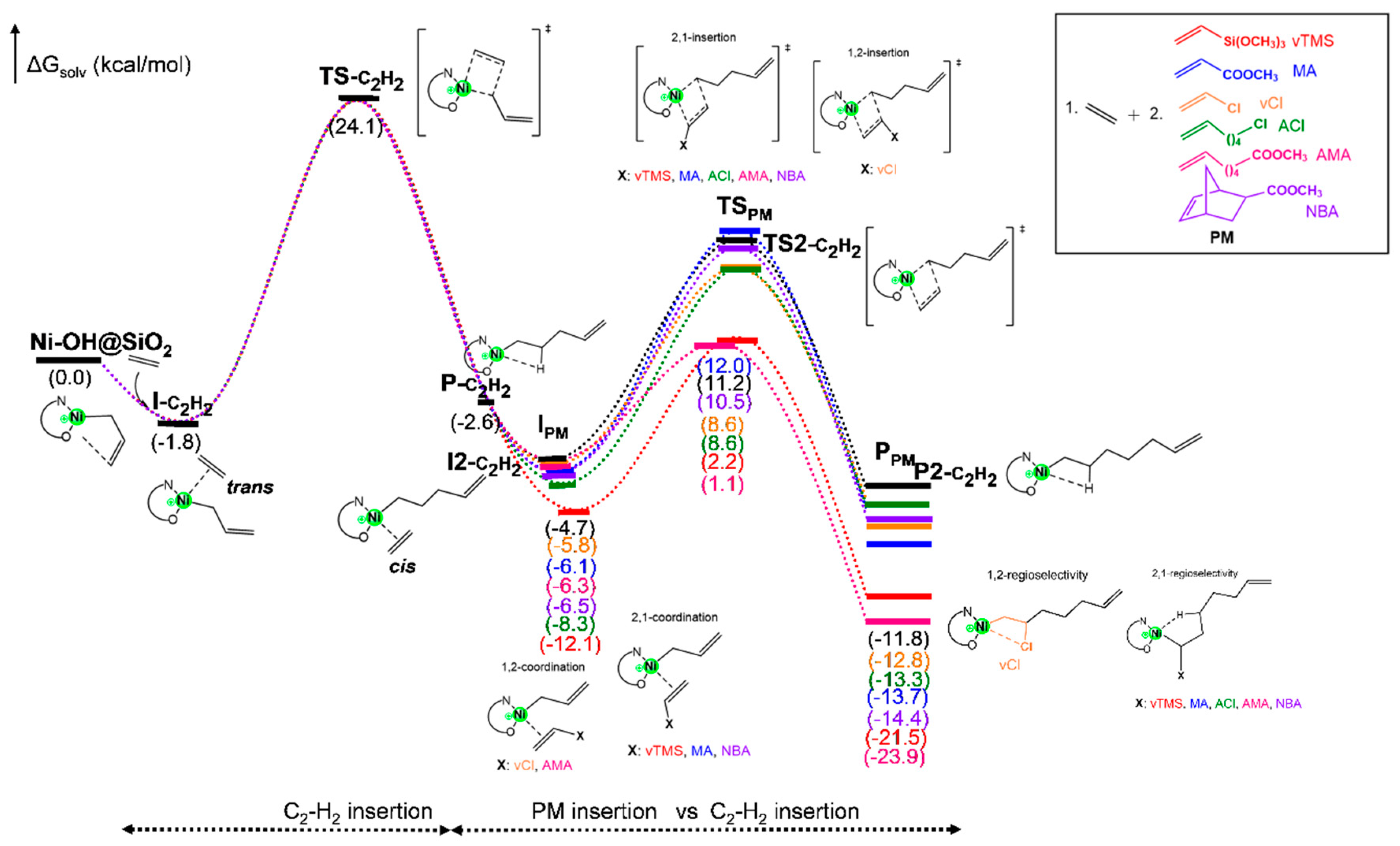
The reaction mechanism for the ethylene copolymerization with polar monomers (PM) was studied based on the chain initiation and propagation steps by ethylene insertion followed by PM insertion. The preferential insertion of ethylene is supported by its lower steric hindrance and higher intrinsic reactivity under mild conditions, as evidenced by experimental studies [
22,
44,
45]. In contrast, polar comonomers typically exhibit reduced reactivity and tend to suppress catalytic activity, particularly at early stages, due to electronic and steric constraints. Therefore, two main precursors are involved in the reaction pathway. These are (i) the Ni–allyl complex (
Figure 1; Ni-OH@SiO
2) and (ii) the Ni–alkyl complex, which is the catalytically active species formed after the insertion of the first monomer (ethylene) into the Ni–allyl precursor (
Figure 1; P
C2H2). This species serves as the reference point for subsequent monomer insertions. To rationally characterize the activity of heterogenization of Ni–α-imino ketone molecular catalyst for ethylene copolymerization reactions, the Gibbs free energy profile is discussed below (
Figure 1). We first investigated the ethylene insertion into the Ni-OH@SiO
2, followed by the PM insertion into the P
C2H2 species, as is shown in
Figure 1. From our previous studies [
21,
46], we found that the ethylene uptake is more stable
trans to the ketone moiety (monomer
trans to the O atom) by almost 2.0 kcal/mol (I
C2H2). Then, the ethylene insertion (TS
C2H2) has a barrier of 25.9 kcal/mol exergonically, forming a stable β-agostic complex P
C2H2. We further investigated the polar monomer (PM) insertion vs. the ethylene insertion in the P
C2H2 species.
3.1. Insights into the Feasibility of Incorporation of PMs into Ni-OH@SiO2 Catalyst
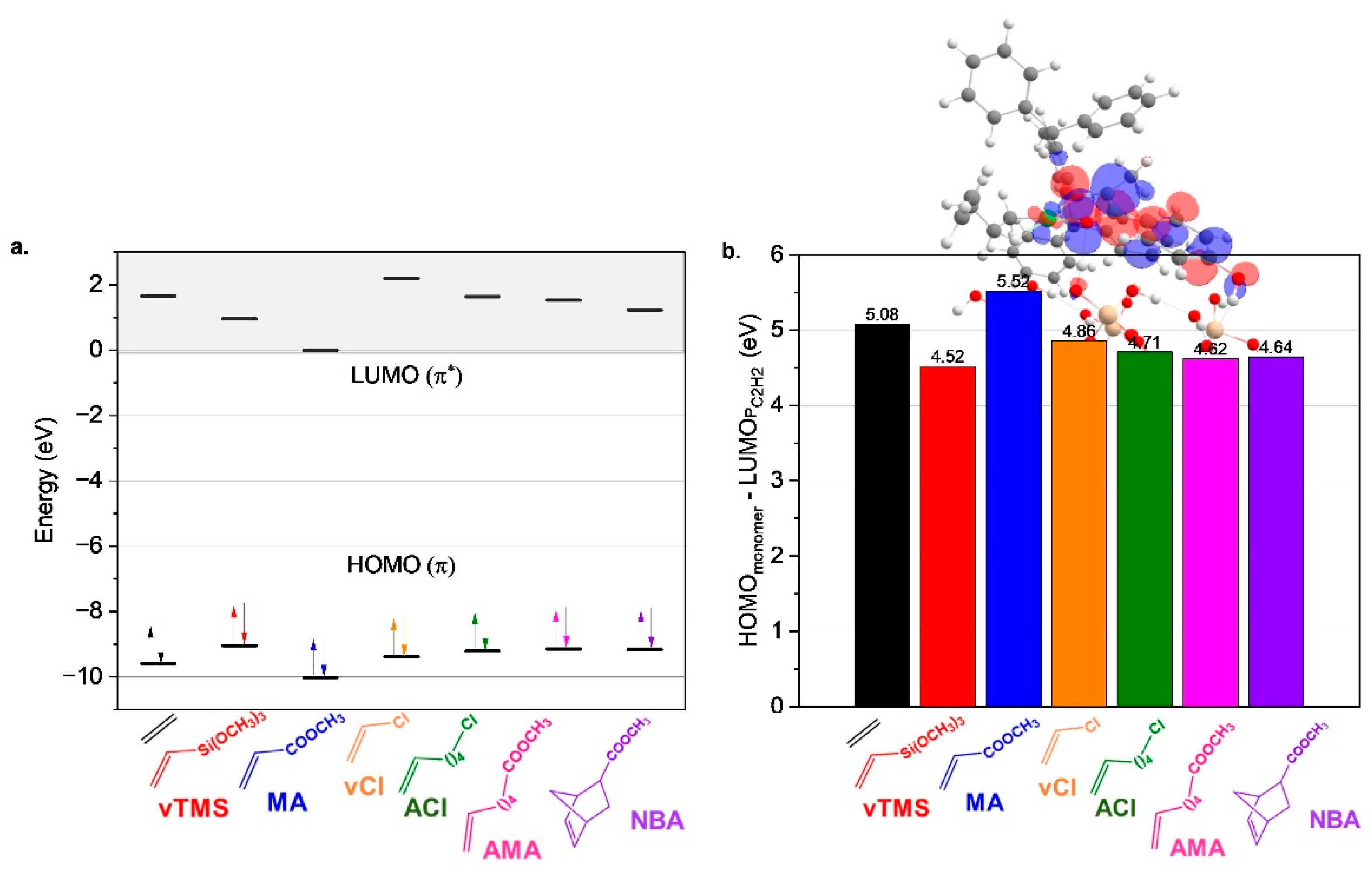
A total of six polar monomers (PMs) were investigated for ethylene copolymerization (
Figure 1). The monomer coordination properties were predicted based on the energies of the HOMO (π C=C bonding orbital) and LUMO (π* C=C antibonding orbital) of ethylene and the polar monomers (
Figure 2a). The HOMO energies of the polar monomers differ significantly from that of ethylene, exhibiting higher values (−9.05 to −9.38 eV vs. −9.60 eV), with the notable exception of methyl acrylate (MA), which has the lowest HOMO energy (−10.03 eV). This suggests that most polar monomers exhibit stronger σ-donation to the catalyst, forming stronger carbon–metal π-bonds during the copolymerization process. Among the monomers, vinyltrimethylsilane (vTMS) is predicted to show the strongest π-complexation due to its highest HOMO energy (−9.05 eV), whereas MA exhibits the weakest coordination. Vinyl chloride (vCl), with a HOMO energy of −9.38 eV, is predicted to have the second weakest performance after MA. For monomers with similar HOMO energies, such as alkyl chloride (ACl), alkyl methacrylate (AMA), and norbornene acetate (NBA) (~−9.2 eV), comparable σ-donation to the metal center is anticipated.
Insights into the back-donation from the catalyst to the monomer were obtained from the LUMO energies of the monomers (
Figure 2a), which is critical for stabilizing the π-complex. MA has the lowest LUMO energy (0.01 eV), indicating it could facilitate substantial back-donation compared to other polar monomers, whose LUMO energies range between 0.97 and 2.19 eV. Particularly, vCl has the highest LUMO energy (2.19 eV), predicting the weakest back-donation. The back-donation trend is as follows: vCl (2.19 eV) < ethylene (1.66 eV) ≈ ACl (1.64 eV) < AMA (1.53 eV) < NBA (1.23 eV) < vTMS (0.97 eV). This trend highlights that vTMS is expected to exhibit the most favorable back-donation among the polar monomers.
In addition to these insights, we analyzed the interaction between the HOMO and LUMO of the monomers and alkyl product formed after the first ethylene insertion (P
C2H2), respectively.
Figure 2b shows that the LUMO of P
C2H2 is mainly localized on the nickel center and the imine ketone ligand, providing an important descriptor of coordination strength. The HOMO-LUMO energy difference in MA is the largest (5.52 eV), indicating the most challenging coordination. In comparison, the HOMO-LUMO energy difference of vTMS is the smallest (4.52 eV), suggesting the easiest coordination among the polar monomers. The gaps for the other monomers follow the trend: ethylene (5.08 eV) > vCl (4.86 eV) > ACl (4.71 eV) > AMA ≈ NBA (~4.60 eV) > vTMS (4.52 eV). The study predicts that vTMS will have the strongest coordination properties among polar monomers due to favorable HOMO and LUMO energies, while MA will have weak coordination behavior. However, all polar monomers are predicted to coordinate more effectively with the catalyst than ethylene, except for MA, which is consistent with its low incorporation experimentally observed in supported Ni-based catalysts, such as in the study by Zhang et al. [
44], where MA incorporation was limited to ~0.5–0.7 mol%. This highlights the diverse electronic properties of the polar monomers and their potential to influence the copolymerization process.
Lastly, we have examined the coordination of polar monomers (PMs) into P
C2H2 (I
PM species in
Figure 1). The coordination of the PMs taking place at the
cis site of the ketone moiety consistently yielded more stable π-complexes. Consequently, this study focused exclusively on the favorable
cis insertion of monomers. Among the polar monomers, comparative energy data are reported in
Table S1, in which it is shown that the 1,2-coordination mode to the nickel center was preferred for vCl and AMA comonomers. Interestingly, despite AMA forming a 1,2-coordinated π-complex, the transition state for insertion is more stable in the 2,1 orientation. In contrast, the 2,1-coordination mode was favored for vTMS, MA, ACl, and NBA. The formation of I
PM species was exergonic for all PMs (
Figure 1). These findings are consistent with the experimental evidence [
47], in which MA undergoes 2,1-insertion exclusively, whereas other polar monomers like vCl insert via the 1,2 pathway. Notably, all I
PM species exhibited more exergonic incorporation than the ethylene monomer, which is consistent with our previous HOMO-LUMO analysis (
Figure 2b) and reflects their enhanced thermodynamic favorability.
Experimentally, the homogeneous counterpart of the Ni-OH@SiO
2 catalyst demonstrated higher incorporation rates of special polar monomers (SPMs) such as AMA (6.3%), NBA (5.5%), and ACl (3.4%) compared to FPMs (functionalized polar monomers) such as MA (0.6%), with vTMS showing the highest incorporation (10.8%) [
22]. Consistent with these findings, our calculations predict the following order of comonomer incorporation based on Gibbs free energy changes:
vTMS (−9.5 kcal/mol) > ACl (−5.7 kcal/mol) > NBA (−3.9 kcal/mol) > AMA (−3.7 kcal/mol) > MA (−3.5 kcal/mol) > vCl (−3.2 kcal/mol). While NBA, AMA, MA, and vCl exhibit similar ΔG° values, subtle differences in their functional group properties influence their incorporation. For example, variations in inductive effects, electron delocalization, and steric hindrance impact the monomer’s interaction with the catalyst.
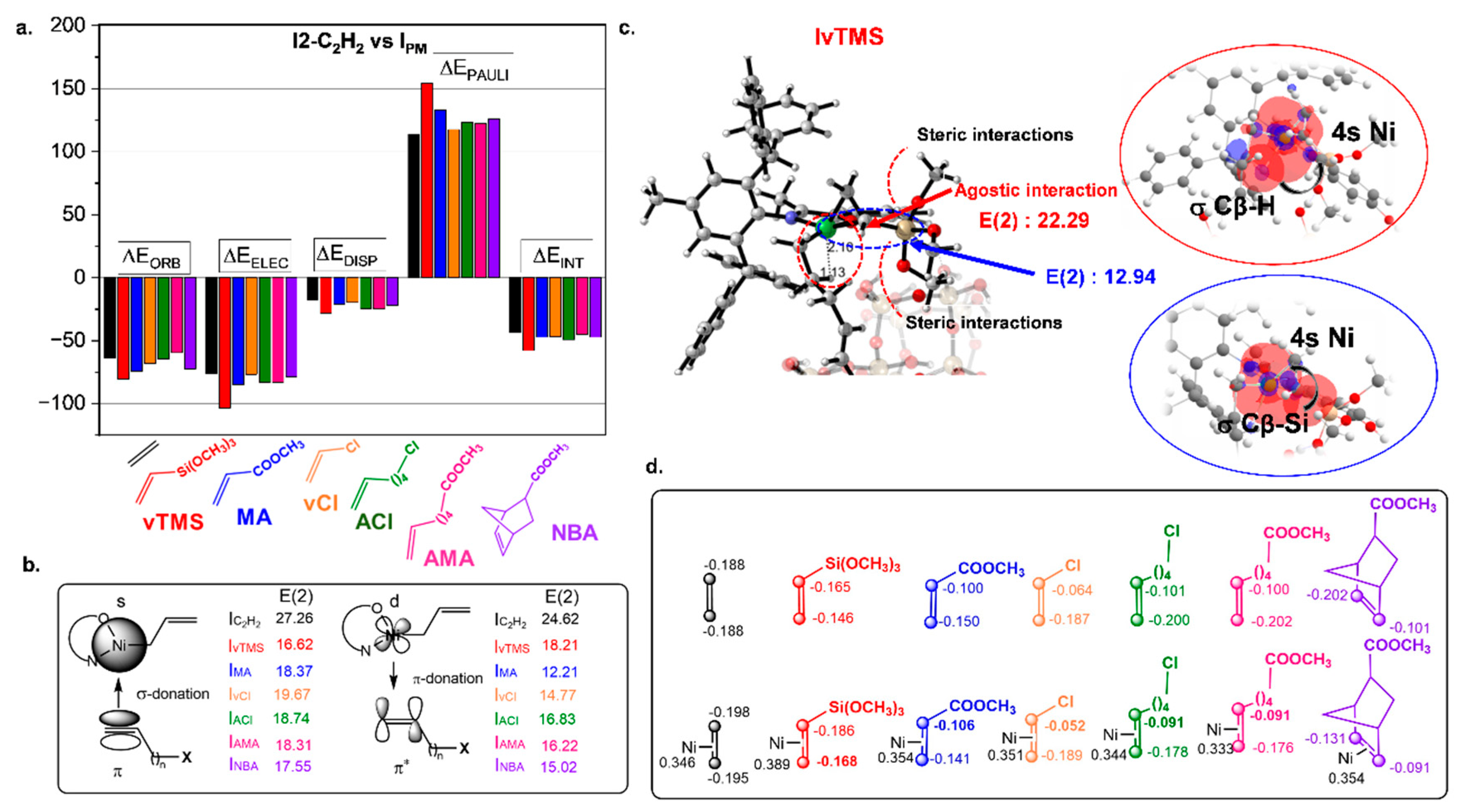
To elucidate the origin of monomer incorporation, we conducted an ALMO-EDA study according to Equation (1), revealing the nature of catalyst-monomer interactions in the π-complexes (
Figure 3a). The I
vTMS complex emerged as the most thermodynamically favorable intermediate, with the highest interaction energy (ΔE
INT = −57.9 kcal/mol). This interaction is predominantly governed by electrostatic (ΔE
ELEC = −103.4 kcal/mol) and orbital (ΔE
ORB = −80.6 kcal/mol) contributions, with Pauli repulsion (ΔE
PAULI = 154.5 kcal/mol) and dispersive effects (ΔE
DISP = −28.4 kcal/mol) playing relatively smaller roles. A similar trend was observed for other PMs, where electrostatic and orbital interactions were the primary contributors to the catalyst–monomer binding.
NBO analysis further supports this conclusion, highlighting donor-acceptor interactions in terms of stabilization energy [E(2)] (
Figure 3b). The polarization of the vinyl groups was assessed through CM5 atomic charges (
Figure 3d), which revealed that the polar groups induce an asymmetric π-electron distribution, as reflected in the following order of polarization: vTMS (Δe = 0.018 e) < MA (Δe = 0.035 e) < NBA (Δe = 0.040 e) < AMA (Δe = 0.085 e) < ACl (Δe = 0.087 e) < vCl (Δe = 0.137 e). For most monomers (e.g., MA, vCl, ACl, AMA, NBA), the vinyl carbons become less negative upon π-complex formation, indicating stronger σ-donation to the nickel center [higher E(2) values for σ-donation compared to π-donation]. This behavior underscores the role of the metal as an electron acceptor, stabilizing the π-complex and priming the monomer for subsequent insertion.
For instance, the highest polarization observed in the IvCl complex is due to stronger σ-donation [E(2) = 19.67 kcal/mol] compared to π-donation [E(2) = 14.77 kcal/mol]. This behavior is attributed to the strong electron-withdrawing inductive effect of chlorine, which reduces electron density on the vinyl carbon and alters the distribution of orbital interactions. The presence of the electronegative chlorine atom enhances the asymmetry of the π-system, increasing its overall polarizability and favoring σ-type interactions with the Ni center. A similar trend is noted in ACl, where the longer spacer weakens the vinyl carbon polarization (0.087 e vs. 0.137 e for vCl) but promotes stronger π-donation [E(2) = 16.83 kcal/mol].
For AMA (Δe = 0.085 e), the ester group contributes moderate electronegativity and polarizability, facilitating electron delocalization. In comparison, MA shows a lower degree of polarization (Δe = 0.035 e) due to its simpler structure. In NBA, the slightly greater polarization (Δe = 0.040 e) relative to MA can be attributed to its rigid structure, which confines electron density and concentrates it around the vinyl and ester groups. This promotes efficient electronic interactions, leading to stronger π-donation from the nickel center to the NBA π* orbital (E(2) = 15.02 kcal/mol) compared to MA (E(2) = 12.21 kcal/mol).
In the I
vTMS complex, an agostic interaction was identified between the nickel center and the β-hydrogen of the already inserted chain (
Figure 3c). This interaction arises from the overlap between the σ Cβ−H orbital and the nickel 4s orbital [E(2) = 22.29 kcal/mol], which contributes to stabilizing the π-complex. However, the observed Ni–Hβ distance of 2.10 Å is significantly longer than typical agostic distances (1.7–1.8 Å), suggesting that the interaction between the monomer and the nickel center partially suppresses optimal charge transfer where the nickel center exhibits high electrophilicity (+0.389 e). In addition to the agostic effect, a stabilizing σ(C–Si) → Ni electron-donating interaction is also observed [
Figure 3c; E(2) = 12.94 kcal/mol], originating from the SiMe
3 substituent of vTMS. While this σ-donation would generally be expected to reduce the electrophilicity of the metal center, it plays a nuanced role in enhancing the overall stability of the π-complex without significantly altering the nickel’s Lewis acidic nature. The bulky and polarizable SiMe
3 group contributes not only via dispersion forces but also by promoting local orbital complementarity through σ-donation, which facilitates a preorganized geometry favorable for subsequent monomer insertion.
Interestingly, the vinyl group of vTMS exhibits an increase in negative charge when forming the π-complex, compared to its isolated monomer state (
Figure 3d). This behavior underscores a critical aspect of the electronic stabilization within the complex. The redistribution of electron density is facilitated by the nickel center, as reflected in a greater E(2) value for π-donation compared to σ-donation (
Figure 3b). The nickel center not only stabilizes the monomer through coordination but also mediates a transfer of electron density from its d-orbital to the π* orbital of the vinyl group. This interaction exemplifies the metal’s dual role: acting as a stabilizing anchor and promoting charge redistribution to strengthen the complex. Unlike polar monomers containing more electronegative or polarizable substituents, the electronic environment of vTMS enables a unique redistribution of charge under the influence of the nickel center. Furthermore, while the agostic interaction is intramolecular (not directly involving vTMS), it may alter the catalyst’s electronic properties. This adjustment could indirectly influence both the stabilization and the charge redistribution mechanisms within the π-complex.
A comparative analysis of the interaction energy components between the π-complexes formed with ethylene and various polar monomers was performed using ALMO-EDA (see
Figure S1). All comonomers exhibited more favorable interaction energies than the ethylene-only π-complex, indicating enhanced thermodynamic stabilization upon coordination. Among the destabilizing factors, Pauli repulsion was found to contribute significantly (32–45%), particularly for MA, NBA, and vTMS, consistent with their bulkier structures or electron-withdrawing groups. These effects contrast with ethylene’s smaller size and symmetrical geometry, which facilitate more effective orbital overlap with the Ni center and reduced steric disruption. A detailed breakdown of these contributions, including electrostatic and orbital effects, is provided in the
Supporting Information (Figure S1).
In summary, vTMS exhibited the highest incorporation and thermodynamic favorability among the studied polar monomers, driven by strong electrostatic and orbital interactions (
Figure 3a). Differences in functional group properties—such as inductive effects and polarizability—significantly influenced coordination strength and complex stability. vTMS, with its electron-rich alkoxy substituents, enhances orbital overlap with the metal, whereas MA, featuring an electron-withdrawing ester group, increases Pauli repulsion and weakens binding, highlighting the contrasting electronic effects that govern monomer–catalyst interactions. Compared to ethylene, the polar monomers introduce distinct electronic and steric characteristics that modulate the stability and binding efficiency of the π-complexes. Ethylene, due to its smaller size and symmetry, exhibits superior orbital overlap with the nickel center, resulting in stronger σ-donation and π-back-donation interactions (
Figure 3b), which facilitates an effective coordination process. In contrast, polar monomers, despite their enhanced electrostatic contributions, experience greater steric hindrance and electronic polarization at the nickel center. The balance between these factors underpins the competitive incorporation of polar monomers in copolymerization processes. Notably, the ability of vTMS to achieve high incorporation despite these challenges highlights its unique electronic and steric properties, making it a promising candidate for functionalized copolymer synthesis.
3.2. Activation Barrier Analysis for Polar Monomer Insertion into Ni-OH@SiO2 Catalyst
The insertion of ethylene as the second monomer (TS2
-C2H2) presents an energy barrier of 15.9 kcal/mol, leading to the formation of an exergonic β-agostic complex, P2
C2H2 (∆G° = −7.1 kcal/mol) as shown in
Figure 1.
Although no experimental evidence currently confirms the insertion mode of all the polar monomers (PMs) studied here on the Ni-OH@SiO
2 catalyst, our conformational sampling studies predict that the 2,1-insertion mode is the most stable pathway, both kinetically and thermodynamically (
Table S1). This preference is consistent with the catalyst’s ability to stabilize transition states involving polar functional groups. Interestingly, while vCl exhibits thermodynamic favorability for product formation via the 1,2-regioselective pathway, this mode is also kinetically preferred, with a barrier approximately 2.0 kcal/mol lower than the 2,1-insertion (
Table S1). The preference for 1,2-insertion with vCl is attributed to the formation of a highly stable chelate complex (β-Cl product; P
vCl in
Figure 1), which leads to a strong interaction between the comonomer and the nickel center.
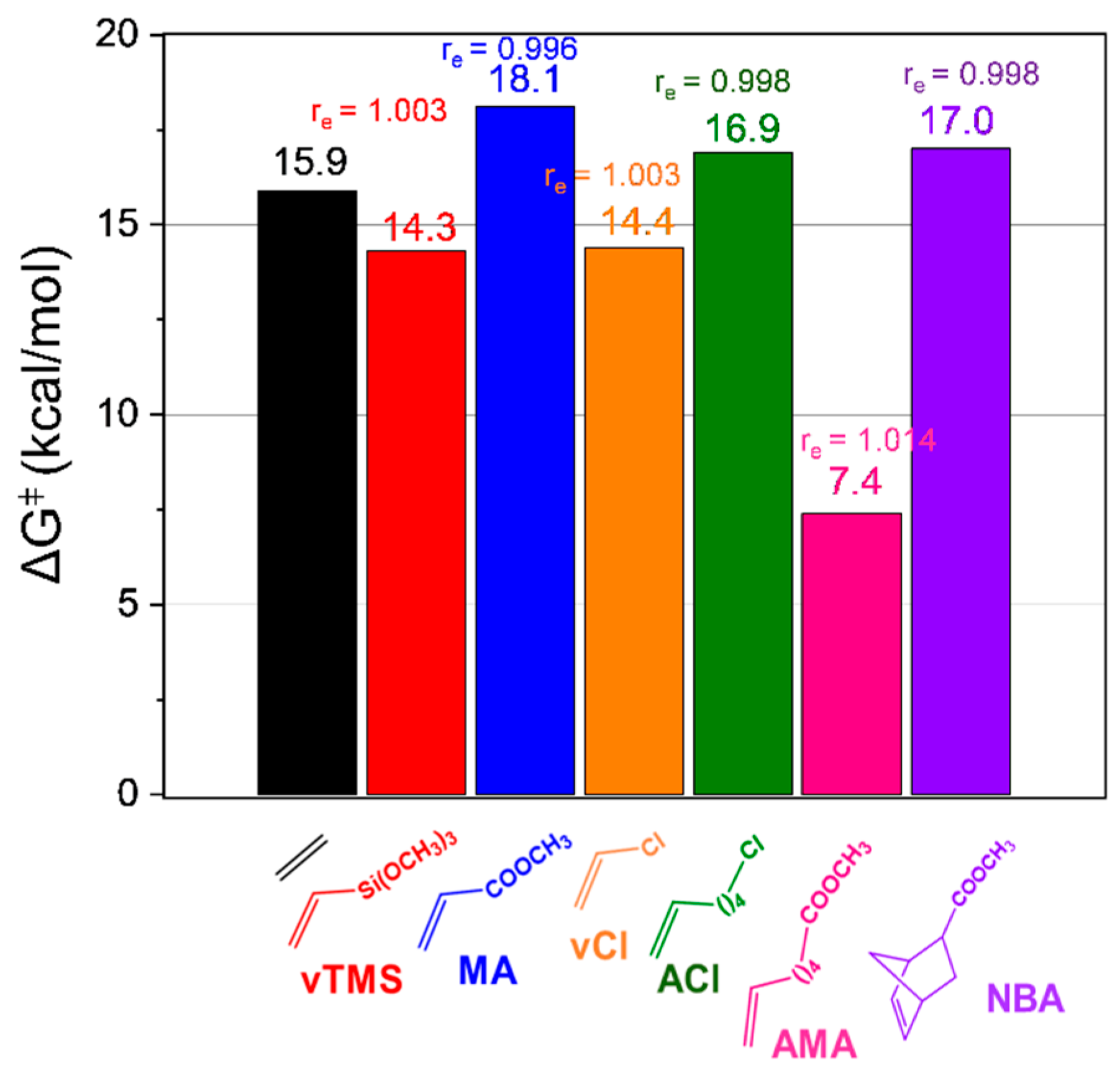
Figure 4 further highlights the activation free energy differences between ethylene homopolymerization and vCl copolymerization. The lower activation free energy for vCl copolymerization (ΔG
ǂ = 14.4 kcal/mol) compared to ethylene homopolymerization (15.9 kcal/mol) indicates that the initial step of copolymerization is more kinetically favorable. This difference can be attributed to the strong electronic interactions between the Cl atom and the nickel. These findings emphasize the inherent trade-offs when incorporating polar monomers into the polymer chain. While polar monomers like vCl exhibit a lower activation barrier, their regioselective behavior and high chelation tendencies may compromise catalyst efficiency and polymer properties. This implies that while the incorporation of vCl is kinetically efficient, the chain growth process may be hindered by competing interactions, such as the stabilization of β-Cl chelate intermediates (P
vCl in
Figure 1).
Figure 4 illustrates that the insertion of the AMA comonomer is both kinetically and thermodynamically more favorable than the homopolymerization pathway. The energy barrier for monomer insertion is notably low at 7.4 kcal/mol, and the process is highly exergonic by −17.6 kcal/mol. This computational prediction aligns well with experimental data for naphthoquinone-based Ni@SiO
2 catalysts, which demonstrate high activity (up to 2.65 × 10
6 g/mol h) during the copolymerization of ethylene with polar comonomers such as 5-hexene acetate [
44] (AMA in this study). The production of high-molecular-weight polymers (Mn up to 630,000) by this catalyst suggests that similar performance can be anticipated for AMA copolymerization on the Ni-OH@SiO
2 catalyst. This strong agreement between experimental and computational results underscores the robustness of the modeling approach in predicting catalyst efficiency and product outcomes [
45,
48].
Nevertheless, the Ni-OH@SiO
2 catalyst demonstrates markedly different behavior when a comonomer with a longer spacer, such as methyl 10-undecenoate, is employed. Experimental evidence indicates complete catalyst deactivation under these conditions. This deactivation is attributed to the strong interactions between the polar comonomer and the hydroxyl groups on the SiO
2 surface, which likely block the active sites of the catalyst, preventing effective polymerization. To gain further insights into this phenomenon, we performed molecular dynamics simulations in the product structure of P
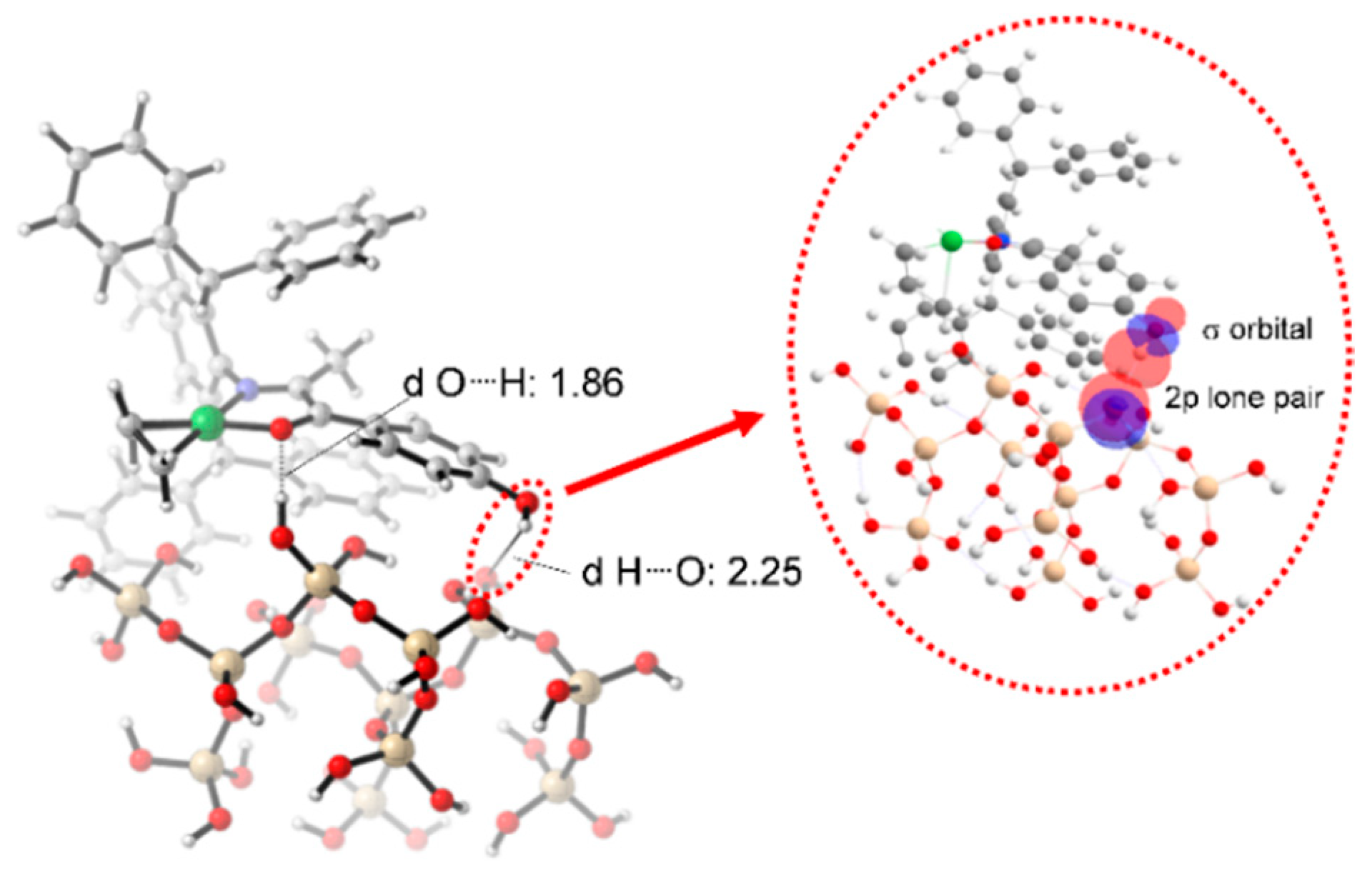
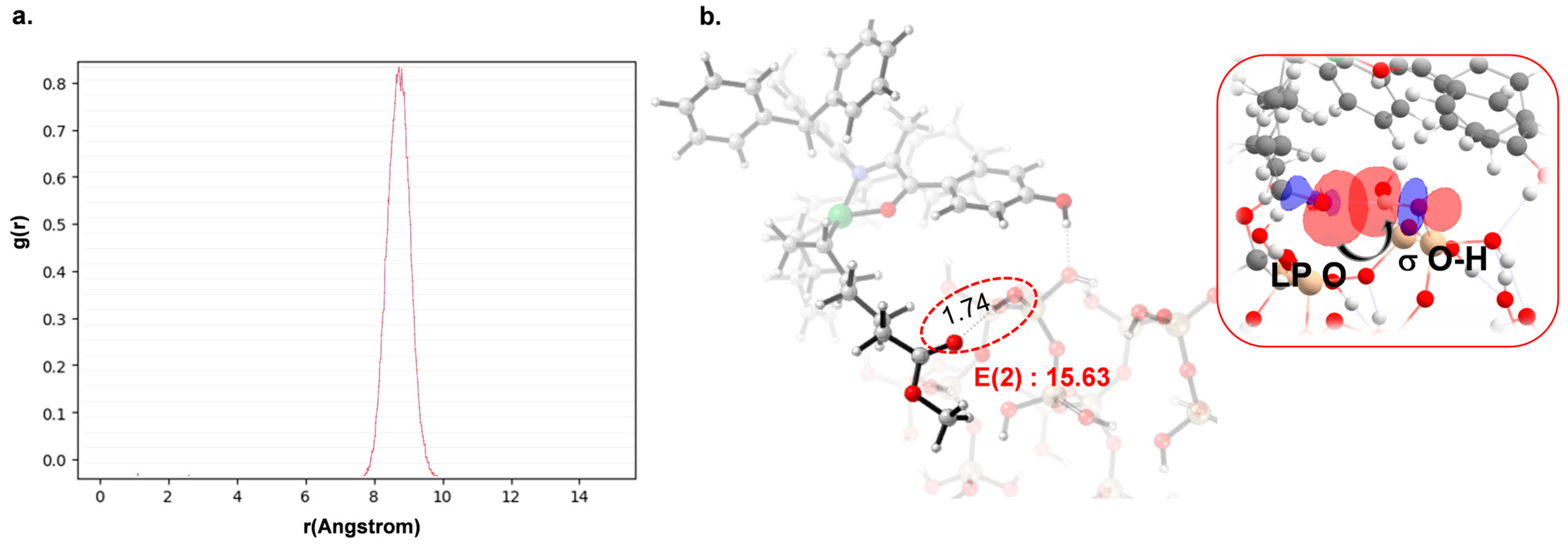
AMA to investigate the nature of the interaction between the AMA polar monomer and the SiO
2 surface (
Figure 5). The Forcite module in BIOVIA Materials Studio [
49] was employed to optimize the system geometry and to conduct the simulations using the Universal Force Field (UFF). Dynamic equilibration was carried out in the NVT ensemble at 373 K for 1000 ps with a time step of 1 fs. This was followed by equilibration in the NPT ensemble for 5 ns. After these steps, a well-equilibrated structure was obtained. The radial distribution function (RDF) between the oxygen atom of the carbonyl group in the ester and the nearest hydrogen atom on the SiO
2 surface was analyzed, as displayed in
Figure 5. The RDF reveals a pronounced peak around 8.5 Å, indicating that this is the most visited distance between O-ester and H-SiO
2 atoms during the simulation. However, the sharp decline in the distribution suggests that this interaction is transient and not persistently structured within the system. For strong interactions, such as hydrogen bonding, a characteristic distance of 1.5–2.0 Å is typically observed. Since no such peak is present, it indicates that there is no strong hydrogen bond formation between O-ester and H-SiO
2. Instead, the interaction appears to be a weak spatial correlation, where these atoms have a high probability of being found at 8.5 Å without forming a stable bond.
Interestingly, NBO analysis in
Figure 5 indicates a stabilization energy of 15.62 kcal/mol between the O-ester and H-SiO
2 atoms, suggesting a significant orbital overlap. However, this interaction does not translate into persistent binding, as evidenced by the fluctuating distances observed in molecular dynamic simulation. The contrast between NBO findings and RDF results suggests that while electronic effects can temporarily stabilize the interaction, thermal fluctuations and dynamic effects prevent long-lasting adsorption. These findings support the hypothesis that the AMA comonomer does not strongly or persistently adhere to the SiO
2 surface, allowing copolymerization to proceed efficiently without significant catalyst deactivation. Moreover, this insight prompts a reassessment of the proposed mechanism of catalyst deactivation observed experimentally with polar monomers containing longer spacers such as 10-undecenoate. Although a significant electronic interaction exists in the product structure, the lack of persistent adsorption under thermal conditions suggests that this factor alone is unlikely to be responsible for catalyst deactivation. Therefore, while our results do not support the strong interaction hypothesis as the cause of deactivation, they open avenues for further investigation into cooperative effects and surface structural reorganizations.
On the other hand, for TS
vTMS, the computational results also indicate a kinetically and thermodynamically favorable insertion compared to ethylene homopolymerization. The activation barrier (∆G
ǂ = 14.3 kcal/mol) and reaction free energy (∆G° = −9.4 kcal/mol) for vTMS insertion are 1.6 and 2.3 kcal/mol lower, respectively, than those for ethylene homopolymerization. These findings suggest that vTMS is well suited for ethylene copolymerization, offering both enhanced catalytic activity and regioselectivity. This prediction aligns with experimental observations [
9] for homogeneous nickel catalysts, further supporting the reliability of our computational results.
On the other hand, comonomers such as ACl, NBA, and MA exhibit moderate activation barriers in the range of 17.0–18.0 kcal/mol, which is higher than the barrier for ethylene homopolymerization. Although these comonomers are kinetically less favorable, the product formation process is more than that of ethylene stable (i.e., more exergonic as is shown in
Figure 1), with ∆G° values of −7.9 kcal/mol for NBA, −7.6 kcal/mol for MA, and −5.0 kcal/mol for ACl, compared to −7.1 kcal/mol for ethylene homopolymerization. The lower exergonicity observed for ACl suggests reduced catalytic activity and the formation of lower-molecular-weight copolymers compared to ethylene homopolymerization. Conversely, the slightly more exergonic processes predicted for NBA and MA suggest that, thermodynamically, these comonomers could yield copolymers with molecular weights comparable to those of ethylene homopolymers. This aligns with experimental evidence [
50] showing that, although nickel-based systems can copolymerize ethylene with acrylates like MA, the resulting polymers typically exhibit lower molecular weights and reduced catalytic activity—limitations that arise from strong electronic interactions and steric hindrance near the nickel coordination site.
In summary, our computational findings demonstrate that the catalytic activity and the resulting polymer properties are strongly influenced by the comonomer’s spacer length, polarity, and insertion energetics. Comonomers such as AMA and vTMS, with lower activation barriers and high thermodynamic stability, are predicted to achieve high incorporation rates and produce high-molecular-weight copolymers. Conversely, comonomers like ACl, NBA, and MA, while thermodynamically favorable, present higher kinetic barriers, potentially limiting their incorporation efficiency. While vCl, their regioselective behavior and high chelation tendencies may compromise catalyst efficiency. These insights are critical for optimizing copolymerization processes, enabling the tailored design of catalysts and monomers for the synthesis of advanced functional materials.
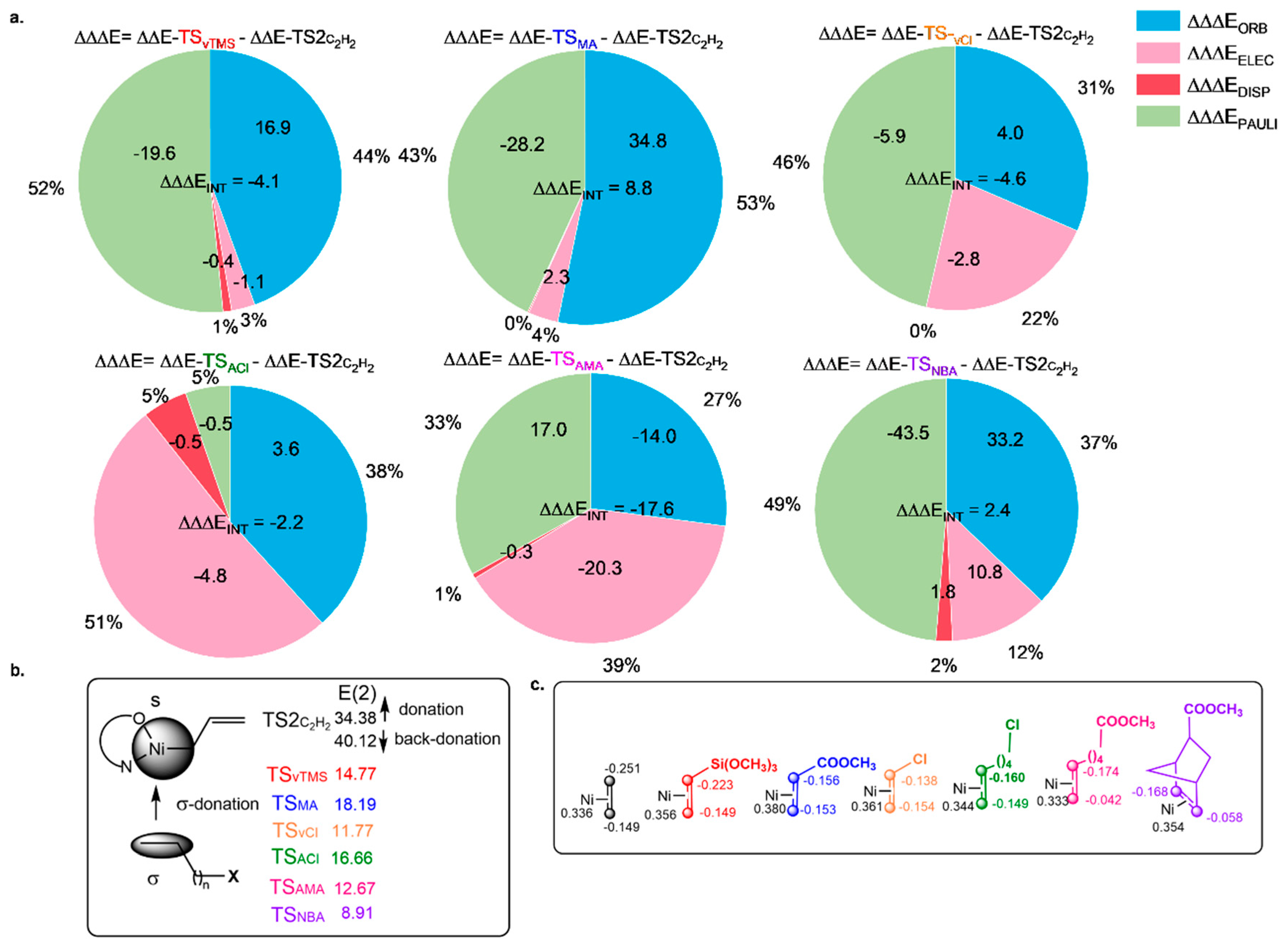
3.3. Nature of Activation Barriers for PM Insertion into Ni-OH@SiO2 Catalysts
To investigate the nature of the energy barriers and the selectivity of the copolymerization with the ethylene homopolymerization reaction, we studied the ALMO-EDA terms on the ground state (π-complex) and transition state structures (∆ΔE
INT = ∆E
TS − ∆E
I) in order to predict the origin of the energy barriers.
Figure 6a highlights the dominant quantitative contributions between the difference between the copolymerization and homopolymerization barriers (∆∆ΔE
INT = Δ∆E
TS-PM − Δ∆E
TS2-C2H2). Positive ΔΔΔE
INT values indicate effects that promote the ethylene polymerization barrier; negative ΔΔΔE
INT values indicate effects that promote the ethylene copolymerization barrier.
In general,
Figure 6a shows that each pie chart includes four different effects on selectivity; among these, it is observed that both types of electronic effects (∆ΔΔE
ORB and ∆ΔΔE
ELEC) are the main stabilizing interactions for all the energy barriers. In contrast, the dispersion interactions are negligible (∆ΔΔE
DISP). Orbital interactions (∆ΔΔE
ORB) are the main effect that promotes the homopolymerization pathway (31–53%), except for the copolymerization with the AMA comonomer, which will be discussed later. Interestingly, in this context, NBO analyses (
Figure 6b) consistently show that the dominant orbital interaction during the transition state corresponds to the σ(C-C) → LP*(Ni) charge transfer with a C–C vinyl bond of around 1.44 Å. This suggests a concerted insertion pathway, analogous in spirit to the Chalk–Harrod mechanism, where the vinyl σ-bond engages directly with a low-lying acceptor orbital on the metal center. The LP* orbital has a significant 4s character, given the geometry and electronic configuration of the Ni(II) center, and serves as a favorable site for charge donation in the absence of strong d–π* back-donation. The prominence of this σ-type interaction, even in sterically or electronically complex systems, indicates a compensatory mechanism: enhanced σ-donation can occur despite steric congestion as the system attempts to stabilize the transition state through orbital realignment. However, this compensation often comes at the cost of increased Pauli repulsion. This is evidenced in comonomers like MA, which exhibits one of the highest NBO stabilization energies (
Figure 6b; E(2) = 18.19 kcal/mol) among the comonomers studied (
Figure 6b). This strong interaction is consistent with the highly electrophilic character of the nickel center in the transition state (
Figure 6c; +0.38 e), which enhances σ-donation from the vinyl group via a strong σ → 4s (Ni) interaction, increasing the Pauli repulsion (
Figure 6a) and the activation barrier. These findings are consistent with previous mechanistic observations by Chen and Brookhart [
9], who reported that polar monomers such as MA and vinyl halides tend to form stable chelates, hindering catalyst turnover. In addition, Zhang et al. [
45] experimentally observed that MA shows limited incorporation into copolymers, even under Pd-SiO
2 supported catalysis, due to its strong electron-withdrawing ester group directly conjugated to the vinyl system, which aligns with our theoretical results.
Similar to MA comonomer, NBA comonomer also exhibits a less stable insertion into the nickel center than the homopolymerization process, which is evidenced by positive ∆∆ΔE
INT values (
Figure 6a). For both comonomers, the Pauli repulsion contribution is the dominant effect (∆∆ΔE
PAULI: 43% for MA and 49% for NBA comonomers), which stems from unfavorable steric and electronic interactions near the nickel center. Interestingly, for NBA comonomer, a lower stabilization energy is observed (
Figure 6b; E(2) = 8.91 kcal/mol), and the charge distribution across the vinyl carbons is highly asymmetric (
Figure 6c; −0.058 e and −0.168 e), which disrupts the electronic balance and reduces the efficiency of orbital interactions with the nickel center. The combined effect of weak orbital stabilization and high Pauli repulsion explains the relatively poor reactivity of NBA in comparison with other polar comonomers. In both cases, MA and NBA, although their electronic profiles differ, the inability to offset destabilizing repulsion through favorable electrostatic or orbital contributions renders their insertion pathways less competitive compared to ethylene. Interestingly, although ethylene is a symmetric molecule in its free state, the vinyl group becomes significantly polarized in the transition state (Δe = 0.102 e). Despite this high polarization, ethylene exhibits the strongest σ → LP(Ni) interaction (
Figure 6b; E(2) = 34.48 kcal/mol) due to its small size, ideal orbital orientation, and lack of steric perturbations. This contrasts with NBA, which also shows high polarization (Δe = 0.110 e) but much weaker orbital stabilization (E(2) = 8.91 kcal/mol), suggesting that the directionality and orbital alignment of the polarized vinyl system are more critical than the absolute magnitude of charge separation alone.
In contrast, ACl, vCl, vTMS, and AMA comonomers exhibit a stronger total interaction energy difference (
Figure 6a; negative ∆∆ΔE
INT values), indicating that the transition state for these comonomer insertions is inherently more stable than that of ethylene. Particularly, although ACl comonomer has a slightly higher activation barrier (ΔG
ǂ = 16.9 kcal/mol) compared to ethylene homopolymerization (ΔG
ǂ = 15.9 kcal/mol), its interaction energy (ΔΔΔE
INT = −2.2 kcal/mol) suggests a more stable transition state. This apparent contradiction can be explained by the need for greater electronic reorganization before insertion (I
ACl species), which increases the initial energy cost. However, once the system reaches the transition state, favorable electrostatic interactions (∆∆ΔE
ELEC: 51%) and reduced Pauli repulsion (∆∆ΔE
PAULI: 5%) stabilize the insertion process, making ACl energetically more favorable overall. This interpretation is further supported by an independent gradient model based on Hirshfeld analysis (IGMH) [
51] (
Figure S2), which reveals significant van der Waals interactions (green isosurfaces) between the ACl monomer and the SiO
2 support and clear electrostatic attraction (blue isosurfaces) between the vinyl group and the Ni center. These non-covalent interactions contribute to the stabilization of the transition state beyond direct orbital interactions, providing a favorable environment that reinforces electrostatic complementarity and minimizes steric repulsion. The involvement of the support highlights the importance of considering the full catalytic system when evaluating monomer–catalyst interactions. This is in contrast to NBA comonomer, which has a similar activation barrier (ΔG
ǂ = 17.0 kcal/mol) but lacks the stabilizing energy terms (
Figure 6a), resulting in a less favorable insertion pathway.
Similar to ACl in structure, vCl exhibits a slightly lower activation barrier (ΔG
ǂ = 14.4 kcal/mol) and lower electrostatic stabilization (∆∆ΔE
ELEC: 22%). The increased Pauli repulsion (∆∆ΔE
PAULI: 46%) arises from the 1,2-insertion mode, which induces spatial proximity between the nickel center and the electron-withdrawing chlorine atom. This interaction perturbs the electronic environment, reducing the efficiency of ΔΔΔE
ORB interactions between the vinyl group and the Ni center (
Figure 6b; E(2) = 11.77 kcal/mol) compared to ethylene. Moreover, as observed with the MA comonomer, the higher positive charge on the nickel center in the vCl transition state (+0.36 e) compared to ethylene (+0.34 e) may indicate a partial donation of electron density from the chlorine atom to the metal. This electron donation enhances the effective Lewis acidity of the nickel center, leading to a strong electrostatic attraction—evidenced by the blue isosurfaces—between the chlorine atom and the nickel (
Figure S2). Altogether, this combination of steric proximity, electronic perturbation, and reduced orbital overlap contributes to a less stabilized transition state despite the modest activation barrier. These findings highlight the importance of designing monomers that prevent post-insertion destabilization, particularly by minimizing unfavorable interactions between polar substituents in FPMs and the metal center.
The insertion of vTMS presents a moderately favorable transition state, with a relative interaction energy of ΔΔΔE
INT = −4.1 kcal/mol. However,
Figure 6a indicates that this stabilization does not arise from stabilizing contributions; ΔΔΔE
ELEC and ΔΔΔE
DISP dispersion terms are only marginally more favorable for vTMS than for ethylene, and ΔΔΔE
ORB is strongly favored for ethylene homopolymerization (
Figure 6b). This apparent discrepancy can be rationalized by the pronounced stabilization of the π-complex in the vTMS system (
Figure 3). Consequently, the overall interaction energy of the transition state appears more favorable, even though the classic stabilizing components are not strongly enhanced.
Lastly, among all the comonomers studied, AMA exhibits the most favorable insertion profile (
Figure 6a). Its energy barrier is highly stabilized, with a ΔΔΔE
INT of −17.6 kcal/mol, and every stabilizing component (ΔΔΔE
ORB, ΔΔΔE
DISP, ΔΔΔE
ELEC) is more favorable than in ethylene (
Figure 6a).
Figure 6b confirms that AMA has the strongest σ(C-C) → LP(Ni) interaction (E(2) = 24.88 kcal/mol) among the polar comonomers. This efficiency is supported by the pronounced polarization of the vinyl group (−0.174 e and −0.042 e), which creates a highly directional donor orbital that aligns well with the vacant orbital on Ni. The presence of an aliphatic spacer between the acetate group and the vinyl moiety minimizes electronic perturbation of the π-system and avoids steric congestion at the metal center. These features combine to produce a remarkably low activation barrier (~7.4 kcal/mol), consistent with experimental observations of high incorporation and molecular weight in AMA-based copolymers [
44]. In contrast, MA places the electron-withdrawing ester group in direct conjugation with the vinyl system, which increases electron density near the nickel center and enhances Pauli repulsion. The superior performance of AMA over its parent monomer MA aligns with the design principle highlighted by Chen and Brookhart [
9], who emphasize the importance of spatially separating the polar group from the reactive vinyl moiety. In AMA, the aliphatic spacer effectively insulates the π-system from electronic perturbations, minimizing Pauli repulsion and enhancing orbital overlap with the nickel center—features reflected in its exceptionally low barrier and high E(2) interaction energy. In addition, the lower charge on the nickel center in the AMA system (
Figure 6c; +0.33 e) reflects reduced electrophilicity, which has been associated with enhanced copolymerization tolerance toward polar groups. This is in agreement with observations in supported Ni@SiO
2 catalysts [
44], where neutral nickel centers enabled higher molecular weights and better performance in ethylene copolymerization with AMA comonomer systems.
In summary, ALMO-EDA analysis in the activation barriers reveals structure–reactivity relationships governing the copolymerization behavior of polar comonomers with nickel catalysts. AMA, with minimal Pauli repulsion, enhanced electrostatic and orbital interactions, and optimal vinyl polarization, surpasses ethylene in favorability. The use of aliphatic spacers in AMA decouples the functional group from the reactive site, promoting efficient interaction with the metal center. These results highlight the importance of tuning both electronic and steric features to design comonomers that not only coordinate well with the catalyst but also promote smooth and favorable insertion pathways.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}